

Abstract
Acute intermittent hypoxia (AIH) induces phrenic long-term facilitation (pLTF) by a mechanism that requires spinal serotonin (5-HT) receptor activation and NADPH oxidase (NOX) activity. Here, we investigated whether: 1) spinal nitric oxide synthase (NOS) activity is necessary for AIH-induced pLTF; 2) episodic exogenous nitric oxide (NO) is sufficient to elicit phrenic motor facilitation (pMF) without AIH (i.e. pharmacologically); and 3) NO-induced pMF requires spinal 5-HT2B receptor and NOX activation. In anesthetised, mechanically ventilated adult male rats, AIH (3×5min episodes; 10% O2; 5min) elicited a progressive increase in the amplitude of integrated phrenic nerve bursts (i.e. pLTF), which lasted 60 min post-AIH (45.1 ± 8.6% baseline). Pre-treatment with intrathecal (i.t.) injections of a neuronal NOS inhibitor (nNOS-inhibitor-1) near the phrenic motor nucleus attenuated pLTF (14.7 ± 2.5%), whereas an inducible NOS (iNOS) inhibitor (1400W) had no effect (56.3 ± 8.0%). Episodic i.t. injections (3×5μl volume; 5mins) of a NO donor (sodium nitroprusside; SNP) elicited pMF similar in time-course and magnitude (40.4 ± 6.0%, 60 min post-injection) to AIH-induced pLTF. SNP-induced pMF was blocked by a 5-HT2B receptor antagonist (SB206553), a superoxide dismutase mimetic (MnTMPyP), and two NOX inhibitors (apocynin and DPI). Neither pLTF nor pMF were affected by pre-treatment with a PKG inhibitor (KT-5823). Thus, spinal nNOS activity is necessary for AIH-induced pLTF, and episodic spinal NO is sufficient to elicit pMF by a mechanism that requires 5-HT2B receptor activation and NOX-derived ROS formation, which indicates AIH (and NO) elicits spinal respiratory plasticity by a nitrergic-serotonergic mechanism.
Keywords: intermittent hypoxia, nitric oxide, respiratory plasticity
1.0 Introduction
Nitric oxide (NO) is critical for many forms of neuroplasticity, including hippocampal LTP (Bliss and Collingridge, 1993), cerebellar LTD (Shibuki and Okada, 1991) and Aplysia long-term sensory motor facilitation (Antonov et al., 2007). NO also plays complex, but poorly understood roles in the neural control of breathing. For example, NO inhibits carotid body chemoreceptor responses to hypoxia (Prabhakar et al., 1993; Chugh et al., 1994; Summers et al., 1999), but excites neurons in the nucleus of the solitary tract where those chemoafferent neurons terminate (Gozal and Gozal, 1999; Gozal et al., 2000; Torres et al., 1997). However, little is known concerning the role of NO in hypoxia-induced respiratory plasticity. Thus, we tested the hypothesis that NO is necessary for phrenic long-term facilitation (pLTF), a form of serotonin-dependent respiratory motor plasticity induced by acute intermittent hypoxia (AIH) (Bach and Mitchell, 1996; Mitchell et al., 2001; Mahamed and Mitchell, 2007; MacFarlane et al., 2008).
Key steps in the mechanism of pLTF include: spinal serotonin receptor activation (Bach and Mitchell, 1996; Fuller et al., 2001; Baker-Herman and Mitchell, 2002; MacFarlane et al., 2011), new synthesis of brain-derived neurotrophic factor (BDNF) and activation of its high affinity receptor, TrkB (Baker-Herman et al., 2004), followed by ERK MAP kinase signalling (Hoffman et al., 2012; Figure 7). Other molecules regulate pLTF, including NADPH oxidase (NOX; MacFarlane et al., 2008, 2009) and serine-threonine protein phosphatases (Wilkerson et al., 2008; MacFarlane et al., 2008). These molecules constitute a “regulatory cassette” for pLTF (Dale-Nagle et al., 2010).
Figure 7.
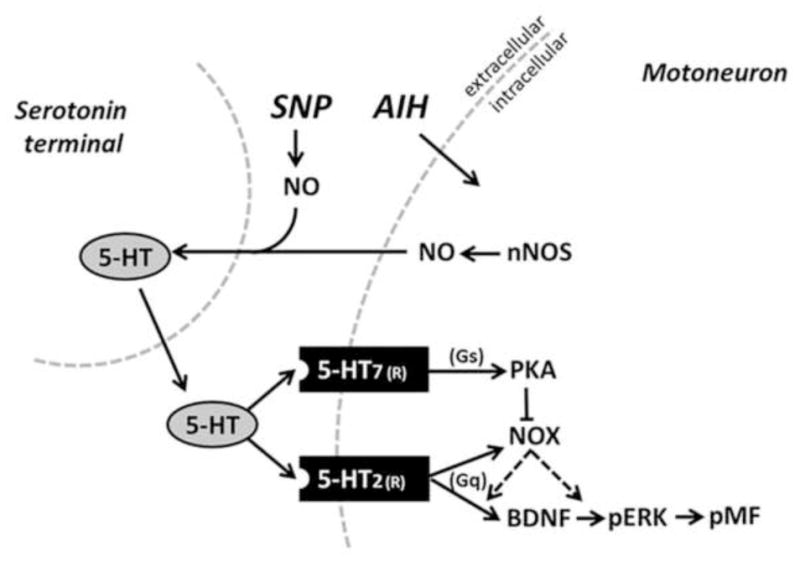
Schematic of proposed signaling mechanisms involved in pMF. AIH stimulates spinal nNOS, increasing NO formation, which could lead to an increase in 5-HT release from serotonin terminals and extracellular 5-HT accumulation. Following activation of the Gq-coupled 5-HT2 receptor, NOX-derived ROS formation could then function to either increase BDNF synthesis or ERK phosphorylation (pERK) leading to pMF At high NO concentrations (via the NO donor SNP), greater 5-HT accumulation activates the less abundant Gs-coupled 5-HT7 receptors on phrenic motor neurons, thereby activating PKA. PKA could inhibit NOX activity via a mechanism of “cross-talk inhibition”, and ultimately inhibits pMF. Thus, we hypothesize that the key role of NOS/NO in AIH-induced pLTF is through regulation of serotonin release and accumulation in the extracellular space.
Pre-conditioning with chronic intermittent hypoxia (CIH) enhances phrenic (Ling et al., 2001) and ventilatory LTF (McGuire et al., 2004) by a serotonin-dependent mechanism; however, it is not known if enhanced pLTF results from central vs peripheral mechanisms. CIH reveals a novel form of carotid chemosensory long-term facilitation (Peng et al., 2003), amplifies central neural integration of chemoafferent inputs (Ling et al., 2001) and strengthens spinal synaptic pathways to phrenic motor neurons (Fuller et al., 2003). Thus CIH preconditioning elicits both peripheral chemosensory and central neural plasticity. Episodic serotonin receptor activation elicits chemosensory LTF by a NOX-dependent mechanism (Peng et al., 2006). Similarly, episodic spinal serotonin receptor activation (particularly 2B receptors) elicits phrenic motor facilitation (pMF) by a NOX-dependent mechanism (MacFarlane et al., 2009; 2011). Thus, carotid chemosensory and spinal respiratory plasticity result from similar cellular mechanisms after CIH pre-conditioning.
CIH decreases carotid body neuronal nitric oxide synthase (nNOS) expression (Marcus et al., 2010), and AIH-induced ventilatory LTF is attenuated in nNOS knock-out mice (Kline et al., 2002). Further, NO triggers serotonin release in the central nervous system (Harkin et al., 2003; Inan et al., 2004; Bryan-Lluka et al., 2004). Thus, NO may be a critical regulator of AIH-induced pLTF. To determine the role of NO in pLTF, we tested the hypotheses that: 1) spinal nNOS activity is required for pLTF; 2) episodic NO release (via sodium nitroprusside; SNP) is sufficient to elicit pMF without AIH; and 3) that SNP-induced pMF requires spinal 5-HT2B receptor activation and NOX activity.
2.0 Experimental procedures
Experiments were performed on 3–4 month old male Sprague Dawley rats (Harlan, colony 218A). All experiments were approved by The Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin-Madison.
2.1 Surgical preparation
Rats were briefly anesthetized with isoflurane, tracheotomized and pump ventilated (normalized for each rat to 7.5 ml/kg tidal volume and 70 breaths/min ventilator frequency; Rodent Ventilator, model 683; Harvard Apparatus, South Natick, MA, USA). Isoflurane anesthesia was maintained (3.5% in 50% O2, balance N2) for the duration of the surgical procedure, followed by conversion to urethane anesthesia (1.8 mg/kg) via a tail vein catheter. The adequacy of anesthesia depth was confirmed by the lack of response to a toe-pinch given prior to paralysis via an injection of pancuronium bromide. Surgery was performed on a custom-made temperature-controlled stainless-steel surgical table connected to an adjustable water bath (Isotemp 1006S, Fisher Scientific, Pittsburgh, PA, USA) to maintain body temperature constant. Rectal temperature was monitored continuously with a temperature probe (Fisher Scientific, Pittsburgh, PA, USA), and maintained constant by adjusting the temperature of the water bath. The concentration of inspired O2 was monitored throughout experiments using a fuel-cell O2 sensor (TED 60T, Teledyne Analytical Instruments, CA, USA). A tail vein catheter (24 gauge, Surflo, Elkton, MD, USA) was inserted to allow delivery (1.5–2ml/hr; Cole-Palmer, Vernon Hills, IL, USA) of fluids consisting of a 1:1 lactated Ringers:hetastarch solution to assist in maintenance of blood pressure (6% Hetastarch; Hospira Inc., IL, USA) and base excess (Lactated Ringers, Baxter, IL, USA). A small amount (1:20) of sodium bicarbonate (8.4% Hospira Inc., IL, USA) was also added to the infusion solution. Rats received an initial 1ml intravenous injection of lactated ringers over a 5 minute period to minimize early changes in base excess.
Rats were vagotomized bilaterally and a polyethylene catheter (PE50 ID: 0.58mm, OD: 0.965mm; Intramedic, MD, USA) was inserted into the right femoral artery to monitor blood pressure (Gould Pressure Transducer, P23, USA). Blood samples were analyzed for partial pressure of O2 (PO2) and CO2 (PCO2) and pH with a blood gas analyzer (ABL 800, Radiometer, Copenhagen, Denmark); base excess, calculated by the analyzer, was used as an indicator of metabolic acid-base status.
The left phrenic and hypoglossal (XII) nerves were dissected and exposed via a dorsal approach, cut distally, and de-sheathed. Nerves were submerged in mineral oil and placed on bipolar silver recording electrodes. Once electrical signals were detected, the rats were assessed for adequate depth of anesthesia by checking for transient increases in blood pressure and/or respiratory neural output following toe-pinch. Rats were then paralyzed with pancuronium bromide (~1.2 ml i.v., 1mg/ml). The rats received another toe-pinch to test for anesthetic depth immediately prior to and at the end of the experimental protocol. We did not observe increased blood pressure or respiratory nerve activity in any of the rats, consistent with previous studies demonstrating the efficacy of urethane anesthesia for many hours longer than the duration of our experimental protocols (Maggi and Meli, 1986). End tidal CO2 was monitored using a flow-through capnograph (Novametrix, model 1265, Wallingford, CT, USA) and maintained ~40–45mmHg for 1hr to allow stabilization of the preparation and nerve signals. Nerve activity was amplified (gain, 10,000; A-M systems, Everett, WA, USA), bandpass-filtered (100 Hz to 10 kHz), rectified and integrated (CWE 821 filter; Paynter, Ardmore, PA, USA; time constant, 50msec). The signal was then digitized and recorded using WINDAQ data acquisition system (DATAQ Instruments, Akron, OH, USA). Data analysis was performed using custom designed software based on a Labview platform (LabVIEW, National Instruments, Austin, TX, USA).
Depending on the experiment, 1 or 2 silicone intrathecal catheters enabled localized injections of various pharmacological agents (see below) into the CSF of the cervical spinal cord. In brief, after dorsal C2 laminectomy an incision was made in the dura and catheter(s) (2 French, ID: 0.3mm, OD: 0.6mm; Access Technologies, Skokie, IL, USA) were inserted and advanced ~2mm caudally. The tip(s) of the catheter(s) were positioned proximal to the region just rostral (~C3) to the cervical sections containing the phrenic motor nucleus. Prior to insertion of the catheter(s), they were primed with the drug of interest (or their vehicle) using a 25μl glass micro-syringe (Hamilton Co., Reno, NV, USA). A 2μl air bubble occupied the tip of the catheter to create a barrier between its contents and the CSF to prevent unintended premature leakage/diffusion into the CSF. All drugs were prepared as outlined below.
2.2 Neurophysiological experiments
Stable nerve activity was established while the rat was ventilated with a hyperoxic inspired gas mixture (FIO2 ~ 0.50; PaO2, >200 mmHg), and sufficient levels of inspired CO2 to maintain arterial PCO2 (PaCO2) above apnea (typically between 40–45mmHg). After nerve recordings had stabilized, the apneic threshold for phrenic and XII activity was determined by progressively lowering the inspired CO2 until fictive breathing had ceased. End-tidal PCO2 (which approximates the arterial level) was monitored and then progressively increased in 1mmHg increments every 1–2 minutes until nerve activity resumed (i.e. the recruitment threshold). Baseline nerve activity was established with PaCO2 set 2 mmHg above the CO2 recruitment threshold. This procedure allows a standardized level of baseline respiratory drive for each rat.
Rats were separated into the following experimental treatment groups that received: 1) 3×5min hypoxic episodes (~10% O2) with 5min intervals of baseline conditions (i.e. ~50% O2); 2) bolus (12μl) i.t. injections of non-selective (L-NAME, 10mM) and selective nitric oxide synthase inhibitors (iNOS: 1400W, 10mM; nNOS: nNOS-inhibitor-1, 100μM) prior to AIH; 3) episodic i.t. injections of the NO donor sodium nitroprusside (SNP: 3×5μl, 1μM - 1mM); 4) AIH pre-treated with a PKG inhibitor (KT-5823, 1mM); 5) Episodic SNP pre-treated with KT-5823; 6) episodic SNP pre-treated with the 5-HT2B (SB206553, 300μM) and 5-HT7 receptor antagonist (SB-269970, 5mM); and 7) episodic SNP pre-treated with the superoxide dismutase mimetic (MnTMPyP, 5.5mM), or the NOX inhibitors apocynin (100μM) and DPI (1mM). Additional rats were used as time controls to test for effects of vehicle and drugs. Injections were given after a stable baseline nerve activity and 20 minutes prior to AIH or episodic SNP.
Approximately 20 min post-injection, a baseline blood sample was taken and the rat was exposed to AIH (or episodic SNP, see below) and nerve activity was monitored for a further 60 min while maintaining baseline levels of arterial blood gases. Blood samples (60μl heparinized capillary tube; Radiometer, Copenhagen, Denmark) were analyzed before (baseline), during the first hypoxic episode, and at 15, 30 and 60min post-AIH to ensure that blood gases met the criteria outlined below. Blood gas values at the various time points are given in Table 1. In rats that received episodic i.t. injections of the NO donor, SNP, blood samples were taken prior to the first injection, and also at 15, 30 and 60min post-injections. At the end of the experiment the rat received a lethal dose of urethane via the tail vein.
Table 1.
Body temperature (Tb), blood gas values and integrated phrenic nerve burst amplitude (during acute hypoxia) for i.t treated rats.
| Group | Tb | PaO2 | PaCO2 | pH | MAP | Integrated Phrenic amplitude (%) | |
|---|---|---|---|---|---|---|---|
| Baseline | AIH + vehicle | 37.5 ± 0.2 | 301.8 ± 9.9 | 46.6 ± 0.5 | 7.337 ± 0.015 | 109.0 ± 3.3 | 0 |
| AIH + L-NAME | 37.8 ± 0.2 | 288.5 ± 6.4 | 47.2 ± 1.2 | 7.326 ± 0.007 | 111.8 ± 4.8 | 0 | |
| AIH + nNOS-1 | 37.6 ± 0.1 | 286.0 ± 7.2 | 47.2 ± 0.7 | 7.344 ± 0.009 | 105.1 ± 2.6 | 0 | |
| AIH + 1400W | 37.5 ± 0.2 | 310.6 ± 13.0 | 47.3 ± 0.5 | 7.355 ± 0.001 | 104.6 ± 6.3 | 0 | |
| Hypoxia | AIH + vehicle | 37.4 ± 0.2 | 36.7 ± 1.5* | 46.4 ± 0.7 | 7.330 ± 0.013* | 84.5 ± 6.1* | 110.8 ± 16.0* |
| AIH + L-NAME | 37.7 ± 0.2 | 42.0 ± 1.5* | 47.6 ± 1.2 | 7.304 ± 0.010* | 72.9 ± 12.8* | 62.8 ± 8.4*# | |
| AIH + nNOS-1 | 37.5 ± 0.1 | 36.9 ± 1.5* | 48.0 ± 0.6 | 7.322 ± 0.009* | 75.2 ± 6.5* | 80.6 ± 10.7*# | |
| AIH + 1400W | 37.4 ± 0.2 | 38.7 ± 0.9* | 47.3 ± 0.9 | 7.324 ± 0.007* | 79.6 ± 14.8* | 106.6 ± 27.5* | |
| 60 mins | AIH + vehicle | 37.4 ± 0.2 | 280.2 ± 7.0 | 47.0 ± 0.7 | 7.336 ± 0.013 | 90.6 ± 3.9 | N/A |
| AIH + L-NAME | 37.8 ± 0.2 | 268.2 ± 9.0 | 47.4 ± 1.0 | 7.319 ± 0.006 | 96.5 ± 6.6 | N/A | |
| AIH + nNOS-1 | 37.6 ± 0.1 | 277.5 ± 6.3 | 47.3 ± 0.7 | 7.338 ± 0.009 | 86.0 ± 2.9 | N/A | |
| AIH + 1400W | 37.5 ± 0.2 | 292.9 ± 11.9 | 47.2 ± 0.8 | 7.351 ± 0.007 | 87.7 ± 7.8 | N/A |
Values are means ± 1SEM.
significant difference from baseline;
significant difference from AIH+vehicle treated group at the respective time point; p<0.05. Tb, body temperature; PaO2 and PaCO2, arterial pressure of oxygen and carbon dioxide, respectively; MAP, mean arterial pressure; amplitude of integrated phrenic nerve burst amplitude expressed as a % change from baseline nerve burst amplitude.
2.3 Drugs
The following drugs were used: apocynin, DPI, 1400W, sodium nitroprusside and KT 5823 (Sigma-Aldrich, MO, USA); SB206553 (Cayman Chemicals MI, USA); L-NAME and MnTMPyP (Alexis, PA, USA), nNOS-inhibitor-1 (Calbiochem, CA, USA), SB269970 and KT 5720 (Tocris Bioscience, MO, USA). All drugs (except sodium nitroprusside, which was dissolved in aCSF) were dissolved in dimethylsulphoxide (DMSO) and then diluted in artificial cerebral spinal fluid (aCSF) equating to 20% DMSO in aCSF or less, depending on the solubility of individual drugs. Aliquots of some drugs in DMSO were occasionally frozen and used the following day after being thawed and diluted in freshly made aCSF. aCSF was always bubbled for at least an hour with a gas mixture consisting of 95% O2 and 5% CO2.
2.4 Experimental protocols
To test whether NOS was necessary for AIH-induced phrenic LTF, rats received a 12μl i.t. injection of L-NAME (10mM; N=6; non-specific inhibitor), 1400W (10mM; N=6; iNOS inhibitor), or nNOS inhibitor-1 (100μM; N=7; nNOS inhibitor) 20min prior to AIH. In other rats (N=5), only the vehicle (12μl of 20% DMSO in aCSF) was injected prior to AIH as a control experiment to test for the effects of vehicle on phrenic nerve activity. There were no differences in the magnitude of pLTF following AIH between vehicle and untreated rats (N=4), so these rats were combined to form one group (i.e. N=9 total). A separate time control (TC, i.e. without AIH) was also performed for each inhibitor (N=3 total) to confirm that neither one by itself affected persistent spontaneous nerve activity.
To test whether the NO donor SNP could elicit pMF without AIH, rats received episodic i.t. injections (3×5 μl at 5min intervals) of SNP at the following concentrations: 1μM (N=5), 10 μM (N=5), 100 μM (N=8) or 1 mM (N=5). Other rats (N=2) served as a control group in which they received episodic i.t. injections of the vehicle for SNP (3×5μl injections of aCSF). A separate group of rats (N=5) was pre-treated with an i.t. bolus injection (1mM; 12μl) of the PKG inhibitor KT 5823 to determine whether PKG activity was necessary for SNP-induced pMF. In another group of rats (N=5), we determined if PKG activity is necessary for AIH-induced pLTF. A time control (TC) rat (N=1) which received only the PKG inhibitor was also used to test for effects of this drug alone (data not shown).
Since SNP-induced pMF was not PKG-dependent, we determined if SNP-induced pMF required 5-HT2B receptor activation. Rats (N=6) received an i.t. injection (12μl, 300μM) of the 5-HT2B receptor antagonist, SB206553, 20min prior to episodic SNP (3×5μl, 100μM). At the highest concentration of SNP (i.e. 1mM), pMF was no longer observed, which we propose was due to 5-HT7 receptor activation (MacFarlane et al., 2009). Thus, a group of rats that received episodic (3×5μl) injections of 1mM SNP were pre-treated with the 5-HT7 receptor antagonist, SB 269970 (12μl, 5mM, N=6) to determine if pMF could be restored. Rats were also pre-treated with the PKA inhibitor KT5720 prior to episodic i.t. injections of 1mM SNP to determine if restoration of SNP-induced pMF involved PKA activity. Neither the PKA inhibitor or 5-HT7 receptor antagonist alone had any effect on spontaneous or long-lasting phrenic nerve activity.
To test whether SNP-induced pMF required ROS and NOX activity, rats were pre-treated with the SOD mimetic, MnTMPyP (5.5mM, 12μl; N=6), or the NOX inhibitors apocynin (12μl, 600μl; N=6) and DPI (12μl, 1mM; N=6). Time controls were also performed to test for the effects of each drug alone. In all experiments, neither the NOS inhibitors, 5-HT receptor antagonists, PKG inhibitor, NADPH oxidase inhibitors, nor vehicle injections by themselves affected baseline phrenic nerve activity. None of these drugs (or their vehicles) affected baseline respiratory burst frequency, XII nerve burst amplitude or blood pressure, consistent with pharmacologically active drug distribution restricted to the cervical spinal cord.
Throughout the experimental protocol, arterial blood gases and blood pressure were monitored and corrected as necessary to ensure that they were maintained near baseline levels. PaCO2 was adjusted by manipulating inspired CO2. Decreases in base excess were corrected gradually by adjusting the rate of i.v. infusion (solution containing hetastarch, sodium bicarbonate and lactated ringers solution, see above). Data from rats were included in the analysis only if they complied with the following criteria: (1) PaO2 during hypoxia was between 35 and 45 mmHg; (2) PaO2 during the hyperoxic baseline and recovery periods was >180 mmHg; and (3) PaCO2 remained within 1 mmHg of baseline at each sampling time point (see Table 1).
2.5 Immunohistochemistry
Immunohistochemistry was performed on 4 rats injected with the retrograde tracer Cholera toxin B fragment (CtB, List Biologicals, Campbell CA, USA) to identify phrenic motor neurons in the cervical spinal cord. Each rat received an intrapleural injection of CtB for retrograde labeling of spinal phrenic motor neurons using a technique similar to that described previously (Mantilla et al., 2009; MacFarlane et al., 2011). In brief, each rat was anaesthetized with isoflurane (~1.5% in O2), placed supine on a surgical table while breathing into a face mask. The lateral sides of the rib cage were shaved and gently palpated in order to identify the fifth intercostal space at the anterior axillary line. A sterilized 27G needle attached to a 50μl Hamilton syringe was used to inject 25μl of a 0.2% CtB (dissolved in sterile injectable saline) between the ribs into the pleural space on one side and another 25μl on the other. The needle was modified so that it could be inserted no further than 6mm below the surface of the skin. After the injections, the animals were monitored closely for signs of respiratory distress associated with pneumothorax. Isoflurane was discontinued and the rats were allowed to recover and monitored for a further 30 minutes. All rats recovered well and showed no sign of respiratory distress.
At 3 days post-injection, the rats were again anesthetized with isoflurane, euthanized (0.3ml Beuthanasia-D, i.c., Schering-Plough, NJ. USA) and transcardially perfused with heparinized saline (4 IU/ml) followed by 4% paraformaldehyde solution (freshly made in 0.1M phosphate buffer, pH adjusted to 7.4). The brain and cervical spinal cord were removed and stored in 4% PFA overnight at 4°C, then transferred to a cryoprotectant (20% sucrose in PBS) until it lost buoyancy (typically overnight). Serial coronal sections (30μm thick) were prepared using a freezing microtome (Leica SM2000R) and stored in a cryoprotectant solution (30% ethylene glycol, 30% glycerol in PBS 0.1M). Sections were prepared for double labeling with CtB and nNOS. For double labeling with CtB and the target molecule of interest, free floating sections were washed with PBS 0.1M, incubated in a blocking solution (5% normal donkey serum, 0.2% tween 20 in PBS for 30 min) and incubated overnight at 4°C with goat anti CtB antibody (1:4000, Calbiochem) and rabbit anti-nNOS (1:1000, Upstate #07-571, Billerica, MA, USA). After 3×5min washes, the sections were incubated with a secondary antibody (Donkey anti-goat, Alexa 594 1:2000 for CtB; donkey anti-rabbit Alexa 488 1:500for nNOS; Molecular probes, Eugene, OR, USA) at room temperature for 2hr 30min. The sections were washed several times in PBS and mounted on slides (superfrost) using an anti-fade solution (Prolong Gold antifade reagent, Invitrogen, OR, USA). Negative controls in which primary or secondary antibodies were excluded from the incubation period were also performed, including pre-absorption using a peptide (nNOS) to confirm specificity of the primary antibody.
The sections were examined using a confocal microscope (Leica SPE) with Leica acquisition software. Z-stacks were taken (30μm thick, 2μm steps) and images were rendered with the max intensity finished using the Z project from ImageJ Software.
2.6 Statistical analysis
Peak integrated nerve amplitude and frequency (bursts per minute) of phrenic and XII nerve activity were averaged in 1min bins at each recorded data point (baseline, 15, 30 and 60 minutes time points post-episodic injections (SNP) or AIH. Values were expressed as a % change from baseline. For changes in phrenic and XII nerve activity, within rat comparisons were made with baseline; whereas comparisons were also made between TC treated rats at all time points. Statistical comparisons were made for time and drug treatment using two-way, repeated measures ANOVA. The Bonferroni post hoc test was used to identify statistically significant individual comparisons. Differences were considered significant at p < 0.05. All values are expressed as mean ± 1 SEM.
3.0 Results
3.1 nNOS, but not iNOS is necessary for AIH-induced pLTF
AIH elicits a progressive increase in the amplitude of integrated phrenic nerve activity (i.e. pLTF; Figure 1A). At 60 minutes post-AIH, phrenic burst amplitude was 39.7 ± 8.6% above baseline, and 49.4 ± 8.4% in a separate group of rats that received a single i.t. bolus injection of vehicle (12μl of aCSF) 20 min prior to AIH. Since there was no difference between these groups, their results were combined (control AIH: 45.1 ± 8.6% at 60 min post-AIH; versus time controls, TC: 5.5 ± 4.4%; Figure 1A; p < 0.05). pLTF was blocked by pre-treatment with L-NAME (16.8 ± 4.2%; 10mM) and nNOS-inhibitor-1 (14.7 ± 2.5%; 100μM), whereas 1400W (56.3 ± 8.0%; 10mM) had no effect compared to control AIH treated rats. Representative traces of each treatment group are shown (Figure 1B–E).
Figure 1.

AIH-induced pLTF (A) following pre-treatment with NOS inhibitors. Representative neurograms of individual rats treated with (B) vehicle, (C) L-NAME (10mM; a non-selective NOS inhibitor), (D) 1400W (10mM; selective for iNOS), and (E) nNOS-1 (100μM; selective for nNOS). AIH consisted of 3×5 min hypoxic (~10% O2) episodes at 5 min intervals. Injections were given 15 min prior to baseline blood sampling; the first hypoxic episode was administered after 5 min additional baseline recording. L-NAME and nNOS-1 inhibited AIH-induced pLTF, whereas the iNOS inhibitor had no effect. *significant difference from baseline; #significant difference from AIH-treated groups 60 min post-AIH (p<0.05). Prior to each neurogram are 10 sec expanded recordings illustrating integrated breaths. Grey regions in B and D illustrate phrenic nerve activity exceeding baseline post-AIH. Open arrows indicate time of injections; solid arrows indicate blood sampling; horizontal dashed arrows at end of traces indicates baseline amplitude. Note, that time control (TC) rats were not exposed to AIH, but did receive intrathecal injections NOS inhibitors (N=3).
Although neither of the NOS inhibitors affected baseline integrated phrenic nerve activity, L-NAME (62.8 ± 8.4%) and nNOS-inhibitor-1 (80.6 ± 10.7%) significantly attenuated the magnitude of the short-term hypoxic phrenic response (Table 1; 110.8 ± 16.0%) whereas 1400W had no effect (106.6 ± 27.5%) compared to control AIH treated rats. None of the inhibitors affected the magnitude of the XII response to hypoxia (data not shown), confirming that the drug effects were localized to the cervical spinal cord. PaCO2 was maintained constant for all treatment groups (Table 1), and they were equally hypoxemic during AIH). Baseline blood gas values were the same for each group (Table 1), and they were equally hypotensive during AIH.
3.2 Episodic intrathecal injections of sodium nitroprusside (SNP) elicits dose-dependent pMF
Episodic i.t. injections (3×5μl, 5min intervals) of the NO donor SNP (without AIH) near the phrenic motor nucleus caused a progressive increase in the amplitude of integrated phrenic nerve activity (Figure 2A) lasting at least 60 minutes post-injection (Figure 2C). The magnitude of SNP-induced pMF was dose-dependent, increasing only up to 100μM (40.4 ± 6.0% above baseline; Figure 2A). XII nerve activity was unaffected (Figure 2B) by SNP, although there was a non-significant trend at the highest dose (1mM) tested. There was no SNP effect on baseline phrenic or XII nerve activity (Figure 2C). Episodic i.t. injections of vehicle (i.e. aCSF; time control, TC) did not cause pMF (Figure 2A and D). Further, episodic SNP at the highest dose (1mM), failed to elicit pMF (4.4 ± 9.7%; Figure 2A).
Figure 2.
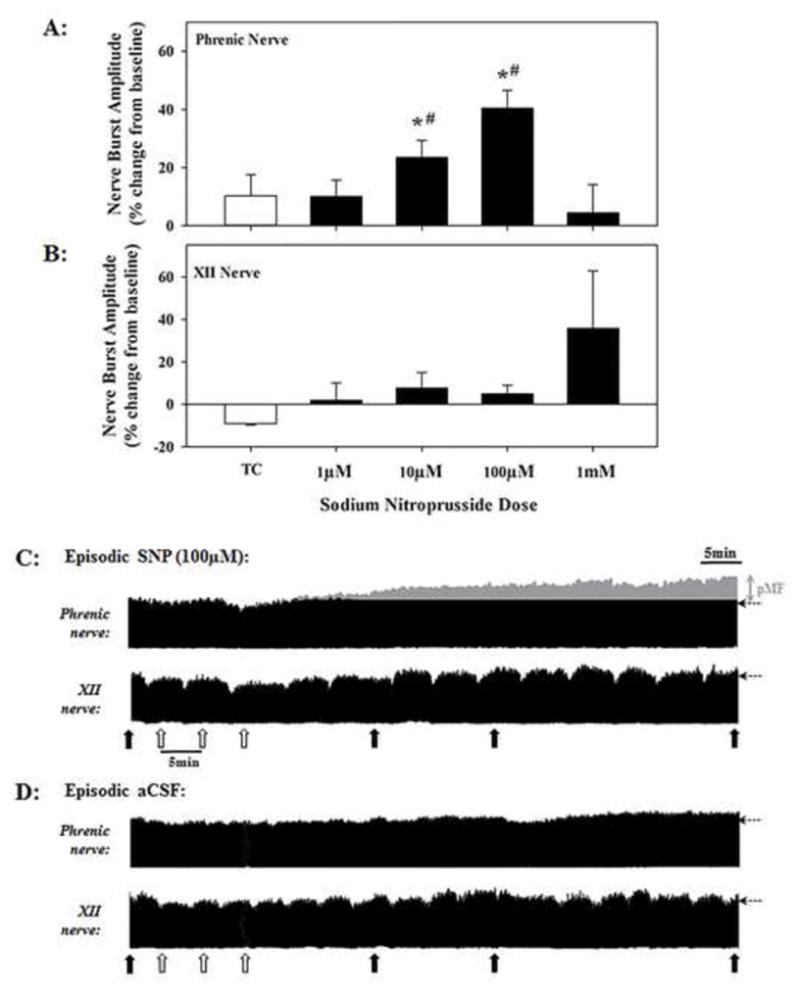
Dose-dependent effects of episodic intrathecal injections (3×5μl, 5 min intervals) of the nitric oxide (NO) donor sodium nitroprusside (SNP) on phrenic (A) and hypoglossal (XII) (B) nerve activity. Injections delivered to CSF via catheter placed near the phrenic motor nucleus (~C4–C5). Solid bars in A and B represent average values of integrated phrenic and XII nerve activity at 60 min post-SNP injections SNP-induced pMF occurred after 10μM injections, reaching a peak at 100μM. The highest SNP dose (1mM) failed to elicit pMF. *indicates significant difference from vehicle (i.e. time control, TC); #indicates significant difference from 1mM treated rats. Representative phrenic and XII neurograms from individual rats that received episodic intrathecal injections of (C) 100μM SNP or (D) vehicle (aCSF). In C and D, open arrows indicate timing of injections; solid arrows indicate blood sampling; horizontal dashed arrows at the end of each trace indicate baseline amplitude; grey region illustrates phrenic nerve activity exceeding baseline after SNP. (i.e. pMF). There was no SNP effect on XII nerve activity.
3.3 SNP-induced pMF requires differential activation of 5-HT2B and 5-HT7 receptors
At the SNP dose that elicited maximal pMF (100μM), pre-treatment with an i.t. injection of the 5-HT2B receptor antagonist, SB206553 (300μM), blocked pMF (6.9 ± 6.0%) (Figure 3A and C). In contrast, at the highest dose of SNP (1mM) in which pMF was no longer present (Figure 3D and E), pre-treatment with an i.t. bolus injection of the 5-HT7 receptor antagonist, SB269970 (5mM) restored pMF (46.7 ± 10.6%, Figure 3D and F). Finally, pre-treatment with the PKA inhibitor, KT 5720, prior to 1mM episodic SNP also restored pMF (37.6 ± 6.9%; Figure 3D). Neither the 5-HT2B (11.5 ± 3.0%, TC, Fig. 3A), or the 5-HT7 receptor antagonists or the PKA inhibitor alone (13.4 ± 5.2%, TC, Fig. 3D) affected integrated phrenic nerve activity at any time.
Figure 3.
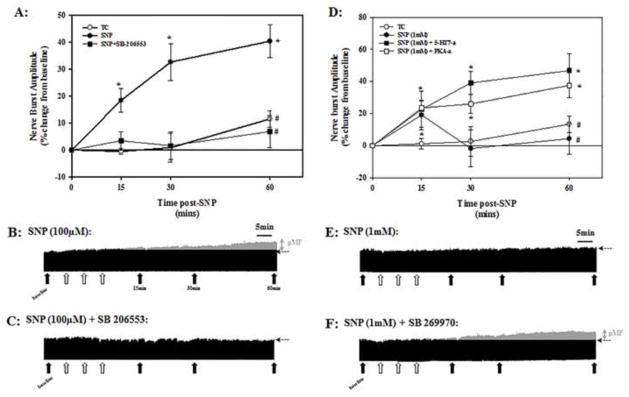
Effects of selective 5-HT2B and 5-HT7 receptor antagonists on pMF following episodic intrathecal (i.t.) SNP. Pre-treatment with the 5-HT2B receptor antagonist (SB 206553; 300μM) blocked SNP-induced (100μM) pMF (A); neurograms from individual rats are shown in B and C. SB 206553 was injected via a second i.t. catheter 20 min prior to episodic SNP (3×5ul injections; 5 min intervals). Additional experiments were performed to determine if the absence of pMF after 1mM episodic SNP resulted from 5-HT7 receptor activation and PKA activity (D). Values in A and D are averaged integrated phrenic nerve burst amplitude at 15, 30 and 60 min post-SNP. Grey areas in each neurogram (B and F), represent nerve activity above pre-SNP baseline. Solid arrows indicate blood sampling; open arrows indicate individual SNP injections. *indicates significant difference from TC at respective time post SNP; #indicates significant difference from episodic SNP group at 60 min post-SNP (p<0.05). Time controls (TC) in (A) are rats that only received an i.t. injection of the 5-HT2B receptor antagonist prior to episodic vehicle, and (B) a combined group of rats that only received the 5-HT7 or PKA antagonist prior to episodic vehicle.
3.4 SNP-induced pMF requires spinal ROS formation and NADPH oxidase activity
Pre-treatment with the SOD mimetic MnTMPyP (5.5mM i.t.) or the NOX inhibitors apocynin (600μM, i.t.) or DPI (1mM, i.t.) prior to episodic 100μM SNP injections prevented pMF (Figure 4). Phrenic nerve amplitude 60min post-SNP in MnTMPyP (-3.1 ± 8.0%), apocynin (8.3 ± 4.9%) and DPI (15.5 ± 2.7%) pre-treated rats were also not significantly different from TC-treated rats (7.8 ± 3.7%).
Figure 4.
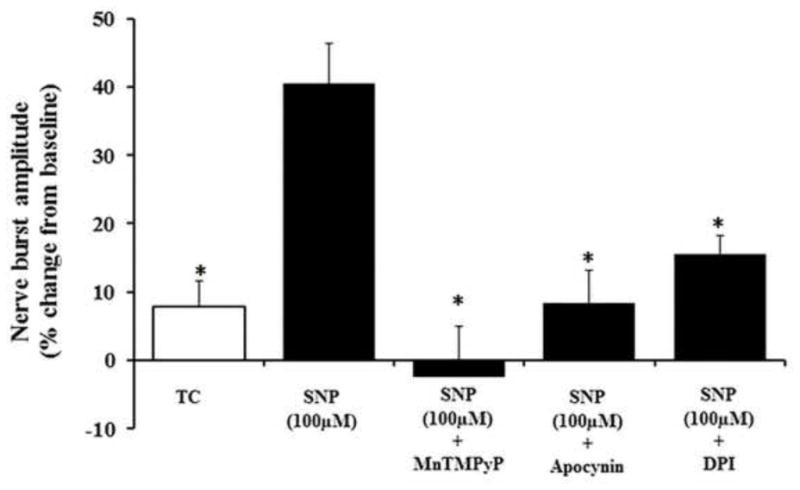
Effects of antioxidant or NOX inhibitor pre-treatment on SNP-induced pMF. Rats were pre-treated before episodic SNP (100μM) with the superoxide dismutase mimetic, MnTMPyP, or the NADPH oxidase inhibitors, apocynin and DPI. Values are means ± SEM at 60 min post-episodic SNP; *indicates significant difference from episodic SNP-induced pMF.
3.5 PKG activity is not necessary for either AIH-induced pLTF or SNP-induced pMF
Pre-treatment with an i.t. injection of the PKG inhibitor, KT-5823 (1mM), prior to AIH or episodic SNP (100μM) had no effect on pLTF or pMF (49.1 ± 11.3% and 35.9 ± 9.3%, respectively; Figure 5A and B). KT-5823 alone had no effect on phrenic nerve activity (data not shown).
Figure 5.
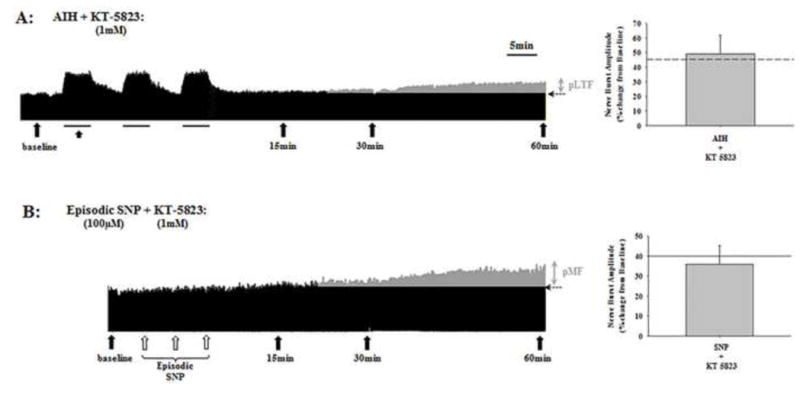
Representative neurograms of AIH-induced pLTF (A) and SNP-induced pMF (B) following pre-treatment with the PKG inhibitor, KT-5823. To the right of each neurogram is the mean average of pLTF and pMF at 60 min post-hypoxia or SNP, respectively; for comparison, dashed (A) or solid (B) lines in the bar graphs illustrate magnitude of AIH-induced pLTF and SNP-induced pMF, respectively. Filled arrows indicate blood sampling; open arrows in B indicate individual SNP injections; horizontal dashed arrows at the end of each trace indicate baseline nerve activity. Grey areas in A and B indicates nerve activity above baseline after AIH or SNP injections. PKG inhibition had no effect on AIH-induced pLTF or SNP-induced pMF.
3.6 nNOS immunoreactivity in retrogradely labeled phrenic motor neurons
Retrogradely labeled phrenic motor neurons (Cholera toxin-B immuno-positive) were identified in clusters in the ventral-lateral gray matter in coronal sections of ~C4. A representative section indentifying CtB-positive phrenic motor neurons co-labeled with nNOS is provided in Figure 6. nNOS labeling was punctate both within and around the phrenic motor neurons (Figure 6A–C)
Figure 6.
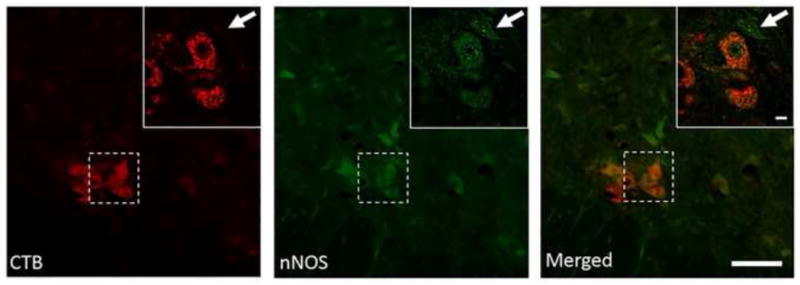
Immunohistochemical analysis of nNOS expression (green) in cholera toxin-B (CTB) labeled phrenic motor neurons (red) in the ~C4 spinal cord. Note the expression nNOS within CTB-positive neurons (double-labeled) and also neighboring cells (arrow). Low magnification also shows diffuse expression in the phrenic motor nucleus region; a higher magnification (inset) also shows expression specifically within phrenic motorneurons. Lower magnification represents are shown as a Z-stack (16 images, 2μm steps, rendered as max intensity; scale bar = 100μm) whereas higher magnification of the region of interest (inset) are a one stack image taken in the middle of the Z-stack to highlight nNOS expression within the cytoplasm (Scale bar = 10μm).
4.0 Discussion
Here, we showed that: 1) spinal nNOS is necessary for AIH-induced pLTF; 2) episodic SNP (and presumably NO) is sufficient to elicit pMF; 3) PKG, a canonical downstream effector of NO signaling, is not necessary for SNP-induced pMF or AIH-induced pLTF; 4) SNP-induced pMF requires 5-HT2B receptor activation and NOX activity; and 5) high SNP doses cause 5-HT7 receptor activation which constrains pMF via cross-talk inhibition between competing pathways. Collectively, these data demonstrate that complex nitrergic mechanisms regulate pLTF, and that these mechanisms are both 5-HT- and NOX-dependent. We suggest that NO increases extracellular 5-HT concentrations, thereby triggering 5-HT-induced pMF (MacFarlane and Mitchell, 2009).
4.1 nNOS, but not iNOS is necessary for AIH-induced pLTF
NOS is involved in multiple forms of neuroplasticity, including hippocampal LTP (Bliss and Collingridge, 1993; Son et al., 1996), cerebellar long term depression (Shibuki and Okada, 1991) and 5-HT-dependent long-term sensory-motor facilitation in Aplysia (Antonov et al., 2007). NO also modulates peripheral (Prabhakar et al., 1993; Chugh et al., 1994; Summers et al., 1999) and central (Gozal and Gozal, 1999; Gozal et al., 2000; Torres et al., 1997) mechanisms of respiratory control; specifically, NO inhibits carotid body O2 sensitivity (Prabhakar et al., 1993; Chugh et al., 1994; Summers et al., 1999), but is excitatory to neurons in the nucleus of the solitary tract (Gozal and Gozal 1999; Gozal et al., 2000; Torres et al., 1997). Although these reports provide insights concerning neuromodulatory actions of NO on respiratory control during hypoxia, limited evidence is available linking NO and respiratory plasticity. nNOS inhibition blunts AIH-induced ventilatory LTF (vLTF) in unanesthetized mice; further, vLTF is abolished in nNOS knock-out mice (Kline et al., 2002). Sympathetic nerve activity during normoxia and acute hypoxia is increased in adult rats following CIH, an effect associated with reduced carotid body nNOS expression (Marcus et al., 2010). Although NOS is clearly involved in peripheral chemo-sensation and neuroplasticity, its role in CNS respiratory plasticity is unknown. Thus, our finding that spinal nNOS (but not iNOS) inhibition blocks pLTF demonstrates for the first time that spinal nNOS is necessary for this form of respiratory plasticity. To determine if NO is sufficient (without AIH) for long-lasting pMF, we locally applied the NO donor SNP to the cervical spinal cord.
4.2 Episodic intrathecal SNP injections elicit pMF
Episodic intrathecal exogenous SNP application elicited pMF for 60 minutes post-injection. SNP-induced pMF was phenotypically similar to AIH-induced pLTF (c.f. Figure 1B & 3C), demonstrating that episodic NO formation is sufficient to elicit pMF. The magnitude of SNP-induced pMF was dose-dependent and increased up to 100μM, but then decreased at the highest dose tested (1mM; Figure 2A). This dose-response curve is consistent with previous findings that episodic intrathecal 5-HT or 5-HT2A receptor agonist (DOI) injections also elicits a similar response curve (MacFarlane and Mitchell, 2009; MacFarlane et al., 2011). Similarly, episodic 5-HT2B receptor agonist (BW723C86) injections also elicited pMF in a progressively increasing dose-response, but failed to reveal a bell-shaped curve (MacFarlane et al., 2001). In the current study, pre-treatment with a 5-HT2B receptor antagonist (SB206553) prevented SNP-induced pMF, suggesting a requirement for spinal NO-5-HT coupling. We also confirmed that SNP-induced pMF requires spinal NOX activity (Figure 4), again similar to AIH-induced pLTF (MacFarlane et al., 2009) and 5-HT-induced pMF (MacFarlane and Mitchell, 2009; MacFarlane et al., 2011). Thus, NO elicits pMF by a mechanism that requires (concurrent) 5-HT receptor activation and NOX-derived ROS formation. Similarities between mechanisms of AIH-induced pLTF and 5-HT- and SNP-induced pMF lead us to suggest that NO signalling regulates spinal 5-HT release, thereby eliciting AIH-induced pLTF (Figure 7).
Since the small intrathecal injections of drugs used in this study did not change phrenic burst frequency or XII nerve activity, pMF is unlikely to have resulted from unintended drug spread to brainstem respiratory neurons. However, while intrathecal L-NAME or nNOS-inhibitor-1 injections (Table 1) did not affect spontaneous phrenic nerve activity, they attenuated phrenic responses to hypoxia, suggesting that NOS activity modulates bulbo-spinal synaptic transmission during hypoxia. Although we are uncertain how NOS/NO modulate descending respiratory drive, it may modulate the release of multiple neurotransmitters (e.g. glutamate and 5-HT), and it has been shown to modulate neuronal activity in the nTS during acute hypoxia (Gozal and Gozal, 1999; Gozal et al., 2000; Torres et al., 1997).
4.3 Opposing serotonergic mechanisms during SNP/NO-induced pMF
Similarities in the dose-response curves of SNP and 5-HT (MacFarlane and Mitchell, 2009) may reflect similar 5-HT-dependent mechanisms. The lack of pMF at higher 5-HT concentrations results from coincident activation of 5-HT receptor subtypes that promote (5-HT2) or undermine (5-HT7) pMF (MacFarlane and Mitchell, 2009). Since SNP-induced pMF requires 5-HT2 receptor activation, we determined if the lack of pMF at the highest dose of SNP also arises from coincident 5-HT7 receptor activation. At the highest SNP dose, which failed to elicit pMF (Figure 3B), 5-HT7 receptor inhibition restored pMF (Figure 3), indicating that the magnitude of pMF arises from competition between 5-HT receptors. This interesting interaction is most likely an expression of “cross-talk” inhibition between pathways to pMF (MacFarlane and Mitchell, 2009; Dale-Nagle et al., 2010). The capacity to elicit spinal hyperalgesia also varies with the availability of NO (Aley et al., 1998). Although NO-dependent neuromodulation often involves PKG activity (Straub et al., 2007) we found no apparent role for PKG in SNP-induced pMF or AIH-induced pLTF.
nNOS/NO may elicit pMF by regulating extracellular 5-HT levels. nNOS and NO inhibits 5-HT uptake (Bryan-Lluka et al., 2004; Chanrion et al., 2007; Harkin et al., 2003; Inan et al., 2004) and the function of 5-HT1 auto-receptors on raphe neurons (Chiavegatto and Nelson, 2003; Pitsikas et al., 2005). Both effects should increase extracellular 5-HT levels and 5-HT receptor activation. Thus, low NO levels may favor pMF via 5-HT2 receptor activation whereas higher levels cause greater extracellular 5-HT accumulation, activating the lower affinity, less abundant 5-HT7 receptors that constrain pMF (Figure 7; Hoffman and Mitchell, 2008). Further work is necessary to determine how nNOS/NO affect 5-HT accumulation, receptor activation and downstream signaling cascades that give rise to pMF.
NOX is inversely regulated by PKC and PKA activity (Klann and Thiels, 1999) and LTP requires ROS activation of PKC (Klann et al., 1998). PKC activity is necessary for AIH-induced pLTF (M. Devinney and G.S. Mitchell, unpublished) and is a downstream effector of metabotropic Gq protein-coupled 5-HT receptors (eg. 5-HT2). On the other hand, PKA is a downstream effector of metabotropic Gs protein-coupled 5-HT receptors (e.g. 5-HT7) and inhibits NOX activity (Kim et al., 2007). Thus, since PKA inhibition restored pMF at the highest SNP dose tested (Figure 3D), we propose that NO-induced pMF (and AIH-induced pLTF) depends, in part, on the level of NOX activity established by the balance of activating (PKC) and inhibiting (PKA) protein kinases.
4.4 Sources of NO
Although phrenic motor neurons express nNOS, and may be the relevant cellular source of NO for pLTF, diffuse expression around and presence within non-CtB labeled neighboring cells does not allow a clear distinction. In sensory-motor facilitation of the gill-withdrawal reflex in Aplysia (Antonov et al., 2007) and the excitatory response of B4 neurons in Lymneae stagnalis to 5-HT (Straub et al., 2007), the relevant NO originates from interneurons instead of the sensory or motoneurons that express the plasticity. One hypothesis concerning AIH-induced nNOS activation is via increased NMDA receptor modulation of intracellular Ca2+, which in turn can modulate NOS activity. NMDA receptors also increase Ca2+-dependent nNOS activation (Garthwaite, 2008; Garthwaite, 1991). Hippocampal LTP requires post-synaptic NMDA receptor activation and increased intracellular Ca2+ (Nicoll and Malenka, 1999; Kandel, 2000), and NMDA receptor activation is necessary for AIH-induced pLTF (McGuire et al., 2005; Ling, 2008). Since iNOS is Ca2+-insensitive (Knowles, 1997; Stuehr, 1999), and plays no apparent role in AIH-induced pLTF (Figure 1), Ca2+ induced nNOS activity most likely elicits pLTF. Finally, since episodic SNP elicits pMF (Figure 2), then increased nNOS activation likely occurs during AIH, thereby inducing pLTF. Future experiments are necessary to determine a role of Ca2+ in AIH-induced pLTF, including possible links between NMDA receptor-activation and nNOS activity. In addition, further experiments are crucial to aid in our understanding of which cells in specific are responsible for generating the relevant NO necessary for pLTF. Thus, we can’t rule out the possibility that spinal inter neurons (or other cell types), for example, may also play an important role in pLTF since they also seem to contain nNOS.
4.5 Conclusions
Spinal nNOS activity is necessary for AIH-induced pLTF. We speculate that NO regulates pLTF by modulating extracellular 5-HT accumulation near phrenic motor neurons. Thus, NO exerts dose-dependent regulation of the balance between opposing serotonergic receptors and their downstream kinases (5-HT2B/PKC vs. 5-HT7/PKA). Further work is necessary to determine whether nNOS plays a role in pLTF metaplasticity (Ling et al., 2001). Collectively, our data reveal a nitrergic-serotonergic interaction that gives rise to AIH-induced pLTF. This interaction is important for an understanding of the role played by NO in other forms of 5-HT-dependent neuroplasticity.
Highlights.
Neuronal nitric oxide synthase is necessary for phrenic long-term facilitation (pLTF) following acute intermittent hypoxia.
Episodic spinal NO is sufficient to elicit facilitation of phrenic nerve activity (pMF).
NO-induced pMF involves 5-HT2B receptors and spinal NADPH oxidase activity.
Acknowledgments
Supported by NIH HL80209. Vinit was supported by the C. Neilsen Foundation and MacFarlane by a Parker B. Francis Foundation Fellowship. We thank Brad Hodgeman for technical assistance, Safraaz Mahamed for his custom designed data analysis software, and Benoît Maury from the CYMAGES platform for its help with the confocal microscope.
Glossary
- NO
nitric oxide
- NOS
nitric oxide synthase
- NOX
NADPH oxidase
- BDNF
brain-derived neutrophic factor
- ROS
reactive oxygen species
- pLTF
phrenic long-term facilitation
- pMF
phrenic motor facilitation
- AIH
acute intermittent hypoxia
- SNP
sodium nitroprusside
- CIH
chronic intermittent hypoxia
- 5-HT
serotonin
- XII
hypoglossal
- C3–4
cervical spinal section 3–4
- i.t
intrathecal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aley KO, McCarter G, Levine JD. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J Neurosci. 1998;18(17):7008–7014. doi: 10.1523/JNEUROSCI.18-17-07008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov I, Ha T, Antonova I, Moroz LL, Hawkins RD. Role of nitric oxide in classical conditioning of siphon withdrawal in Aplysia. J Neurosci. 2007;27(41):10993–101002. doi: 10.1523/JNEUROSCI.2357-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman T, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22(14):6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7(1):48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–91. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bryan-Lluka LJ, Papacostas MH, Paczkowski FA, Wanstall JC. Nitric oxide donors inhibit 5-hydroxytrptamine (5-HT) uptake by the human 5-HT transporter (SERT) Brit J Pharmacol. 2004;143:63–70. doi: 10.1038/sj.bjp.0705904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanrion B, Mannoury la Cour C, Bertaso F, Lerner-Natoli M, Freissmuth M, Millan MJ, Bockaert J, Marin P. Physical interaction between the serotonin transporter and neuronal nitric oxide synthase underlies reciprocal modulation of their activity. Proc Natl Acad Sci USA. 2007;104(19):8119–8124. doi: 10.1073/pnas.0610964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavegatto S, Nelson RJ. Interaction of nitric oxide and serotonin in aggressive behavior. Horm Behav. 2003;44(3):233–241. doi: 10.1016/j.yhbeh.2003.02.002. [DOI] [PubMed] [Google Scholar]
- Chugh DK, Katayama M, Mokashi A, Bebout DE, Ray DK, Lahiri S. Nitric oxide-related inhibition of carotid chemosensory nerve activity in the cat. Respir Physiol. 1994;97(2):147–156. doi: 10.1016/0034-5687(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–66. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Glutamate, nitric oxide and cell-cell signaling in the nervous system. Trends Neursci. 1991;14(2):60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27(11):2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Gozal E. Episodic hypoxia enhances late hypoxic ventilation in developing rat: putative role of neuronal NO synthase. Am J Physiol Reg Int Comp Physiol. 1999;276:17–22. doi: 10.1152/ajpregu.1999.276.1.R17. [DOI] [PubMed] [Google Scholar]
- Gozal D, Gozal E, Simakajornboon N. Signaling pathways of the acute hypoxic ventilatory response in the nucleus tractus solitarius. Respir Physio. 2000;121(2–3):209–221. doi: 10.1016/s0034-5687(00)00129-8. [DOI] [PubMed] [Google Scholar]
- Harkin A, Connor TJ, Walsh M, St John N, Kelly JP. Serotonergic mediation of the antidepressant-like effects of nitric oxide synthase inhibitors. Neuropharmacol. 2003;44(5):616–623. doi: 10.1016/s0028-3908(03)00030-3. [DOI] [PubMed] [Google Scholar]
- Hoffman MS, Mitchell GS. Spinal PKA inhibits phrenic long-term facilitation following acute intermittent hypoxia. Soc for Neurosci Program No: 571.8 2008 [Google Scholar]
- Hoffman MS, Nichols NL, MacFarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation, but not TrkB synthesis. J Appl Physiol. 2012;113(8):1184–1193. doi: 10.1152/japplphysiol.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inan SY, Yalcin I, Aksu F. Dual effects of nitric oxide in the mouse forced swimming test: possible contribution of nitric oxide-mediated serotonin release and potassium modulation. Pharmacol Biochem Behav. 2004;77(3):457–464. doi: 10.1016/j.pbb.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Kandel ER. Cellular mechanisms of learning and the biological basis of individuality. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. New York: McGraw-Hill; 2000. pp. 1247–1279. [Google Scholar]
- Kim J-S, Diebold BA, Babior BM, Knaus UG, Bokoch GM. Regulation of NOX1 activity via protein kinase A-mediated phosphorylation of NoxA1 and 14–3–3 binding. J Biol Chem. 2007;282(48):34787–34800. doi: 10.1074/jbc.M704754200. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: effects of ketanserin and 5-carboxamidotryptamine. Am J Physiol. 1999;277(3 Pt 2):R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273(8):4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23(3):359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short term potentiation in breathing. J Physiol. 2002;539:309–315. doi: 10.1113/jphysiol.2001.014571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles RG. Nitric oxide biochemistry. Biochem Soc Trans. 1997;25(3):895–901. doi: 10.1042/bst0250895. [DOI] [PubMed] [Google Scholar]
- Ling L. Serotonin and NMDA receptors in respiratory long-term facilitation. Respir Physiol & Neurobiol. 2008;164(1–2):233–241. doi: 10.1016/j.resp.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21(14):5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neurosci. 2008;152(1):189–97. doi: 10.1016/j.neuroscience.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Wilkerson JER, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol & Neurobiol. 2008;164:263–271. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587(Pt 22):5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Satriotomo I, Windelborn JA, Mitchell GS. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J Physiol. 2009;587(9):1931–1942. doi: 10.1113/jphysiol.2008.165597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neurosci. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi CA, Meli A. Suitability of urethane anesthesia for physiopharmacological investigations. Part 3: Other systems. Experientia. 1986;42(5):531–537. doi: 10.1007/BF01946692. [DOI] [PubMed] [Google Scholar]
- Mahamad S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnea? Exp Physiol. 2007;92(1):27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182(2):244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Li YL, Bird CE, Schultz HD, Morgan BJ. Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Resp Physiol Neurobiol. 2010;171(1):36–45. doi: 10.1016/j.resp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Integr Comp Physiol. 2004;286(2):R334–R341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Phrenic long-term facilitation requires NMDA receptors in the phrenic motonucleus in rats. J Physiol. 2005;567(Pt. 2):599–611. doi: 10.1113/jphysiol.2005.087650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by endogenous central serotonin. Respir Physiol. 1980;42:171–188. doi: 10.1016/0034-5687(80)90113-9. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1998;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body of intermittent hypoxia: implications for recurrent apneas. PNAS. 2003;100(17):10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576(1):289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsikas N, Tsitsirigou S, Zisopoulou S, Sakellaridis N. The 5-HT1A receptor and recognition memory. Possible modulation of its behavioural effects by the nitrergic system. Behav Brain Res. 2005;159(2):287–293. doi: 10.1016/j.bbr.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Chang CH, Agani FH, Haxhiu MA. Nitric oxide in the sensory function of the carotid body. Brain Res. 1992;625(1):16–22. doi: 10.1016/0006-8993(93)90132-7. [DOI] [PubMed] [Google Scholar]
- Straub VA, Grant J, O’shea M, Benjamin PR. Modulation of serotonergic neurotransmission by nitric oxide. J Neurophysiol. 2007;97:1088–1099. doi: 10.1152/jn.01048.2006. [DOI] [PubMed] [Google Scholar]
- Summers BA, Overholt JL, Prabhakar NR. Nitric oxide inhibits L-type Ca2+ current in glomus cells of rabbit carotid body via a cGMP-independent mechanism. J Neurophysiol. 1999;81(4):1449–1457. doi: 10.1152/jn.1999.81.4.1449. [DOI] [PubMed] [Google Scholar]
- Susswein AJ, Chiel HJ. Nitric oxide as a regulator of behavior: new ideas from Aplysia feeding. Prog Neurobiol. 2012;97(3):304–317. doi: 10.1016/j.pneurobio.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Okada D. Endogenous nitric oxide release required for long-term synaptic depression in the cerebellum. Nature. 1991;349:326–328. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Mammalian nitric oxide synthases. Biochem Biophys Acta. 1999;1411(2–3):217–30. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- Torres JE, Kreisman NR, Gozal D. Nitric oxide modulates in vitro intrinsic optical signal and neural activity in the nucleus tractus solitaries of the rat. Neursci Lett. 1997;232(3):175–178. doi: 10.1016/s0304-3940(97)00598-3. [DOI] [PubMed] [Google Scholar]