

Abstract
Background
We have previously demonstrated that suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, improves survival in a lipopolysaccharide (LPS)-induced lethal endotoxemia model. The goal of this study was to investigate the impact of SAHA on survival in a more clinically relevant cecal ligation and puncture (CLP)-induced septic shock model, and to elucidate changes in cytokine responses and organ injury.
Methods
C57BL/6J mice were subjected to CLP, and 1 hour later given either SAHA dissolved in dimethyl sulfoxide (DMSO) or DMSO only, intra-peritoneal. Survival was monitored for 10 days. In a second study, livers were harvested for evaluation of acute liver injury, and peritoneal fluid and blood samples were collected for cytokine assays. In addition, RAW264.7 and bone marrow-derived macrophages (BMMs) were utilized to assess effects of SAHA on cytokine responses.
Results
SAHA treated animals displayed a significant improvement in survival. In addition, SAHA also significantly attenuated cytokine levels in blood and peritoneal fluid compared to vehicle animals, as well as in culture supernatant of macrophages stimulated with bacterial components (LPS or Pam3CSK4). Moreover, SAHA treated animals showed a significant decrease in acute liver injury.
Conclusions
SAHA treatment improves survival, reduces “cytokine storm”, and prevents distant organ damage in a lethal septic model.
INTRODUCTION
Sepsis causes more than 225,000 deaths annually in the United States and is a tremendous burden for health-care systems 1. Sepsis activates numerous cellular and subcellular pathways that result in organ damage, and no pharmacological agents have been shown to effectively block these cascades and change the outcomes. The multicenter study that failed to show efficacy of recombinant activated protein C is the latest attempt in the quest for a truly effective pharmacological treatment for sepsis 2, 3.
The innate immune system is the first line of defense against pathogenic invasion. Numerous genetic and epigenetic regulatory factors modulate how the immune cells react to the pathogens. This in turn shapes the overall inflammatory response for any given individual 4. Histone acetylation is an essential epigenetic mechanism that determines the amplitude of immune signaling, by controlling the chromatin structure, accessibility of transcription factors to the DNA, and the subsequent gene transcription. This process regulated by the opposing actions of two families of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). Histone acetylation relaxes the chromatin structure (DNA unwinding) and promotes gene transcription, whereas histone deacetylation compacts the chromatin structure favoring gene silencing. Beside histones, numerous non-histone proteins, such as α-tubulin, heat shock protein (HSP) 90, steroid receptors, and regulators of nuclear import, are also reversibly modified by acetylation 5.
Dysregulated HDAC activity has been linked to the pathogenesis of cancer as well as inflammatory and autoimmune diseases. We have also recently discovered that histone deacetylase inhibitors (HDACI) can be used to modulate the inflammatory response. A number of Toll-like receptor (TLR) target genes that encode key inflammatory molecules, such as cytokines, chemokines and other secreted inflammatory mediators, are under the control of the HDAC/HAT regulatory system 6. Only limited information exists concerning the actual inflammatory mechanisms that are altered by different HDACI, but this potentially offers an attractive model for pharmacological intervention with a favorable risk-benefit profile. By targeting different molecules/transcription factors and controlling the fate of the inflammatory signals, HDACI hold great promise as novel therapeutic agents for the future 4.
Suberoylanilide hydroxamic acid (SAHA) or vorinostat was the first HDACI approved by the U.S. Food and Drug Administration for the treatment of cutaneous T cell lymphoma (CTCL) 7. SAHA was also found to exhibit antiinflammatory properties in vitro and in vivo 8. However, it remains unknown whether treatment with SAHA could improve survival from lethal septic shock. Our lab was the first to demonstrate that administration of SAHA improved survival in a rodent model of lipopolysaccharide (LPS)-induced lethal endotoxemia 9, 10. In this model, it decreased acute lung injury, and rapidly modulated several key genes involved in inflammation, with an attenuation of inflammatory mediators derived from both cellular and humoral arms of the innate immune system 11. However, LPS infusion does not fully replicate the complexities of a poly-microbial infection. The current study was therefore designed to investigate the impact of SAHA on survival in a more clinically relevant cecal ligation and puncture (CLP) model of sepsis, and to characterize its influence on cytokine responses and organ damage.
MATERIALS AND METHODS
Cells and Reagents
RAW 264.7 murine macrophages (American Type Culture Collection, Manassas, VA) and mouse bone marrow-derived macrophages (BMMs) were cultured in Dubelcco’s modified Eagle’s medium (DMEM; Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 2mM glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin (Invitrogen, Grand Island, NY) at 37 °C and 5% CO2. BMMs were obtained as described previously 12, 13. Bone marrow cells were flushed out from bone cavity of femurs and tibias with cold phosphate-buffered saline (PBS; Corning, Manassas, VA), and single-cell suspension was obtained by passing through 70 μm cell strainers (BD Biosciences, San Jose, CA). Isolated bone marrow cells were cultured for 6 days in the presence of 10 ng/mL macrophage colony-stimulating factor (M-CSF; R&D Systems, Minneapolis, MN), and medium was changed into DMEM without M-CSF 24 h before treatments 12. All drugs were added at the same starting point. Where indicated, cells were incubated with 1 μg/mL LPS (Sigma Chemical Co, St. Louis, MO), 1 μg/mL Pam3CSK4 (EMD Millipore Corporation, Billerica, MA), or 10 uM SAHA (Enzo Life Sciences, Farmingdale, NY) 14, 15.
Sepsis Model: Cecal Ligation and Puncture (CLP)
Male C57BL/6J mice (18–26 gm) were purchased from The Jackson Laboratory and housed for 3 days before manipulations. The CLP murine model16, modified by our laboratory, was used to induce fecal peritonitis. In brief, the peritoneal cavity was opened under inhaled isoflurane anesthesia. Cecum was eviscerated, ligated below the ileocecal valve using a 5-0 suture, and punctured through and through (2 holes) with a 20 gauge needle. The punctured cecum was squeezed to expel a small amount of fecal material and returned to the peritoneal cavity. The abdominal incision was closed in two layers with 4-0 silk suture. Animals were resuscitated by subcutaneous injection of 1 mL of saline. Sham-operated animals were handled in the same manner, except that the cecum was not ligated or punctured. This protocol was approved by the Animal Review Committee at the Massachusetts General Hospital.
Administration of SAHA and Experimental Design
In the survival experiment, mice received intra-peritoneal SAHA dissolved in DMSO (50 mg/kg) or vehicle DMSO 1 h after CLP (n=10/group). Mortality was recorded for up to 10 days post-procedure.
In the non-survival experiment, animals were randomly assigned to the following three groups (n = 10/group): (a) Sham-operated animals (SHAM); (b) vehicle treated animals after CLP (CLP+DMSO), and (c) SAHA treated animals after CLP (CLP+SAHA). Sham-operated animals were subjected to laparotomy and intestinal manipulation, but the cecum was neither ligated nor punctured. At the time of sacrifice [24 and 48 h after CLP (n= 3–7/group/time point)], abdominal cavity was opened and irrigated with 1 ml normal saline, which was collected for analysis, and blood samples were collected by cardiac puncture. Liver tissue was harvested 24 h after CLP, which were fixed in 10% buffered formalin for later histological analysis.
Cytokine Measurements
Concentrations of tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 in the peritoneal fluid, plasma, or cell culture supernatant were measured using the Quantikine Enzyme-Linked Immunosorbent Assay (ELISA) Kit (R&D Systems, Minneapolis, MN) according to manufacturer’s instructions.
Cell Viability Assay
Cell viability was measured using an annexin V-PE detection kit (Abcam, Cambridge, MA). Macrophages were treated with or without LPS and SAHA for 3, 6, and 9 h. Cells were then harvested, washed in ice-cold phosphate-buffer saline, resuspended in binding buffer, and incubated with annexin V-PE for 5 min at room temperature. Cells were analyzed by fluorescence-activated cell sorting (FACS), and viable cells were defined as negative for annexin V-PE and DAPI staining 17. Experiments were carried out in triplicate.
Phenotypic Characterization of BMMsby FACS
BMMs were harvested on day 6 when they were fully differentiated, blocked with anti-mouse CD16/32 antibody (eBiosicence, San Diego, CA), and stained with anti-mouse F4/80 Antigen APC and anti-mouse CD11b FITC (eBiosicence, San Diego, CA) 12. Data were acquired on an LSRII (BD Biosciences, San Jose, California) and analyzed with FlowJo (Tree Star, Ashland, OR).
Histological Analysis
Twenty-four hours after CLP, liver samples were harvested for histological analysis. Each sample was fixed by immersing in 10% buffered formalin. The samples were then embedded in paraffin, sliced into 5-μm sections and stained with hematoxylin and eosin (H&E) for histological assessment by a pathologist blinded to their group allocation. Hepatocellular necrosis, hemorrhage/congestion, parenchyma inflammation, sinusoidal inflammation, and degenerative changes were evaluated objectively on a scale of 0–3, with 0 meaning “absent,” 1 meaning “mild,” 2 meaning “moderate,” and 3 meaning “severe.” The total injury score was expressed as the sum of the scores for all of these parameters 18.
Statistical Analysis
Results were expressed as mean ± SEM. Student’s t-test was used to compare the differences between two groups. Differences between 3 or more groups were assessed using one way analysis of variance (ANOVA) followed by Bonferroni post hoc testing for multiple comparisons. Kaplan-Meier method was used for survival, and differences were analyzed using log-rank test. All analyses were performed using GraphPad Prism. P values of 0.05 or less were considered significant.
RESULTS
SAHA improves survival in CLP-induced lethal septic model
In this CLP-induced lethal sepsis model, all mice in the DMSO vehicle group died in less than 3 days. However, SAHA-treated animals displayed significantly higher long-term survival compared to the DMSO vehicle group (40% versus 0% survival, P < 0.01; Figure 1).
Figure 1. SAHA protects mice against severe sepsis and septic shock-induced lethality.
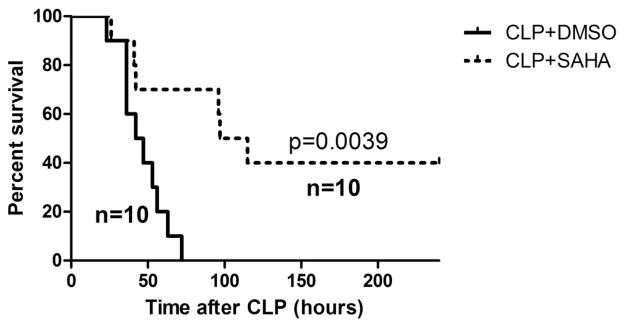
Mice were intraperitoneally administered 50mg/kg SAHA or vehicle DMSO 1 h after CLP (n=10 animals/group). Treatment with SAHA significantly improved long-term survival compared to DMSO vehicle group (40% versus 0% survival, P < 0.01).
SAHA decreases cytokine levels in peritoneal fluid and circulation in vivo
In sham group, TNF-α levels in the peritoneal fluid and blood were low at all time points (−8.7 ± 0.7 and −11.3 ± 2.3 pg/mL, respectively), and so were IL-6 levels in peritoneal fluid and blood (6.3 ± 2.0 and 14.9 ± 6.3 pg/mL, respectively). SAHA significantly blunted TNF-α increase 24 h after CLP in the peritoneal fluid (463.3 ± 48.4 versus 98.1 ± 36.2 pg/mL, P < 0.01; Figure 2), as well as in the serum (298.3 ± 24.6 versus 91.4 ± 46.5 pg/mL, P < 0.05; Figure 2). In addition, SAHA suppressed increased IL-6 levels 48 h after CLP in the peritoneal fluid (646.4 ± 81.8 versus 236.9 ± 103.2 pg/mL, P < 0.05; Figure 3), as well as in the serum (583.8 ± 83.9 versus 320.3 ± 47.4 pg/mL, P < 0.05; Figure 3).
Figure 2. SAHA attenuates TNF a levels in peritoneal fluid and blood during severe sepsis and septic shock.
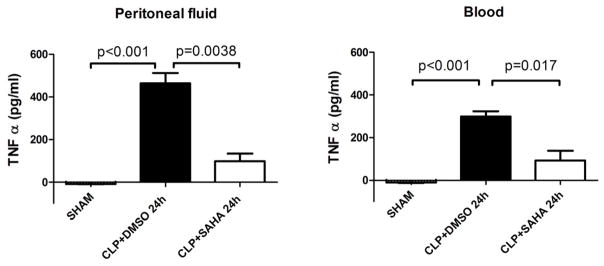
The peritoneal fluid and blood were collected 24 h after CLP and assayed for TNF-α levels by ELISA kits (means ± SEM, n = 3–4 animals/group).
Figure 3. SAHA attenuates IL-6 levels in peritoneal fluid and blood during severe sepsis and septic shock.
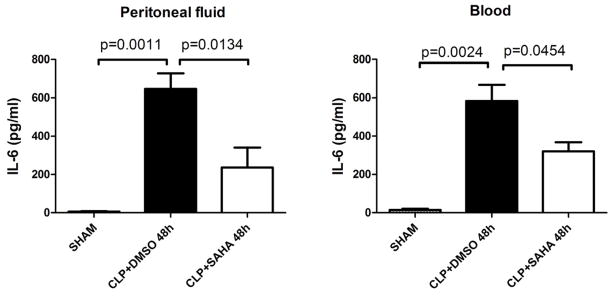
The peritoneal fluid and blood were collected 48 h after CLP and assayed for IL-6 levels by ELISA kits (means ± SEM, n = 3–7 animals/group).
SAHA decreases TNF-α level in culture supernatant of RAW264.7 Macrophages in presence of LPS or Pam3CSK4 in vitro
TLRs constitute a family of non-polymorphic receptors that are devoted to pathogen recognition. SAHA significantly suppressed TNF-α production (induced by TLR4 ligand LPS) in RAW264.7 macrophages, as measured in the supernatant at 2, 5, and 8 hours (656.8 ± 27.3 versus 404.0 ± 29.3 pg/mL, 780.8 ± 31.3 versus 579.8 ± 23.8 pg/mL, 843.8 ± 26.5 versus 650.6 ± 26.2 pg/mL, respectively, P < 0.01; Figure 4). Similar findings were observed at 2, 5, 8 hours when the macrophages were stimulated by TLR1/2 ligand Pam3CSK4 (714.4 ± 20.9 versus 320.6 ± 9.1 pg/mL, 789.0 ± 31.2 versus 418.6 ± 20.9 pg/mL, 874.2 ± 27.0 versus 486.6 ± 26.6 pg/mL, respectively, P < 0.01; Figure 4). The lower TNF-α levels were not due to a decrease in the number of viable RAW264.7 cells (Figure 5A and 5B).
Figure 4. SAHA decreases enhanced TNF-α production stimulated by LPS or Pam3CSK4 in culture supernatant of RAW264.7 macrophages.
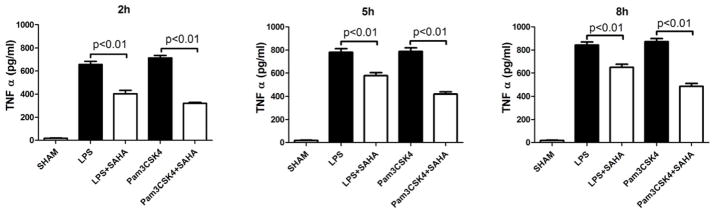
Concentrations of TNF-α and IL-6 in culture supernatant of RAW264.7 Macrophages were determined by ELISA at 2, 5, and 8 h after LPS or Pam3CSK4 treatment in the absence or presence of SAHA. Untreated macrophages served as control (means ± SEM, n = 3).
Figure 5. Effects of LPS and SAHA on cell viability of RAW264.7 macrophages at 3 h and 6 h.


(A) Representative plots show cell viability of RAW264.7 macrophages at 3 and 6 h treated with LPS or Pam3CSK4 in the absence or presence of SAHA. Cells were analyzed by FACS, and viable cells were defined as negative for annexin V-PE and DAPI staining indicated in the lower left corner of each panel. (B) Quantification of cell viability (means ± SEM, n = 3).
SAHA decreases LPS-induced cytokine production by bone marrow derived macrophages in vitro
After differentiation with M-CSF for 6 days, percentage of mature BMMs (double positive for CD11b and F4/80 Antigens) was 91.14% (Figure 6). Mature BMMs were then harvested for further experiments. SAHA attenuated LPS-stimulated TNF-α production in the BMM supernatant at 6 h (757.9 ± 19.0 versus 619.9 ± 22.6 pg/mL, P < 0.01; Figure 7). Moreover, IL-6 levels at 3 and 6 h were also significantly suppressed by SAHA (150.8 ± 23.6 versus 17.8 ± 6.0 pg/mL, 740.2 ± 37.0 versus 50.1 ± 24.5 pg/mL, respectively, P < 0.01; Figure 8).
Figure 6. Phenotypic characterization of BMMs by FACS.
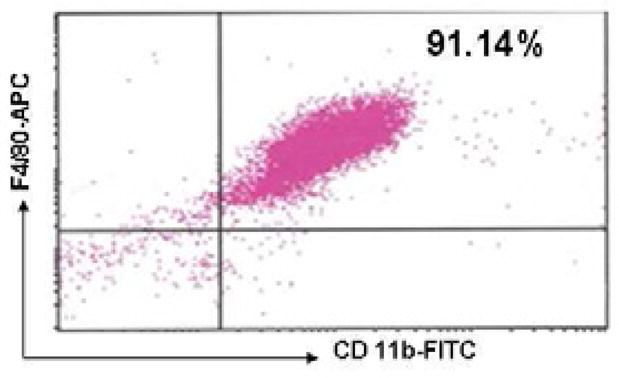
Representative plots show percentage of mature BMMs, which are double positive for CD11b-FITC and F4/80-APC indicated in the up right corner of the panel.
Figure 7. SAHA decreases TNF-α levels in culture supernatant of BMMs treated with LPS.
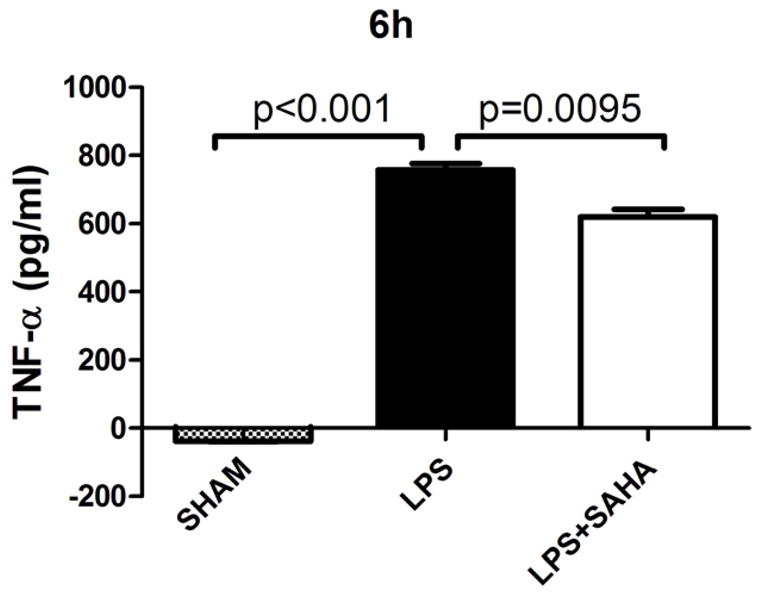
TNF-α concentration in culture supernatant of BMMs were assayed by ELISA at 6 h after LPS treatment with or without SAHA. Untreated BMMs served as control (means ± SEM, n = 3).
Figure 8. SAHA decreases IL-6 levels in culture supernatant of BMMs treated with LPS.
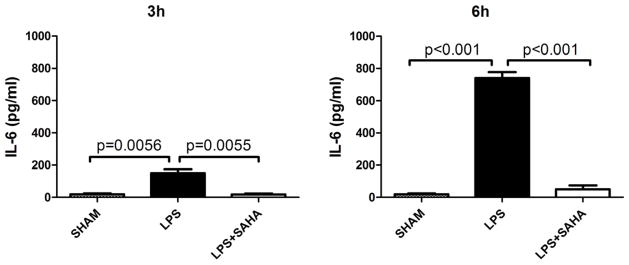
TNF-α levels in culture supernatant of BMMs were determined by ELISA at 6 h after LPS treatment with or without SAHA. Untreated BMMs served as control (means ± SEM, n = 3).
SAHA decreases acute liver injury
Sham-operated animals had normal liver histology, but liver sections isolated 24 h after CLP showed increased parenchyma inflammation and degenerative changes. These pathological changes were attenuated by SAHA treatment, with significantly lower acute liver injury score (5.1 ± 0.8 versus 2.8 ± 0.2, P < 0.05; Figure 9).
Figure 9. SAHA protects animals from acute liver injury 24h after CLP (H&E, magnification 40×).
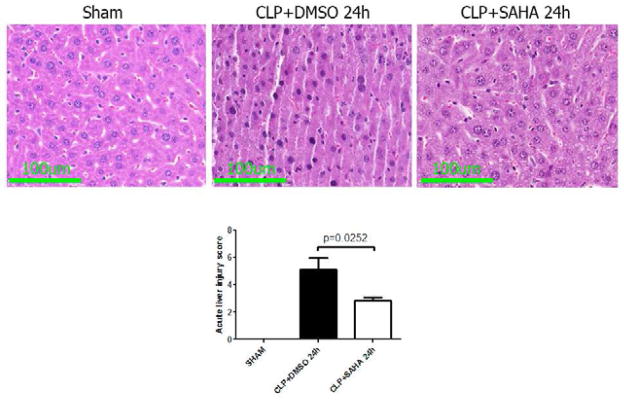
Mice were intraperitoneally administered 50 mg/kg SAHA 1 h after CLP. 24 h after CLP, liver tissues were obtained. Representative images were chosen from different experimental groups. Semiquantitative pathology scores in liver were determined by a scoring system as described in Materials and Methods (means ± SEM, n = 4–6 animals/group).
DISCUSSION
We have investigated the therapeutic effect of SAHA, a HDACI, in a lethal CLP-induced sepsis model, and discovered some important findings: (1) SAHA significantly improves long-term survival; (2) it significantly attenuates local and systemic proinflammatory cytokines in vivo, as well as LPS or Pam3CSK4-stimulated cytokines production from RAW264.7 macrophage cell line or primary BMMs in vitro; (3) it attenuates acute liver injury.
Several experimental animal models have been developed to study the underlying mechanisms that are typically involved in septic patients. CLP in rodents is currently considered as the gold standard in sepsis research, as it creates polymicrobial sepsis in well-controlled experimental settings 16. Our present study shows that SAHA treatment improves long-term survival in CLP model similar to what we have previously demonstrated in LPS models. This adds further evidence supporting the use of HDACI as a potential pharmacological treatment for lethal sepsis.
A largely unopposed early proinflammatory response occurs in severe sepsis, which can lead to organ damage and death. Cytokines attract immune cells such as macrophage and T-cells to the site of infection, where these cells produce more cytokines to perpetuate the response. Normally, this feedback loop is kept in check but in severe sepsis, the exaggerated immune response can get out of control 19. Cytokine storms have the potential to cause significant cellular damage as they can potentiate the destructive ability of macrophages, plasma cells, complement system and T lymphocytes, as well as retard the healing effects of other cell types (such as M2 macrophages, regulatory T lymphocytes, and Th2 lymphocytes). In our study, SAHA significantly attenuated the local and systemic proinflammatory cytokines in the peritoneal fluid as well as the circulation following CLP. It also suppressed LPS (TLR4 ligand) or Pam3CSK4 (TLR1/2 ligand)-stimulated cytokines production from the macrophage cell line as well as primary macrophages differentiated from bone marrow. This provides further evidence supporting the anti-inflammatory properties of SAHA and explaining in part how SAHA improves survival in lethal sepsis.
TLR signaling and associated downstream regulators play a crucial role in the innate immune system as the first line of defense against pathogens. TLR4 and TLR2 are the main sensors for pathogen-associated molecular patterns recognition from gram-negative and gram-positive bacteria respectively. LPS binds to host TLR4, triggering an inflammatory reaction characterized by releasing a large number of inflammatory mediators like TNF-α and IL-6, while recognition of Pam3CSK4 is mediated by TLR2 that cooperates with TLR1 through their cytoplasmic domain to induce the signaling cascade, leading to nuclear factor-kappa B (NF-kB) activation and excessive proinflammatory cytokine production 20. Downstream TLR signaling occurs via two major pathways: the myeloid differentiation factor 88 (MyD88)-dependent pathway and MyD88-independent pathway. The MyD88-dependent pathway has been shown to be responsible for expression of proinflammatory cytokines, such as TNF-α and IL-6, while the MyD88-independent pathway mediates the induction of type I interferons and interferon-inducible genes. MyD88 interacts with TLRs, leading to activation of the mitogen activated protein kinase (MAPK) cascade and NF-κB, as well as expression of numerous proinflammatory mediators. Genetic deficiency of MyD88 partially protected mice from the lethal effects of polymicrobial sepsis, and hyperinflammatory response to sepsis was strongly attenuated in the absence of MyD88 21. Our team has previously demonstrated that SAHA can reduce MyD88 expression at both gene and protein levels after LPS insult in the lung tissue, suggesting that inhibition of TLR4-MyD88-dependent pathway could be one explanation for the anti-inflammatory effects of SAHA 9. Currently, it remains elusive as to how acetylation affects expression of MyD88. Accumulating evidence suggests that acetylation of signal transducer and activator of transcription (STAT)-1 could be involved in down-regulation of MyD88 expression through a cross-talk between STAT-1 and NF-kB signaling 22. Acetylated STAT-1 inhibits transcriptional activity of the NF-kB p50/p65 heterodimer, consequently decreasing NF-kB nuclear localization, DNA binding, and expression of NF-kB target genes 23. Therefore, SAHA may suppress TLR4-MyD88-dependent pathway by inhibiting MyD88 expression through interaction of acetylated STAT-1 and NF-kB.
In addition, we have shown that anti-inflammatory properties of SAHA involve acetylation of HSP 90 15. In LPS-stimulated macrophages, SAHA acetylates HSP 90, resulting in dissociation and degradation of its client protein interleukin-1 receptor associated kinase 1 (IRAK1), subsequently decreasing nuclear translocation of NF-κB, and leading to attenuation of key pro-inflammatory cytokines. Meanwhile, SAHA attenuates hypoxia-inducible factor (HIF)-1α-mediated inflammatory pathway in macrophages, and suppresses hypoxia-induced release of proinflammatory nitric oxide and TNF-α 24.
Excessive production of proinflammatory cytokines in sepsis promotes migration of leukocytes, lymphocytes, and platelets to the infected areas, leading to endothelial damage, increased microvascular permeability, platelet aggregation, local blood flow reduction, and ischemia/reperfusion (I/R) injury, which finally results in multiple organ damage. We have previously found that during LPS-induced endotoxemia, SAHA treatment significantly reduces acute lung injury 10. Here, we show that SAHA can also attenuate acute liver injury during severe polymicrobial sepsis. Our group has previously demonstrated that SAHA significantly decreases LPS-induced enhanced expression of the pro-inflammatory MAPK phosphorylated p38, phosphorylated extracellular signal-regulated kinase (ERK), myeloperoxidase, and IL-6 in the liver, while increases levels of the anti-inflammatory IL-10 25. Attenuation of MAPK and ERK activation, and subsequently decreased NF-κB-dependent gene transcription, as well as alteration of inflammatory and anti-inflammatory markers may account for SAHA’s protective role in reducing acute liver injury during our polymicrobial severe sepsis and septic shock model.
This study has certain limitations that must be acknowledged. For logistical reasons, we only measured selected cytokines and explored limited pathways. Many more mechanisms are likely to be influenced by SAHA treatment. Similarly, we only studied liver injury in this study. Also, we only tested a single dose of SAHA that has previously been found to be effective without performing a detailed dose response study.
In conclusion, we have demonstrated that SAHA, a HDACI, can improve survival in a lethal polymicrobial sepsis model. The survival advantage is associated with an attenuation of local and systemic proinflammatory cytokines, as well as protection against distant organ injury. Although the fundamental molecular and cellular signaling events still requires further investigation, HDACI may represents a novel and promising therapeutic strategy for severe sepsis and septic shock.
Acknowledgments
This work was funded by a grant from NIH RO1 GM084127 to HBA.
ABBREVIATIONS
- BMMs
bone marrow-derived macrophages
- CLP
cecal ligation and puncture
- CTCL
cutaneous T cell lymphoma
- DMSO
dimethyl sulfoxide
- ELISA
Enzyme-Linked Immunosorbent Assay
- ERK
extracellular signal-regulated kinase
- FACS
fluorescence-activated cell sorting
- HDAC
histone deacetylase
- HDACIs
HDAC inhibitors
- HIF
hypoxia-inducible factor
- HSP
heat shock protein
- H&E
hematoxylin and eosin
- IFN
interferon
- IRAK1
interleukin-1 receptor associated kinase 1
- IL
interleukin
- LPS
lipopolysaccharide
- M-CSF
macrophage colony-stimulating factor
- MyD88
myeloid differentiation factor 88
- NF-kB
nuclear factor-kappa B
- Pam3CSK4
Pam3-Cys-Ser-Lys4
- SAHA
suberoylanilide hydroxamic acid
- STAT
signal transducer and activator of transcription
- TNF-α
tumor necrosis factor-α
- TLR
Toll-like receptor
Footnotes
Data presented at the 8th Annual Academic Surgical Congress in New Orleans, LA (February, 2013).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2012;306:2594–605. doi: 10.1001/jama.2011.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 3.Wenzel RP, Edmond MB. Septic shock--evaluating another failed treatment. N Engl J Med. 2012;366:2122–4. doi: 10.1056/NEJMe1203412. [DOI] [PubMed] [Google Scholar]
- 4.Suliman BA, Xu D, Williams BR. HDACi: molecular mechanisms and therapeutic implications in the innate immune system. Immunol Cell Biol. 2012;90:23–32. doi: 10.1038/icb.2011.92. [DOI] [PubMed] [Google Scholar]
- 5.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 6.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32:335–43. doi: 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;148:246–54. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32:517–23. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Liu B, Gu X, Kochanek AR, Fukudome EY, Liu Z, et al. Creating a “pro-survival” phenotype through epigenetic modulation. Surgery. 2012;152:455–64. doi: 10.1016/j.surg.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5080. pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 13.McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science. 2012;335:229–32. doi: 10.1126/science.1214448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, Friggeri A, Yang Y, Park Y-J, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proceedings of the National Academy of Sciences. 2009;106:15819–24. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chong W, Li Y, Liu B, Zhao T, Fukudome EY, Liu Z, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates Toll-like receptor 4 signaling in lipopolysaccharide-stimulated mouse macrophages. J Surg Res. 2012 doi: 10.1016/j.jss.2012.07.023. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–6. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawada J, Zou P, Mazitschek R, Bradner JE, Cohen JI. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. J Biol Chem. 2009;284:17102–9. doi: 10.1074/jbc.M809090200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim TH, Yoon SJ, Lee SM. Genipin attenuates sepsis by inhibiting Toll-like receptor signaling. Mol Med. 2012;18:455–65. doi: 10.2119/molmed.2011.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Weighardt H, Jusek G, Mages J, Lang R, Hoebe K, Beutler B, et al. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. Eur J Immunol. 2004;34:558–64. doi: 10.1002/eji.200324714. [DOI] [PubMed] [Google Scholar]
- 22.Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206:2027–35. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer OH, Baus D, Knauer SK, Stein S, Jager E, Stauber RH, et al. Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev. 2006;20:473–85. doi: 10.1101/gad.364306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chong W, Li Y, Liu B, Liu Z, Zhao T, Wonsey DR, et al. Anti-inflammatory properties of histone deacetylase inhibitors: a mechanistic study. J Trauma Acute Care Surg. 2012;72:347–53. doi: 10.1097/TA.0b013e318243d8b2. discussion 53–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, et al. Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. J Surg Res. 2010;163:146–54. doi: 10.1016/j.jss.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]