

Abstract
A chemically reactive hybrid diblock polypeptide gelator poly(ethylene glycol)-block-poly(dl-allylglycine) (PEG-b-PDLAG) is an exceptional material, due to the characteristics of thermo-reversible organogel formation driven by the combination of a hydrophilic polymer chain linked to a racemic oligomeric homopeptide segment in a range of organic solvents. One-dimensional stacking of the block copolymers is demonstrated by ATR-FTIR spectroscopy, wide-angle X-ray scattering to be driven by the supramolecular assembly of β-sheets in peptide blocks to afford well-defined fiber-like structures, resulting in gelation. These supramolecular interactions are sufficiently strong to achieve ultra low critical gelation concentrations (ca. 0.1 wt%) in N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO) and methanol. The critical gel transition temperature was directly proportional to the polymer concentration, so that at low concentrations, thermoreversibility of gelation was observed. Dynamic mechanical analysis studies were employed to determine the organogel mechanical properties, having storage moduli of ca. 15.1 kPa at room temperature.
Introduction
Polypeptide-based materials have attracted significant attention for the development of biodegradable, stimuli-responsive materials.1–5 The supramolecular hierarchical assemblies of peptides have precisely defined structures from molecular through to nanometer and micrometer length scales, with high degrees of structural complexity, which could not be easily obtained from non-peptide polymers or low-molecular-weight amphiphiles.6,7 Depending on the amino acids used and the degrees of polymerization obtained, polypeptides can adopt various secondary structures, such as random coils, α-helices and/or β-sheets. While most work has focused on polypeptides composed of natural amino acids, recent attention has been devoted increasingly to the self-assembly behaviors of hybrid polymer structures of varying compositions and structures, and including non-natural, functional synthetic polypeptides under diverse conditions.8
Supramolecular interactions between polypeptide segments9 have been utilized for the self assembly of peptide-based diblock copolymers into stimuli-responsive vesicles,5,10,11 micelles,12–15 hydrogels16–18 and organogels.19–22 Organogels made from polypeptides have potential applications as responsive materials in oil recovery, sensing, electrochemistry, drug delivery and as templates for the fabrication of nanostructures.23–25 Peptide-based organogelators have been studied by many groups, leading to a variety of interesting organogels with special mechanical and physical properties,26 where gel formation is typically driven by hydrophobic interactions among chain segments having selective peptide sequences and is also facilitated by the co-block segment, e.g. PEG, exhibiting thermally responsive insolubility to physically crosslink initially assembled micelles into a gel.19 The gelation mechanisms are different based on the different chemical composition of polypeptides.16,27 The secondary structures of polypeptides, and their stacking interactions, have been suggested as driving forces for gel formation, and are controlled by the compositions and sequences of the component amino acids. Valine, isoleucine and alanine have been reported as crucial for gelation in certain sequences of polypeptides, by aiding in the formation of β-sheets that are further capable of organizing into lamellar-stacking regions.28,29 Polypeptide-based organogelators remain limited, primarily, to certain sequences of natural amino acids. Typically, the synthesis of organogelators with special sequences has required several steps of solid phase synthesis and has experienced difficulty with scaled-up production. Compared to traditional solid phase synthesis, ring opening polymerization (ROP) of N-carboxyanhydrides (NCA) provides a more effective and facile method for the preparation of high molecular weight polypeptides.30,31
Herein, we report a new type of chemically reactive, thermo-reversible organogel formed by a hybrid diblock polypeptide gelator poly(ethylene glycol)-block-poly(dl-allylglycine) (PEG-b-PDLAG). The diblock polymer contains a rigid peptide motif composed of racemic synthetic homopolypeptide and a flexible PEG coil. Although PEG-b-PDLAG has been synthesized previously and studied for its chemical transformation via thiol–ene reactions,32 a tendency toward gelation was not described. Typically, when polypeptides have been prepared by NCA ROP and shown a tendency toward gelation, experimental conditions were then altered to allow for isolation of the polymers and subsequent gelation studies in water.7,33 We, however, found the gel formation in organic solvent in situ to be of interest, especially as this work appears to be the first example of an organogelator formed from a hybrid diblock racemic synthetic polypeptide. The mechanism of gelation is discussed. The critical gelation concentration was as low as 0.1 wt%, which is much lower than most organogelators made from natural or synthetic amino acids. Further, the organogel showed thermoresponsive properties: Above a certain temperature, the gel transformed to solution, and when cooled down, it returned to gel. The thermoresponsive sol–gel transition was reversible and reproducible, providing potential development of these materials as sensors and drug delivery vehicles.
Experimental section
Materials
N,N-Dimethylformamide (DMF), diethyl ether, dichloromethane (DCM), tetrahydrofuran (THF), dimethyl sulfoxide (DMSO), dl-allylglycine, alpha-pinene, triphosgene and methanol were purchased from Sigma-Aldrich Company (USA). mPEG45–NH2 (Mn = 2000 g mol−1), mPEG112–NH2 (Mn = 5000 g mol−1) were purchased from Rapp Polymere (Germany). All other chemicals were used without further purification, unless otherwise noted.
Instrumentation
1H and 13C NMR spectra were recorded on a Varian 300 Inova spectrometer. Chemical shifts were referenced to solvent resonance signals. IR spectra were recorded on an IR Prestige 21 system (Shimadzu Corp., Japan) and analyzed using IRsolution v. 1.40 software.
N,N-Dimethylformamide-based gel permeation chromatography (DMF GPC) was conducted on a Waters Chromatography, Inc. (Milford, MA) system equipped with an isocratic pump model 1515, a differential refractometer model 2414 and a two-column set of Styragel HR 4 and HR 4E 5 mm DMF 7.8 × 300 mm columns. The system was equilibrated at 70 °C in pre-filtered DMF containing 0.05 M LiBr, which served as polymer solvent and eluent (flow rate set to 1.00 mL min−1). Polymer solutions were prepared at concentrations of ca. 3 mg mL−1 and an injection volume of 0.2 mL was used. Data collection and analysis was performed with Empower Pro software. The system was calibrated with poly(ethylene glycol) standards (Polymer Laboratories, Amherst, MA) ranging from 615 to 442 800 Da.
Glass transition temperatures (Tg) were measured by differential scanning calorimetry on a Mettler-Toledo DSC822® (Mettler-Toledo, Inc., Columbus, OH), with a heating rate of 10 °C min−1. Measurements were analyzed using Mettler-Toledo STARe v. 7.01 software. The Tg was taken as the midpoint of the inflection tangent, upon the third heating scan. Thermogravimetric analysis was performed under N2 atmosphere using a Mettler-Toledo model TGA/SDTA851e, with a heating rate of 5 °C min−1. Measurements were analyzed by using Mettler-Toledo STARe v. 7.01 software.
Transmission electron microscopy (TEM) images were collected on a JEOL1200EX operating at 100 kV and micrographs were recorded at calibrated magnifications using a SIA-15C CCD camera. The final pixel size was 0.42 nm per pixel. Samples for TEM measurements were prepared as follows: 4 μL of the dilute solution (with a polymer concentration of 0.1 mg mL−1) was deposited onto a carbon-coated copper grid, and after 2 min, the excess of the solution was quickly wicked away by a piece of filter paper. The samples were then negatively stained with 1 wt% phosphotungstic acid (PTA) aqueous solution or 1 wt% uranyl acetate aqueous solution. After 1 min, the excess staining solution was quickly wicked away by a piece of filter paper and the samples were left to dry under vacuum overnight.
Cryogenic transmission electron microscopy was performed in a Tecnai 12 microscope operated at 120 kV. A small droplet of the solution (1.2 μL) was placed on a 200 mesh lacey carbon film supported on a TEM copper grid within a Vitrobot vitrification system (FEI Inc.). The sample was blotted and quickly plunged into a liquid ethane reservoir cooled by liquid nitrogen. The vitrified samples were transferred to a Gatan 626 cryo-holder and cryo-transfer stage cooled by liquid nitrogen. During observation of the vitrified samples, the cryo-holder temperature was maintained below −176 °C. The images were recorded digitally with a Gatan CCD camera.
Atomic force microscopy (AFM) imaging was performed using a MFP-3D system (Asylum Research, Santa Barbara, CA) in tapping mode using Si3N4 tips (SPM Probes, NSC14/AIBS; length (L), tip radius <10 nm, force constant 5.7 N m−1; resonance frequency, 160 kHz). The samples as DMF solutions (0.05 mg mL−1, 5 μL) were deposited onto freshly cleaved mica and allowed to dry under vacuum before analyses. The average height and diameter values were determined by section analysis, using the IGOR Pro software package.
X-ray diffraction (XRD) was performed on a Bruker D8® Bragg-Brentano X-ray powder diffractometer. The sample was placed in the sample holder of a two circle goniometer, enclosed in a radiation safety enclosure. The X-ray source was a 2.2 kW Cu X-ray tube, maintained at an operating current of 40 kV and 40 mA The X-ray optics were the standard Bragg-Brentano para-focusing mode with the X-ray diverging from a DS slit (1 mm) at the tube to strike the sample and then converging at a position sensitive X-ray Detector (Lynx-Eye, Bruker-AXS). The two-circle 250 mm diameter goniometer was computer controlled with independent stepper motors and optical encoders for the θ and 2θ circles with the smallest angular step size of 0.0001° 2θ. The software suite for data collection and evaluation was windows based. Data collection was automated COMMANDER program by employing a DQL file. Data were analyzed by the program EVA.
Matrix assisted laser desorption ionization (MALDI) experiments were performed on a Voyager DE-STR mass spectrometer (Applied Biosystems, Foster City, CA) under optimized conditions in positive linear mode. Ions were generated by a pulsed nitrogen laser at 337 nm and accelerated through 25 kV. 100 laser shots were used per spectrum. α-Cyano-4-hydroxycin-namic acid (CHCA) was used as the matrix. The sample and matrix were dissolved in methanol at concentrations of 1 and 10 mg mL−1, respectively. The sample solution was mixed with the matrix at a volume ratio of 1 :1. About 0.5 μL of this mixture was deposited on a stainless steel sample holder. After allowing to air-dry, the sample was analyzed using MALDI TOF MS.
Dynamic mechanical analysis (DMA) and static mechanical testing were performed on a Mettler Toledo TT-DMA system. DMA of the 2% (wt/wt) sample in DMF was performed over three hours in compression as 1 mm thick, 10 mm diameter cylinder. Dynamic measurements were recorded over a range of 0.1 to 10 Hz at room temperature with a static stress of 2% and a dynamic range of 1%. Kinetic data presented were obtained as a single exponential decay using Origin Pro 8.6 software. Observation of the loss of volume (for solvent loss kinetics) was obtained using a calibrated digital microscope (Dynolite). The Young's modulus (E) was obtained for the 2% sample from low deformation compression from ca. 0.75% stress to 1.25% stress and is presented in Fig. S5.† The 1% (wt/wt) sample was measured with similar stresses for comparison. DMA for the 1% sample was performed at 1 Hz for 10 cycles for comparison to the 2% sample at t0. The authors acknowledge that the significance of comparison of the Young's modulus over such a small range is somewhat limited, however have chosen to include it for completeness.
Synthesis
Synthesis of dl-Allylglycine NCA.32
In a 250 mL three neck flask, dl-allylglycine (5.0 g) was dissolved in 50 mL THF and triphosgene (7.0 g) were added directly to the reaction flask. Then 15 mL alpha-pinene was added to the reaction mixture and stirred in dry THF (50 mL) for 3 h at 50 °C. The mixture was constantly flushed with a stream of dry nitrogen. The crude product was concentrated and precipitated from hexane, recrystallized three times from ethyl acetate–hexane 1 :1 (v/v), and dried in vacuum; yield: 2.9 g (88%) as a white crystal. 1H NMR (300 MHz, DMSO-d6, ppm): δ 2.51, 2.74 (m, 2H, CH2), 4.38 (m, 1H, CH), 5.29 (m, 2H, H2C=CH2), 5.74 (m, 1H, H2C= CH), 6.35 (br, 1H, NH). 13C NMR (300 MHz, DMSO-d6, ppm): δ 35.3, 57.4, 120.5, 131.9,152.3, 171.5. FT-IR (cm−1): 3410–3270, 1828, 1747, 1436, 1402, 1348, 1292, 1197, 1140, 1112, 1070, 1002, 887, 783, 758. HRMS: calculated [M − H]− for C6H6NO3: 140.0348, found: 140.0351.
Synthesis of polyethylene glycol)-block-poly(dl-allylglycine)
Synthesis of mPEG45-b-PDLAG12
mPEG45–NH2 (Mn = 2000 g mol−1, 70 mg, 0.035 mmol) was added to a solution of dl-allylglycine NCA (150 mg, 1.05 mmol) in dry DMF (10 mL), and the mixture was stirred for 12 h at room temperature under an argon atmosphere in glove box. The polymerization formed gel after 12 h stirring in glove box. The magnetic stir bar was gelled in the flask even at highest speed (800 rpm). The gel was precipitated into diethyl ether under vigorous stirring. The polypeptide block copolymer was collected by filtration and dried in vacuum at room temperature; obtained as a white powder with 88% yield. 1H NMR (300 MHz, DMSO, ppm): δ 2.30, 2.41 (m, CH2), 3.51 (br, CH2CH2O on PEO), 4.38 (m, CH), 5.02 (m, H2C=CH), 5.69 (m, H2C=CH), 8.03 (br, NH). 13C NMR (300 MHz, DMSO-d6, ppm): (the polymer formed gel immediately during the measurement with the concentration of 30 mgmL−1, only the signals assigned to PEG domain could be observed). (Tm) = 48 °C, (Tc) = 17 °C, (Tg) = −50 °C, 98 °C. TGA in N2: 0– 160 °C, 2% mass loss; 160–260 °C, 23% mass loss; 260–380 °C, 34% mass loss, 41% mass remaining above 500 °C.
Synthesis of mPEG112-b-PDLAG12
The mPEO112-b-PDLAG12 was prepared similar as mPEG45-b-PDLAG12. 1H NMR (300 MHz, DMSO, ppm): δ 2.30, 2.41 (m,CH2), 3.51 (br, CH2CH2O on PEO), 4.38 (m, CH), 5.02 (m, H2C=CH), 5.69 (m, H2C=CH), 8.03 (br, NH). (Tm) = 60 °C, (Tc) = 23 °C, (Tg) = −45 °C, 101 °C. TGA in N2: 0–240 °C, 2% mass loss; 280–370 °C, 33% mass loss; 370– 430 °C, 35% mass loss, 30% mass remaining above 500 °C.
Results and discussion
With our general interest in the creation of well-defined polymer nanomaterials with high degrees of functionality and degradability, for fundamental studies and applications with biological systems, we began investigation into the preparation of amphiphilic block copolypeptides having broadly modifiable allyl side chain groups (Scheme 1), but surprisingly, found that spontaneous gelation occurred during the polymerization. Several conditions were screened for the ROP of dl-allylglycine NCA with monomethoxy-monoamino-terminated poly(ethylene glycol) (mPEG–NH2) as a macroinitiator in a glove box under argon atmosphere at room temperature,32 to prepare a series of mPEG-b-PDLAG block copolymers (Table 1). For each polymerization, the DLAG NCA monomer was first dissolved in dry N,N-dimethylformamide (DMF) followed by addition of 2 kDa or 5 kDa mPEG–NH2 macroinitiator (mPEG45–NH2 or mPEG112–NH2) at a 30 :1, monomer : macroinitiator stoichiometry. The reaction mixtures remained transparent and homogeneous for the first 2 h and gelled after stirring overnight (8 h) (for all entries in Table 1). A transparent gel was able to be achieved even at very low concentration (6 mg mL−1, 0.6 wt%) with rapid stirring (800 rpm) (Table 1, entries 3 and 4). When comparatively higher concentration (30 mg mL−1, 3 wt%, Table 1, entries 1 and 2) was imposed, the gel was of sufficient stiffness that the magnetic stir bars were trapped at the bottom of the flasks after stirring overnight (Fig. 1a). The turbidities of the gels also changed when PEG initiators with different molecular weights (mPEG112–NH2 and mPEG45–NH2) were used. The gel initiated by using mPEG112–NH2 (Table 1, entry 1) was more transparent than the one initiated by mPEG45–NH2 (Table 1, entry 2). The gels were precipitated into diethyl ether and dried under vacuum to yield white powders as products. The ability to process the mPEG-b-PDLAG materials as either gels or powders allowed for detailed characterization of the compositions, structures and properties.
Scheme 1.
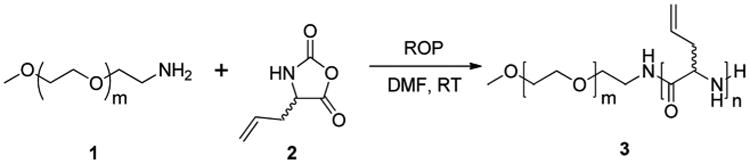
Synthesis of mPEG-b-PDLAG.
Table 1. Synthesis of mPEG-b-PDLAG with different initiators, concentrations and reaction times.
| Entry | Initiator 1 | [1]0 : [2]0 | Concentration (mg mL−1) | Time (h) | DPn | Mn, (kDa) |
|---|---|---|---|---|---|---|
| 1 | mPEG112–NH2 | 1 : 30 | 30 | 12b | 12 | 6.2 |
| 2 | mPEG45–NH2 | 1 : 30 | 30 | 12b | 12 | 3.2 |
| 3 | mPEG112–NH2 | 1 : 30 | 6 | 48b | 11 | 6.1 |
| 4 | mPEG45–NH2 | 1 : 30 | 6 | 48b | 13 | 3.1 |
| 5 | mPEG45–NH2 | 1 : 30a | 30 | 12b | 12 | 3.2 |
2% (wt) 4-methoxyphenol was added as radical inhibitor.
Gel formed after 8 h.
Fig. 1.

(a) Picture of reaction flasks of entry 1 of Table 1 after 12 h at rt (left) and entry 2 of Table 1 after 12 h at rt (right). (b) TEM image of the DMF gel of entry 1 of Table 1, negatively stained with 1 wt% uranyl acetate aqueous solution. (c) AFM height image of DMF gel of entry 1 of Table 1.
The degrees of polymerization and molecular weights of the polymers were determined by a combination of NMR spectroscopy and MALDI-TOF mass spectrometry, whereas gel permeation chromatography studies experienced significant difficulties due to pre-gel aggregate formation. Dissolution of the powders in DMSO-d6 gave a delayed gelation response, requiring ca. 12 h, which allowed for confirmation of the formation of mPEG-b-PDLAG diblock copolymers and determination of the PDLAG degrees of polymerization by 1H NMR spectroscopy measurements (Fig. 2). Comparison of the methylene proton resonances of the PEG chain at ca. 3.4 ppm with the intensities of the PDLAG alkenyl protons resonating at ca. 5.0 ppm (e and e′) or methine proton at 5.7 ppm (d), or the proton signal at 4.3 ppm, allowed for calculation of the DPn of PDLAG. Interestingly, in all the experiments performed, the reaction mixture gelled at ca. 40% conversion, to result in similar PDLAG DPns ≈ 12. MALDI-TOF measurement by using α-cyano-4-hydroxycinnamic acid (CHCA) as the matrix was also carried out to characterize the molecular weight of mPEG112-b-PDLAG12. As shown in Fig. 2b, the distribution with molecular weight centered at ca. 6 kDa was in accordance with the number-average molecular weight calculated by 1H NMR spectroscopy (Mn = 6.2 kDa). Gel permeation chromatography (GPC, using DMF as eluent) was also used to characterize the diblock copolymer, however, significant difficulties were experienced due to pre-gel aggregate formation within each sample (Fig. 3). Both of the diblock copolymers (mPEG45-b-PDLAG12 and mPEG112-b-PDLAG12) were dissolved in DMF solution at a concentration of 3 mg mL−1 followed by 2 min sonication and passing through a 0.2 μ m nylon filter prior to injection into the GPC. Primary peaks at short and long retention times were observed, along with other intermediate peaks, in the chromatograms for both the mPEG45-b-PDLAG12 and mPEG112-b-PDLAG12. For mPEG112-b-PDLAG12 (Fig. 3a), the short retention time peak correlated to an ultra-high molecular weight, Mn = 722 kDa (PDI = 1.43), which was much higher than the molecular weight of the diblock copolymer determined by 1H NMR spectroscopy and was attributed to strong inter-molecular interactions and pre-gel aggregation in DMF. The second peak calculated to a relatively lower molecular weight, Mn = 22.6 kDa, (PDI = 1.07), more in alignment with the molecular weight obtained by 1H NMR and MALDI-TOF, and is expected to correspond to the non-aggregated polymer in DMF. Similarly, the GPC chromatogram for mPEG45-b-PDLAG12 (Fig. 3b) includes two primary peaks, Mn = 2240 kDa (PDI = 1.32) and Mn = 13.5 kDa (PDI = 1.08), corresponding to non-covalent aggregation of diblock polymer and solvated diblock copolymer, respectively, as well as populations of intermediately sized aggregates across the range of the chromatogram. In both of the traces, the peaks with low molecular weight were of low intensity, indicating that the majority of the sample was undergoing significant aggregation by strong intermolecular interactions in DMF. With the shorter PEG segment, both larger sized aggregates and a larger proportion of aggregated sample were observed, suggesting that the gelation is driven by the PDLAG segment.
Fig. 2.
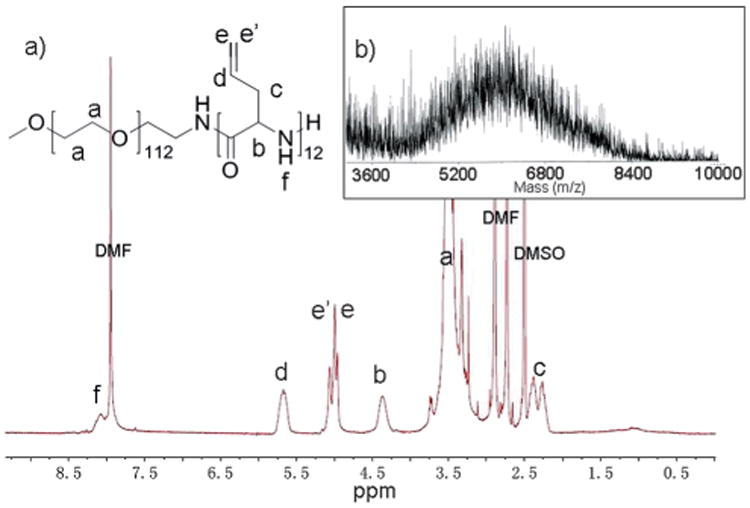
(a) 1H NMR spectrum of mPEG112-b-PDLAG12. (b) MALDI-TOF spectrum of mPEG112-b-PDLAG12.
Fig. 3.
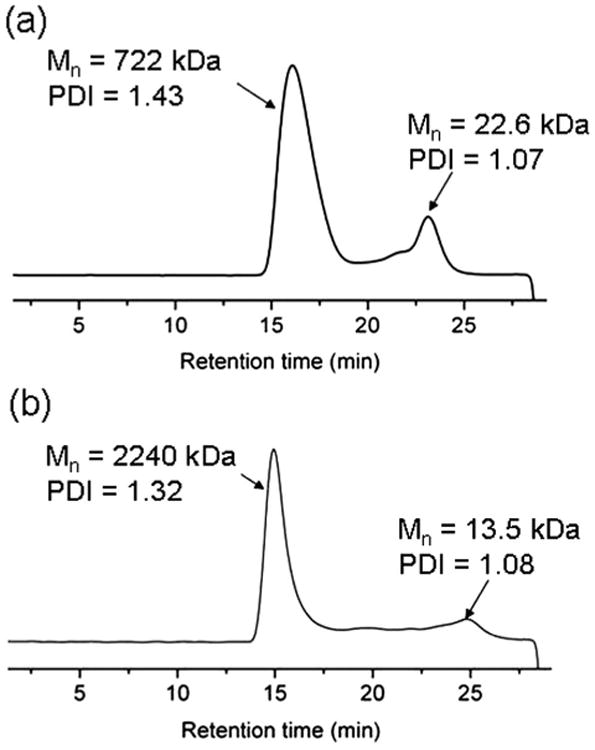
GPC (DMF eluent) traces of (a) of mPEG112-b-PDLAG12, and (b) mPEG45-b-PDLAG12.
To determine whether the PDLAG interactions that led to 3D-network formation and gelation were covalent or non-covalent, to afford an irreversible chemical or reversible physical gel, respectively, the reversibility of gel formation and the influence of radical inhibitor addition were studied. As noted above, the isolation powder sample could be dissolved in DMSO, with delayed gelation occurring, as evidence for reversibility of gel formation. In addition, when 2 wt% 4-methoxyphenol was added as radical inhibitor (Table 1, entry 5) to minimize potential cross-linking between pendant allyl functionalities, there was no significant difference between the monomer conversions, the gelation times and the gels formed with or without the existence of radical inhibitor (Table 1, entry 5 vs. entry 2). Furthermore, 1H NMR spectra of the polymers made from the conditions of Table 1, entry 5 and entry 2 were nearly identical. These results indicated that the driving force for the formation of the gel was not covalent cross-linking between allyl functionalities.
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) were used to investigate the microstructure of the organogel. The TEM samples were prepared and stained by uranyl acetate (Fig. 1b) or phosphotungstic acid (PTA) (Fig. S1†). Micrometer long nano-fibers with a width of ∼12 nm can be observed uniformly throughout the TEM images collected on the mPEG112-b-PDLAG12 organogel samples. The organogel prepared from mPEG45-b-PDLAG12 in DMF was also studied by TEM (Fig. S2†) and a similar fibrous network with micrometer fiber lengths and widths of ∼10 nm were observed. To diminish the evaporation artifacts, cryo-TEM images of the gel state samples were also collected and showed fibrillar network morphology (Fig. S3†). Tapping-mode AFM measurements of mPEG112-b-PDLAG12 gel were also performed. The AFM samples were prepared by spin coating the solution of mPEG112-b-PDLAG12 (0.05 mg mL−1 in DMF) onto freshly cleaved mica and drying under vacuum overnight. AFM height image (Fig. 1c) presented well-defined fibrous structures with an average height of ∼2.5 nm and an average width of ∼18 nm. The AFM height vs. width result suggested that the individual fibrous structures had a ribbon-like rather than a cylindrical morphology. However, an additional factor that could contribute to the fiber flattening is the highly polar mica surface, which could lead to significant collapse of the nano-fibers on the substrate, relative to the hydrophobic carbon surface used for TEM, thereby causing the fibers to exhibit larger than expected widths and smaller than expected heights.
The supramolecular structure of the dried gel was investigated by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) (Fig. 4). The DMF gel made from mPEG112-b-PDLAG12 was dried under vacuum and then measured by ATR-FTIR. The strong amide I band at ∼1627 cm−1 with a weak shoulder at ∼1690 cm−1 can be assigned to parallel and antiparallel β-sheet conformations of the peptide chains respectively.34,35 In addition, the peak in the amide II region at 1520 cm−1 was also consistent with a β-sheet conformation.36 The β-sheet characteristic of the peptide block was further supported by the presence of an amide V band at∼720 cm−1.37 If polypeptides adopt an α helix conformation, low or no signal is observed at ∼700 cm−1, instead a strong amide V band appears typically at 620 cm−1.37 In the FT-IR spectra of mPEG and dl-allylglycine NCA, no peaks were observed at 620 or 700 cm−1, so the peak appearing at 720 cm−1 was assigned to amide V band of PDLAG adopting a β-sheet conformation.
Fig. 4.
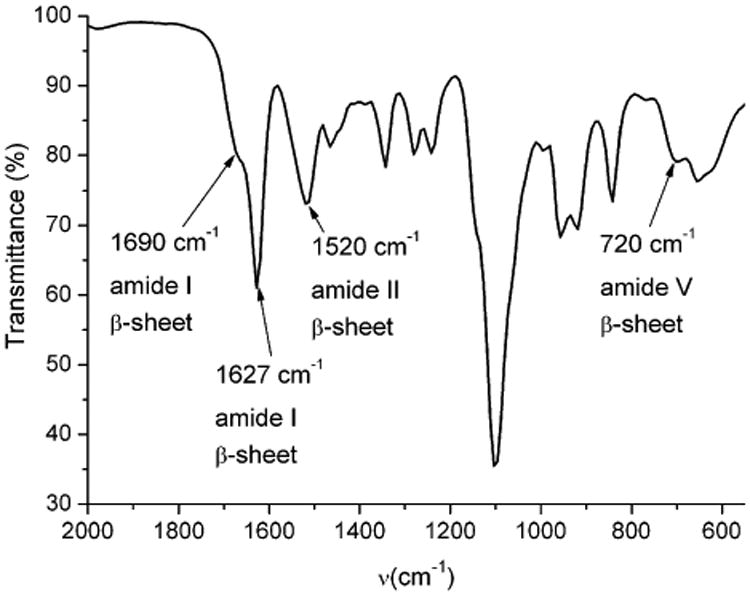
The ATR-FTIR spectrum of dried DMF gel made from mPEG112-b-PDLAG12.
The nanoscale structure of the diblock polymers was also investigated by wide angle X-ray scattering (WAXS). The WAXS pattern for mPEG112-b-PDLAG12 (Fig. 5) gave d spacings of 10.05 and 4.61 Å. A d spacing of 4.61 Å has previously been attributed to antiparallel β-sheet formation in poly(l-alanylglycine)36 and has also been observed in the self-assembled PEG-b-β-sheet-peptides described by Klok and coworkers for solid state organogels,35 and also by Castelletto et al. for hydrogels.38 The strong sharp signal with an associated spacing d = 4.61 Å also included a PEG 120 reflection. Likewise intersheet spacings of 10.05 Å support a β-sheet structure.39–41
Fig. 5.
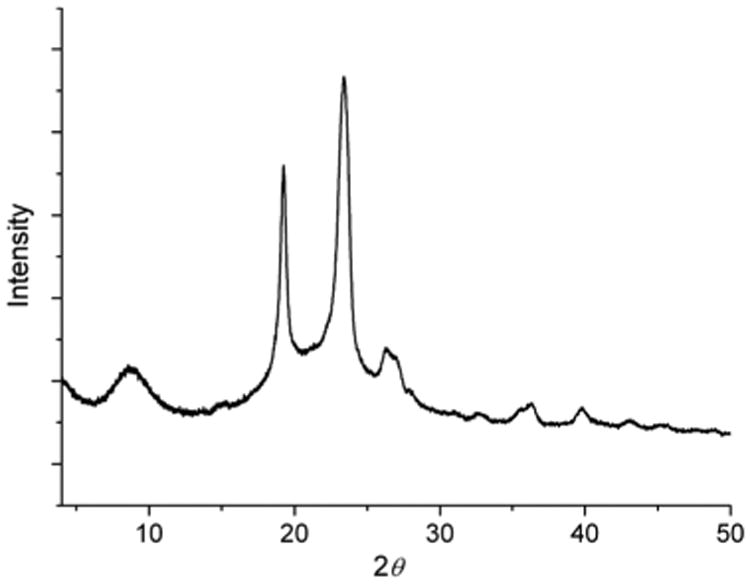
WAXS patterns of mPEG112-b-PDLAG12.
Based on TEM, AFM, ATR-FTIR, and WAXS data, a model for the gelation of the diblock polymer was proposed (Scheme 2). A possible explanation for the well-defined ribbon-like fibers observed in TEM and AFM is that the ribbons were assembled from the one-dimensional stacking of block polymers in a monolayer fashion. Using mPEG45-b-PDLAG12 gel as an example, the radius of gyration of PEG with a molecular weight of 2000 Da can be estimated as 18 Å.35 The axial distance between adjacent α-amino acids is ∼3.5 Å; with the rigid β-sheet feature, the width of peptide domain can be estimated to be 42 Å. Therefore, the width of the self-assembled ribbon can be estimated as ∼114 Å, which is in agreement with the ribbon width observed in TEM (Fig. S2†).
Scheme 2.
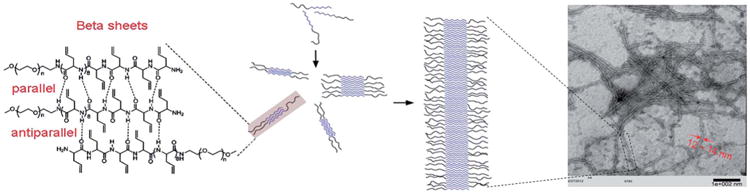
A schematic illustration of the nanofiber formed in the network structure of the DMF gel of mPEG-b-PDLAG12.
In addition, the physical properties of mPEG112-b-PDLAG12 organogel were also investigated (Table 2). The apparent critical gelation concentrations (Cgel, below which the solution exists as a viscous fluid) were measured by inversion tests at room temperature in a variety of organic solvents. We found that the diblock polymer could not be dissolved in non-polar or low polarity organic solvents, such as hexanes, ethyl acetate, dichloromethane, chloroform and tetrahydrofuran, but could be dissolved in relatively highly polar organic solvents such as DMSO, DMF and methanol. After being maintained at room temperature for 8–72 h, transparent gels were obtained. The organogels showed thermal and sonication responsive properties. When elevated to certain temperatures (Tgel), the gels transformed into solution states, while after being cooled down and kept at room temperature for certain periods, the solutions changed back to form gels again. The sol–gel transitions were reversible for a minimum of 3 cycles (Fig. S4†). Besides the thermal stimulus, the 0.1 wt% mPEG112–PDLAG12 DMF gel was transformed to solution after 2 min sonication at room temperature. This transition was also reversible, with the gel reforming after the solution was maintained at room temperature for 8 h. The sol–gel transition temperature (Tgel) measured at Cgel in DMF, DMSO and methanol are summarized in Table 2. Relative to other recently reported polypeptide based organogels,17,20,42 the gels formed from either mPEG112-b-PDLAG12 or mPEG45-b-PDLAG12 diblock copolymer exhibited extremely low critical gelation concentrations, and furthermore, occurred for a racemic homopolypeptide chain segment.
Table 2. Characterization and gelation properties of diblock copolymers.
| Block polymers | Solvent | Cgel [wt%] | Tgela [°C] | Timeb [h] |
|---|---|---|---|---|
| mPEG112-b-PDLAG12 | DMF | 0.1 | 48 | 8 |
| mPEG45-b-PDLAG12 | DMF | 0.2 | 56 | 8 |
| mPEG112-b-PDLAG12 | DMSO | 0.3 | 75 | 12 |
| mPEG112-b-PDLAG12 | Methanol | 0.5 | — | 72 |
| mPEG45-b-PDLAG12 | Methanol | 0.5 | — | 72 |
The sol–gel transition temperature measured with the organogels at Cgel.
Time needed to form stable gel at room temperature.
The mechanical properties of the gel systems were characterized by dynamic mechanical analysis (DMA) for gels at concentrations sufficiently high to produce a robust gel that could withstand the DMA high frequency operation. For the DMF gel made by using mPEG45-b-PDLAG12 at 2 wt% concentration assessment by DMA at 25 °C, operated at 1 Hz, the initial storage modulus (E′) was found to be 15.1 ± 0.2 kPa, with loss modulus (E″) and tan(δ) values of 13.1 ± 0.1 kPa and 0.866, respectively. Static stress–strain in compression provided a Young's modulus (E) of 25.1 ± 0.5 kPa (Fig. S5†). This value is slightly higher than a E= (E′2 + E″2)1/2 assumption of 20 kPa, attributed to the very low strain (0.5%) used in the static configuration preventing the engagement of loss processes and a transition into nonlinear behavior. As the evolution of solvent from the gel, and the frequency of operation, can grossly affect the nature of the material response, a series of experiments whereby frequency was varied from 0.1 to 10 Hz over ca. 3 h is presented in Fig. 6. The gelation cross-over point initially (tan(δ) < 1 → tan(δ) > 1) was found to be at 2.5 Hz with <2.5 Hz expressing gel behavior and >2.5 Hz expressing fluidic response. This finding is important for understanding the shear/deformation rates at which gel behavior crosses back over to fluidic behavior. The total volume of gel studied decreased by 5.1% during this time, allowing the assessment of a less solvated system. Through evaluation of the atmosphere exposed area, determination of the DMF evolution rate was made from kinetic fitting of the change in storage modulus. Single exponential fitting kinetics at 1 Hz indicates a rate constant of 120 ± 10 minutes to an asymptotic limit of 20.7 ± 0.3 kPa. The half-life for this initial phase of solvent evaporation is 80 min, with a rate of mass loss over this period being 0.1% per cm2 per min. Correlating the volume loss at 3 h to the kinetic rate, a calculated critical value for mass loss due to solvent evaporation (before some change of state/surface reordering becomes eminent) is 7.0%. By 160 min (4.8% mass loss) all frequencies observed had reached the gel point.
Fig. 6.
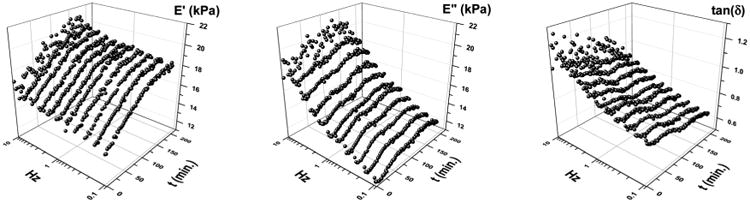
Dynamic mechanical responses from 0.1 to 10 Hz over 3 h.
Conclusions
In conclusion, organogelation characteristics were discovered for a known synthetic hybrid diblock polypeptide, which is of surprisingly low compositional and structure complexity, being based upon a hydrophilic PEG and a hydrophobic racemic homopolypeptide chain segment. Detailed characterization studies indicated that gelation is driven by the supramolecular assembly of β-sheets in the peptide blocks. The one-dimensional stacking of block copolymers in a monolayer fashion, then leads to the construction of well-defined ribbon-like morphologies. These supramolecular interactions are sufficiently strong to achieve ultra-low critical gelation concentrations. Investigation of the mechanical properties of the gels indicated a tunable system, where solvent concentrations can be used to alter the viscoelastic properties with increases in storage modulus 5.4% per 1% solvent lost. The results illustrate a novel and general mode of self-assembly for amphiphilic hybrid diblock synthetic polypeptides and demonstrate an attractive approach to fabricate supramolecular nanomaterials derived from racemic synthetic polypeptides. Moreover, the polypeptide segment carries reactive side chain functionalities, which are being explored for transformation into a variety of functional polymers, together with determination of their gelation characteristics in organic solvents or water.
Supplementary Material
Acknowledgments
This work was supported in part from the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and the National Science Foundation under grant numbers DMR-0906815 and DMR-1105304. The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant no. A-0001. We acknowledge the use of the Laboratory for Biological Mass Spectrometry at Texas A&M University.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm50582k
Notes and references
- 1.Cui HG, Webber MJ, Stupp SI. Biopolymers. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812–1816. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 3.Park MH, Joo MK, Choi BG, Jeong B. Acc Chem Res. 2012;45:424–433. doi: 10.1021/ar200162j. [DOI] [PubMed] [Google Scholar]
- 4.Kramer JR, Deming TJ. J Am Chem Soc. 2012;134:4112–4115. doi: 10.1021/ja3007484. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Hernandez J, Lecommandoux S. J Am Chem Soc. 2005;127:2026–2027. doi: 10.1021/ja043920g. [DOI] [PubMed] [Google Scholar]
- 6.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 7.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 8.Kotharangannagari VK, Sanchez-Ferrer A, Ruokolainen J, Mezzenga R. Macromolecules. 2012;45:1982–1990. [Google Scholar]
- 9.Pati D, Kalva N, Das S, Kumaraswamy G, Sen Gupta S, Ambade AV. J Am Chem Soc. 2012;134:7796–7802. doi: 10.1021/ja300065f. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez AR, Choe UJ, Kamei DT, Deming TJ. Macromol Biosci. 2012;12:805–811. doi: 10.1002/mabi.201200002. [DOI] [PubMed] [Google Scholar]
- 11.Huang J, Bonduelle C, Thevenot J, Lecommandoux S, Heise A. J Am Chem Soc. 2012;134:119–122. doi: 10.1021/ja209676p. [DOI] [PubMed] [Google Scholar]
- 12.Motala-Timol S, Jhurry D, Zhou JW, Bhaw-Luximon A, Mohun G, Ritter H. Macromolecules. 2008;41:5571–5576. [Google Scholar]
- 13.Zhao X, Poon Z, Engler AC, Bonner DK, Hammond PT. Biomacromolecules. 2012;13:1315–1322. doi: 10.1021/bm201873u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang XQ, Li JG, Li W, Zhang A. Biomacromolecules. 2007;8:3557–3567. doi: 10.1021/bm700729t. [DOI] [PubMed] [Google Scholar]
- 15.Ding JX, Xiao CS, Zhao L, Cheng YL, Ma LL, Tang ZH, Zhuang XL, Chen XS. J Polym Sci, Part A: Polym Chem. 2011;49:2665–2676. [Google Scholar]
- 16.Li ZB, Deming TJ. Soft Matter. 2010;6:2546–2551. [Google Scholar]
- 17.Kang EY, Yeon B, Moon HJ, Jeong B. Macromolecules. 2012;45:2007–2013. [Google Scholar]
- 18.Moon HJ, Choi BG, Park MH, Joo MK, Jeong B. Biomacromolecules. 2011;12:1234–1242. doi: 10.1021/bm101518c. [DOI] [PubMed] [Google Scholar]
- 19.Jeong Y, Joo MK, Sohn YS, Jeong B. Adv Mater. 2007;19:3947–3950. [Google Scholar]
- 20.Gibson MI, Cameron NR. Angew Chem, Int Ed. 2008;47:5160–5162. doi: 10.1002/anie.200801056. [DOI] [PubMed] [Google Scholar]
- 21.Kim KT, Park C, Vandermeulen GWM, Rider DA, Kim C, Winnik MA, Manners I. Angew Chem, Int Ed. 2005;44:7964–7968. doi: 10.1002/anie.200502809. [DOI] [PubMed] [Google Scholar]
- 22.Yan XH, Cui Y, He Q, Wang KW, Li JB. Chem Mater. 2008;20:1522–1526. [Google Scholar]
- 23.Vintiloiu A, Leroux JC. J Controlled Release. 2008;125:179–192. doi: 10.1016/j.jconrel.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Scanlon S, Aggeli A, Boden N, Koopmans RJ, Brydson R, Rayner CM. Micro Nano Lett. 2007;2:24–29. [Google Scholar]
- 25.Tang HY, Lee CU, Zhang DH. J Polym Sci, Part A: Polym Chem. 2011;49:3228–3238. [Google Scholar]
- 26.Suzuki M, Hanabusa K. Chem Soc Rev. 2010;39:455–463. doi: 10.1039/b910604a. [DOI] [PubMed] [Google Scholar]
- 27.Couffin-Hoarau AC, Motulsky A, Delmas P, Leroux JC. Pharm Res. 2004;21:454–457. doi: 10.1023/B:PHAM.0000019299.01265.05. [DOI] [PubMed] [Google Scholar]
- 28.Hamley IW, Ansari A, Castelletto V, Nuhn H, Rosler A, Klok HA. Biomacromolecules. 2005;6:1310–1315. doi: 10.1021/bm049286g. [DOI] [PubMed] [Google Scholar]
- 29.Top A, Roberts CJ, Kiick KL. Biomacromolecules. 2011;12:2184–2192. doi: 10.1021/bm200272w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadjichristidis N, Iatrou H, Pitsikalis M, Sakellariou G. Chem Rev. 2009;109:5528–5578. doi: 10.1021/cr900049t. [DOI] [PubMed] [Google Scholar]
- 31.Tang HY, Zhang DH. Polym Chem. 2011;2:1542–1551. [Google Scholar]
- 32.Sun J, Schlaad H. Macromolecules. 2010;43:4445–4448. [Google Scholar]
- 33.Deming TJ. Prog Polym Sci. 2007;32:858–875. [Google Scholar]
- 34.Miyazawa T, Blout ER. J Am Chem Soc. 1961;83:712–719. [Google Scholar]
- 35.Rosler A, Klok HA, Hamley IW, Castelletto V, Mykhaylyk OO. Biomacromolecules. 2003;4:859–863. doi: 10.1021/bm034058s. [DOI] [PubMed] [Google Scholar]
- 36.Panitch A, Matsuki K, Cantor EJ, Cooper SJ, Atkins EDT, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1997;30:42–49. [Google Scholar]
- 37.Miyazawa T, Masuda Y, Fukushima K. J Polym Sci. 1962;62:S62–S64. [Google Scholar]
- 38.Castelletto V, Cheng G, Furzeland S, Atkins D, Hamley IW. Soft Matter. 2012;8:5434–5438. [Google Scholar]
- 39.Serpell LC, Smith JM. J Mol Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- 40.Kirschner DA, Abraham C, Selkoe DJ. Proc Natl Acad Sci U S A. 1986;83:503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao H, Nguyen T, Romano NC, Modarelli DA, Parquette JR. J Am Chem Soc. 2009;131:16374–16376. doi: 10.1021/ja906377q. [DOI] [PubMed] [Google Scholar]
- 42.Choi YY, Jeong Y, Joo MK, Jeong B. Macromol Biosci. 2009;9:869. doi: 10.1002/mabi.200900095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.