

Abstract
Studies suggest that Gr1+CD11b+ cells have immunoregulatory function and these cells may play an important role in autoimmune diseases. In this study, we investigated the regulatory role of Gr1+CD11b+ cells in protecting against type 1 diabetes in NOD mice. Here we showed that temporary B cell depletion induced the expansion of Gr1+CD11b+ cells. Gr1+CD11b+ cells not only directly suppress diabetogenic T cell function, but can also induce Treg differentiation in a TGF-β-dependent manner. Furthermore, we found that Gr1+CD11b+ cells could suppress diabetogenic CD4 and CD8 T cell function in an IL-10-, nitric oxide- and cell contact- dependent manner. Interestingly, single anti-Gr1 monoclonal antibody treatment can also induce a transient expansion of Gr1+CD11b+ cells that delayed diabetes development in NOD mice. Our data suggest that Gr1+CD11b+ cells contribute to the establishment of immune tolerance to pancreatic islet autoimmunity. Manipulation of Gr1+CD11b+ cells could be considered as a novel immunotherapy for the prevention of type 1 diabetes.
Introduction
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of cells that are Gr1+CD11b+ (1). Gr1+CD11b+ cells, as part of a myeloid macropopulation, comprise at least two subsets of polymorphonuclear and monocytic cells with different immunosuppressive properties (2). They have been studied in tumor immunology (3) and other diseases such as graft-versus-host disease (4), sepsis and trauma (5). Recently, the immunosuppressive function of Gr1+CD11b+ cells has also been recognized in autoimmune diseases (6–10). In experimental induced organ specific autoimmune disease, Gr1+CD11b+ cells can be found in the spleen and in target organs, and they may play a role in limiting the T cell response to autoantigens in the target tissue (8). CD11b+Ly-6Chigh cells induced during EAE priming are powerful suppressors of activated T cells (6). When B10.RIII mice are immunized to induce experimental autoimmune uveoretinitis (EAU), Gr1+CD11b+ cells accumulate in large numbers at the peak of disease (9). Iwata and colleagues reported the involvement of Gr1lowCD11b+ cells in autoimmune disorder in MRL-Faslpr mice via the regulation of CCL2/CCR2 signaling (10). In skin transplantation models, adoptive transfer of Gr1+CD11b+ cells and M-CSF induced Gr1+CD11b+ cells can prolong allogeneic graft survival (11, 12). Transplantation tolerance induced by anti-CD28 treatment was association with the accumulation of Gr1+CD11b+ cells in rat kidney allografts (13). Mobilization of bone marrow CD11b+CD115+Gr1+ monocytes could lead to indefinite cardiac allograft survival (14).
In an allogeneic islet transplantation model, adoptive transfer of bone marrow derived Gr1+CD11b+ cells protected recipients from recurrent diabetes (15). Using tumor derived MDSCs, Yin and colleagues showed that CD115+Gr1+ MDSCs efficiently prevents the onset of hemagglutinin-specific TCR T cell-induced diabetes in INS-HA/RAG−/− recipient mice (16). Furthermore, in a spontaneous diabetes model, adoptive transfer of Gr1+CD11b+ cells, generated using GM-CSF and TGF-β stimulated bone marrow cells from transgenic mice expressing proinsulin driven by the class II promoter, protected against diabetes in Non obese diabetic (NOD) mouse (17). However, whether the expansion of endogenous Gr1+CD11b+ cells by monoclonal antibody treatment can control pancreatic islet specific autoimmunity and induce immune tolerance is not known. This is of interest because we found that temporary B cell depletion induced regulatory T and B cells in the hCD20.NOD mouse model (18). Moreover, in the present study, we found that B cell depletion also expanded a subset of Gr1+CD11b+ cells with characteristics of MDSCs. We have further investigated the role of Gr1+CD11b+ cells in beta cell autoimmune tolerance in spontaneous diabetes. We found that Gr1+CD11b+ cells prevented T1D in NOD mice through multiple immune tolerance pathways.
Materials and Methods
Mice
The NOD/Caj mice have been maintained at Yale University for over 20 years. All the mice were kept in specific pathogen–free conditions in a 12-hour dark/light cycle and housed in individually ventilated filter cages with autoclaved food. Human CD20-transgenic NOD (hCD20/NOD) mice were generated as described previously (18). The use of the animals in this study was approved by the Yale University Institutional Animal Care and Use Committee.
Antibodies and reagents
All fluorochrome-conjugated monoclonal antibodies (mAbs) used in this study were purchased from eBioscience. All the hybridoma supernatants containing different mAbs were generously provided by the late Charles Janeway (Yale University). Affinity-purified anti-hCD20 monoclonal antibody 2H7 was prepared as described previously (18). Anti-Gr1 monoclonal antibody (clone RB6-8C5) that binds particularly to the Ly6G component of Gr1, anti-IL-10 (clone JESS-2A5), anti-IL-10R (clone 1B1.3A), and anti-TGF-β (clone 1D11) were purchased from Bio X Cell Inc. Control mouse or rat IgG used in the in vivo studies was purchased from Rockland.
B cell depletion
Temporary B cell depletion in hCD20/NOD mice, using anti-human CD20 monoclonal antibody (clone 2H7), was performed as previously described (18). Briefly, 9 week old hCD20/NOD mice were injected intravenously with 2H7 or control IgG at 0.5 mg/mouse for the first injection (day 0), followed by 3 injections of 0.25 mg/mouse at 3-day intervals.
Anti-Gr1 treatment and its effect on spontaneous diabetes development
Pre-diabetic female NOD mice (9 weeks old) were treated with a single dose of anti-Gr1 mAb (0.25mg/mouse) intravenously. A group of age and sex-matched mice were treated with rat IgG as controls. All the treated mice were observed for diabetes development up to 35 weeks. They were screened for glycosuria twice a week. Diabetes was confirmed by blood glucose measurement greater than 250 mg/dl (13.9 mmol/l).
T cell purification
T cells were isolated using MACS cell isolation kits following the manufacturer’s instructions (Miltenyi Biotech). In some experiments, T cells were also isolated by negative selection to remove B cells (anti-mouse immunoglobulins) and macrophages (clone F4/80) with magnetic beads. CD4+ T cells were purified by removing CD8+ T cells (clone TIB 105), B cells (anti-mouse immunoglobulins), and MHC class II+ cells (clone 10.2.16) using monoclonal antibodies and magnetic beads. CD8+ T cells were purified by removing CD4+ T cells (clone GK1.5), B cells and MHC class II+ cells. The purity of the cells was routinely ≥ 90%, analyzed by FACS.
T cell proliferation and inhibition assays
The following T cells were used in the assays: purified BDC2.5 TCR transgenic CD4 or NY8.3 TCR transgenic CD8 T cells or insulin reactive CD8 T cell clone 6426. The cells (1 × 105/well) were co-cultured with irradiated peritoneal cells (2 × 104/well), as antigen presenting cells, in the presence of BDC2.5 mimotope (19), IGRP peptide (20) or insulin B15–23 peptide (21), respectively, at 37°C, 5% CO2, for 4 days. 3H-Thymidine was added during the last 16 hours of culture. To study the suppressive function of Gr1+CD11b+ cells, FACS sorted Gr1+CD11b+ cells (2 × 104/well) from 2H7 and/or control IgG treated mice were added to the culture. To study the role of soluble immune regulatory molecules, such as IL-10 and nitric oxide, we performed in vitro cultures as described above in the presence or absence of anti-IL-10 (clone JESS-2A5), anti-IL-10R (clone 1B1.3A), and the nitric oxide synthase inhibitor L-NAME (Cayman Chemical), respectively. To test whether the suppressive function was cell-contact dependent, we performed the assays with Gr1+CD11b+ cells in a 0.2µm transwell culture system. All the assays were performed in triplicate for each of the culture conditions.
We also used CFSE dilution assay to study the inhibitory function of Gr1+CD11b+ cells in vivo by transferring CFSE labeled BDC2.5 CD4 or NY8.3 CD8 T cells into NOD recipients treated with anti-Gr1 or control IgG one week earlier.
Regulatory T cell induction in the presence of Gr1+CD11b+ cells
FACS sorted naïve CD4 T cells (CD4+CD25−CD44−/loCD62L+) from NOD mice were stimulated with anti-CD3 (2C11 hybridoma supernatant, 1:200) and anti-CD28 (37.51 hybridoma supernatant, 1:300) and co-cultured with purified Gr1+CD11b+ cells from anti-CD20 and/or control IgG treated mice at ratio of 5:1 in the presence of mouse M-CSF (50 ng/ml, PeproTech), at 37°C, 5% CO2. After 5 days, cells were collected for quantitative PCR analysis of foxp3 mRNA expression (5’-primer: CAGAGTATGACGTGAGAGACCAC, 3’-primer: CAGCTTGTCTACTTCAGTCATGGAG) and intracellular staining of FoxP3 protein expression using an anti-mouse/rat FoxP3 staining kit from eBioscience.
Histopathology and insulitis score
Pancreata were fixed in 10% buffered formalin and then paraffin-embedded. Tissues were sectioned and stained with H&E. Insulitis was scored under light microscopy using the following grading: 0, no insulitis; 1, insulitis affecting less than 25% of the islet; 2, insulitis affecting 25%–75% of the islet; 3, more than 75% islet infiltration. 107 islets from the control group (mice, n=5) and 124 islets from the anti-Gr1 treated group (mice, n=5) were scored.
Statistics
Statistical analysis was performed using GraphPad Prism software.
Results
Expansion of Gr1+CD11b+ cells that express IL-4 and IL-10 after B cell depletion
We have previously shown that temporary B cell depletion by anti-human CD20 monoclonal antibody, 2H7, prevented and reversed T1D in human CD20 transgenic NOD mice (18). To further characterize the mechanisms of immune tolerance induced by anti-CD20 treatment, we performed intracellular immunoregulatory cytokine staining for IL-4 and IL-10. Surprisingly, we found that B cell depletion expanded a population of IL-4 and IL-10 producing cells that were Gr1+ (Figure 1A). These cells were also CD11b+ (Figure 1B). The expansion of Gr1+CD11b+ cells was seen only in mice treated with 2H7 but not in IgG treated control mice (Fig 1B and 1C).
Figure 1.
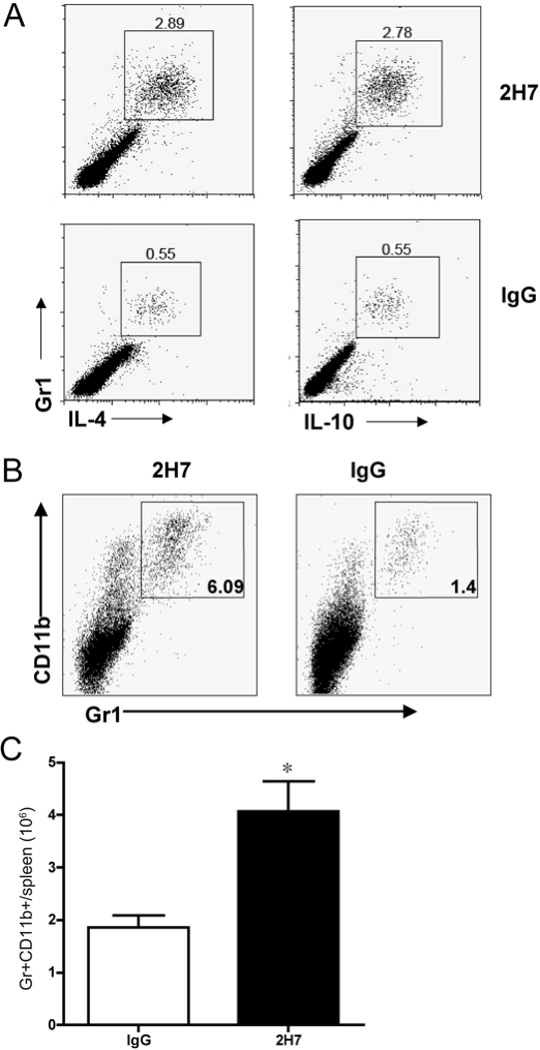
B cell depletion induced regulatory Gr1+ cells that express IL-4 and IL-10. A) Splenocytes from anti-CD20 (2H7) or control IgG treated hCD20.NOD mice (n=5/group) were stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin for 5 hours in the presence of GolgiStop (BD) and then stained with monoclonal antibodies against the surface markers CD4, CD11b and Gr1, followed by intracellular staining with anti-IL-4 and anti-IL-10 or isotype control antibody. One representative set of FACS plots is shown in Figure 1A with gating on CD4− cells. B) Splenocytes from mice treated with 2H7 or control mouse IgG were stained with monoclonal antibodies against the surface markers Gr1 and CD11b. C) Gr1+CD11b+ cell number was enumerated in splenocytes from 2H7 or control mouse IgG treated mice (n=4 each group, p = 0.011, Student’s t test).
Gr1+CD11b+ cells suppress T cell function in an IL-10 dependent manner
To test whether Gr1+CD11b+ cells could suppress T cell function directly in our system, we co-cultured purified diabetogenic CD4 or CD8 T cells with purified Gr1+CD11b+ cells from 2H7 treated hCD20/NOD mice (18), in the presence or absence of antigenic peptides. Interestingly, Gr1+CD11b+ cells suppressed both diabetogenic CD4 and CD8 T cell proliferation with a greater effect on CD8 T cells (Fig. 2A). The suppressive function was not restricted to anti-CD20 treatment as Gr1+CD11b+ cells from control IgG treated mice also showed similar suppression to the diabetogenic CD4 or CD8 T cells (Fig. 2D&E). This suggests that B cell depletion induces the expansion of a subset of pre-existing Gr1+CD11b+ cells with immunosuppressive function in hCD20/NOD mice. To further test the suppressive function of Gr1+CD11b+ cells in vivo, we adoptively transferred CFSE-labeled purified BDC2.5 TCR transgenic CD4+ T cells or purified NY8.3 TCR transgenic CD8+ T cells into NOD recipients that were pre-treated with anti-Gr1 antibody (which would lead the expansion of Gr1+CD11b+ cells, see below) or control IgG. Consistently, the proliferation of both BDC2.5 and NY8.3 T cells, which was indicated by CFSE dilution, was suppressed in the anti-Gr1 treated recipients, compared with the IgG treated mice (Fig. 2B).
Figure 2.

Gr1+CD11b+ cells suppressed diabetogenic T cell proliferation in an IL-10 dependent manner. A) In mixed lymphocyte cultures, purified Gr1+CD11b+ cells (2 × 104/well) suppressed the proliferation of BDC2.5 CD4 T cells (105/well) designated as CD4 or insulin-reactive CD8 T cell clone 6426 (105/well), designated as CD8. One representative data set is shown from at least 3 experiments. B) CFSE labeled BDC2.5 CD4 or NY8.3 CD8 T cells were adoptively transferred into NOD recipients (6 × 106/mouse). NOD recipients were treated with 0.25mg/mouse anti-Gr1 (grey area) or control IgG (dark line) monoclonal antibody at the time of T cell transfer. Cells from pancreatic lymph nodes were collected 7 days later for FACS analysis of CFSE dilution (n=5 each group). C) The addition of neutralizing anti-IL-10 monoclonal antibody (clone JESS-2A5; 10 µg/ml) partially reversed the suppression by Gr1+CD11b+ cells of BDC2.5 CD4 or insulin-reactive CD8 T cell clone 6426 proliferation. One representative data set is shown from at least 3 experiments. Gr1+CD11b+ cells (2 × 104/well) purified from both 2H7 and control IgG treated mice showed comparable suppression of the proliferation of diabetogenic D) BDC2.5 CD4+ (105/well) and E) NY8.3 CD8+ T cells (105/well). One representative data set is shown from 3 experiments. The baseline proliferation, measured by 3H-thymidine incorporation, for all assays was between 100–800 cpm. Error bars represent standard error of the mean. *p<0.05, **p<0.001, ***p<0.0001
We had shown earlier that Gr1+CD11b+ cells express the regulatory cytokine IL-10. To test whether IL-10 was responsible for the suppressive function of Gr1+CD11b+ cells, we cultured purified BDC2.5 CD4+ T cells or NY8.3 CD8+ T cells with antigenic peptides together with purified Gr1+CD11b+ cells from 2H7 treated hCD20/NOD mice in the presence or absence of anti-IL-10 blocking mAb (clone JESS-2A5). As shown in Figure 2C, the addition of anti-IL-10 antibody partially restored the proliferation of T cells. We also tested the role of IL-4 in the same culture system. However, the addition of IL-4 blocking antibody (clone 11B11) could not reverse the suppression (data not shown) although Gr1+CD11b+ cells also expressed IL-4. Similar results were also observed when using purified Gr1+CD11b+ cells from control IgG treated hCD20/NOD mice (data not shown).
The suppressive function of Gr1+CD11b+ cells is nitric oxide and cell-contact dependent
Previous studies have shown the involvement of nitric oxide (NO) in the suppressive function of myeloid-derived suppressor cells (22). To investigate whether NO plays a role in T cell suppression by Gr1+CD11b+ cells in our model system, we performed in vitro T cell culture, as described above, in the presence or absence of the pan-nitric oxide synthase inhibitor L-NAME. Our results showed that in the presence of L-NAME, the suppressive function of Gr1+CD11b+ cells was reversed for CD4 T cells and partially reversed for CD8 T cells (Figure 3A). To test if cell contact was required for suppression of T cells in our system we placed Gr1+CD11b+ cells in a 0.2 micrometer transwell in the above T cell culture system. The presence of the transwell reduced the suppressive function of Gr1+CD11b+ cells (Figure 3B). The NO- and cell contact-dependent suppression were also detected using Gr1+CD11b+ cells that were isolated from control IgG treated hCD20/NOD mice (data not shown).
Figure 3.
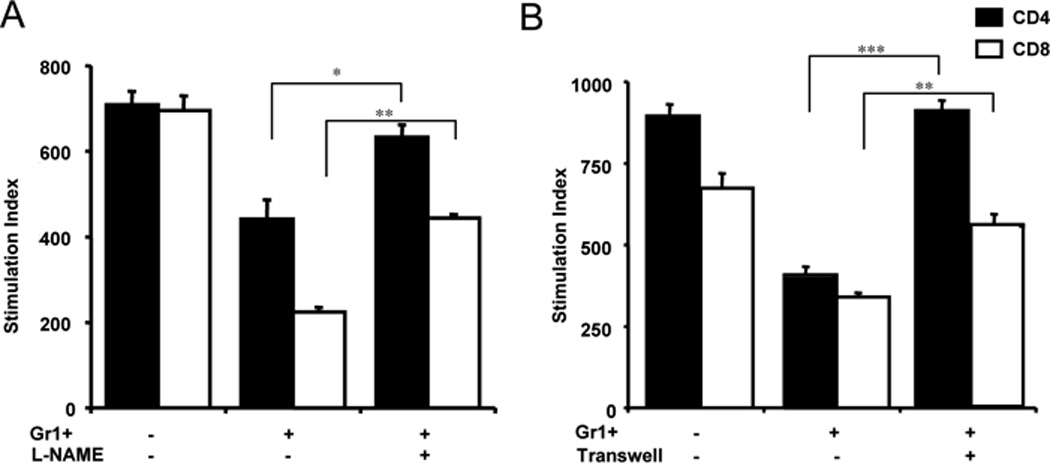
The immunosuppressive function of Gr1+CD11b+ cells is nitric oxide and cell contact dependent. A) T cells were cultured as described in Materials and Methods. The addition of the pan-nitric oxide synthase inhibitor L-NAME (1mM) reversed the suppression of both CD4+ and CD8+ T cell proliferation by Gr1+CD11b+ cells; B) The presence of a transwell abrogated the suppression of T cells by Gr1+CD11b+ cells. CD4+ and CD8+ T cells were co-cultured with purified Gr1+CD11b+ cells in the presence or absence of transwell as described in Materials and Methods. Three independent experiments were performed and one representative data set is shown. *p<0.05, **p<0.001, ***p<0.0001.
The background CPM counts were between 300–800.
Our results, thus far, suggest that B cell depletion induces the expansion of a subset of pre-existing Gr1+CD11b+ cells, which have characteristics of myeloid derived suppressor cells, rather than modulating their function.
Anti-Gr1 treatment prevents diabetes development in NOD mice through transient expansion of Gr1+CD11b+ cells
Since B cell depletion induced Gr1+CD11b+ cells that could contribute to the delay and reduction of diabetes onset in hCD20/NOD mice, we expected that removal of Gr1+CD11b+ cells would accelerate diabetes development in NOD mice. To test this hypothesis, we injected (i.v.) a group of pre-diabetic NOD mice at 9 weeks of age with 0.25mg of depleting anti-Gr1 monoclonal antibody (clone RB6-8C5). To our surprise, instead of accelerating the disease, we found that anti-Gr1 treatment significantly delayed and prevented diabetes development compared to the control rat IgG treated mice (Figure 4A, p<0.05). To test whether anti-Gr1 treatment affected the cellular infiltrate of pancreatic islets, some mice were randomly selected and sacrificed one month after treatment for insulitis analysis. There was significantly less infiltration in mice treated with anti-Gr1 compared with the control rat IgG-treated group as shown in Figure 4B and 4C (p<0.03).
Figure 4.
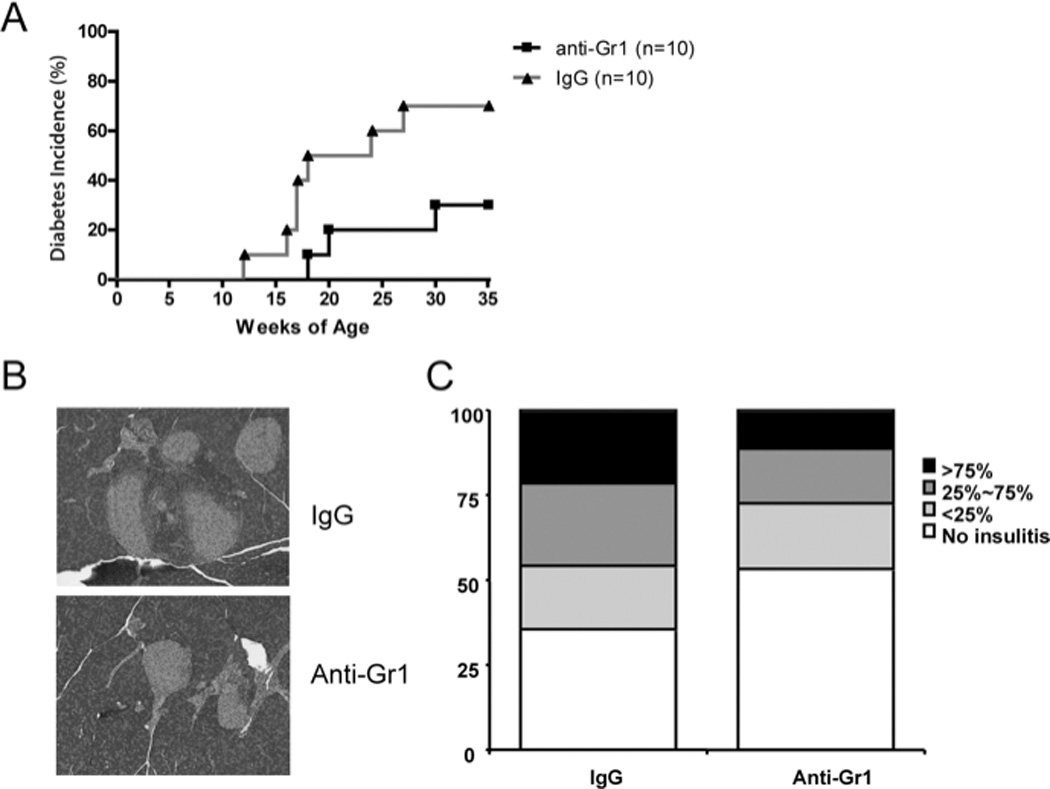
Anti-Gr1 treatment reduced insulitis and prevented type 1 diabetes development in NOD mice. 9 week-old female NOD mice were injected with one dose of 0.25mg anti-Gr1 or control IgG antibody intravenously. Diabetes development was monitored by testing for glycosuria twice a week. Disease was confirmed by a blood glucose level greater than 250 mg/dl. A) Diabetes incidence in mice treated with anti-Gr1 or control IgG antibody (n=10 each group, Log-rank test P=0.0455). B) and C) Insulitis in mice treated with anti-Gr1 or control IgG antibody (n=5 each group), was scored using the system outlined in the methods, χ2 test (p< 0.03).
To examine the efficiency of Gr1+CD11b+ cell depletion, we injected anti-Gr1 antibody (0.25mg/mouse) intravenously in an additional group of NOD mice and analyzed the Gr1+CD11b+ cells in peripheral blood over time using a different anti-Gr1 (clone 1A8, which recognizes a different epitope of Ly6G) and anti-CD11b antibodies. As shown in Figure 5A, one hour after the RB6-8C5 treatment, we observed a significant decrease in the number of Gr1+CD11b+ cells. The depletion of Gr1+CD11b+ cells was maintained for 4 days, at which time Gr1+CD11b+ cells gradually regenerated and increased to a higher level compared to the IgG treated control mice 7 days after treatment. This higher level of Gr1+CD11b+ cells persisted for about 10 days; the cell frequency and number were restored to normal by 18 days after anti-Gr1 treatment. The depletion dynamics demonstrated that anti-Gr1 treatment resulted in a short period of removal of Gr1+CD11b+ cells but was followed by a longer period of transient increase of Gr1+CD11b+ cells, which possibly explains the observed diabetes protection shown in Figure 4. We also observed expansion of Gr1+CD11b+ cells in spleen and lymph nodes from different anatomical sites 7 days after anti-Gr1 treatment (Fig. 5B). Our results support the finding by Bronte and colleagues, who showed that the discontinuation of anti-Gr1 treatment in a tumor model system resulted in increased numbers of myeloid-derived suppressor cells (1).
Figure 5.
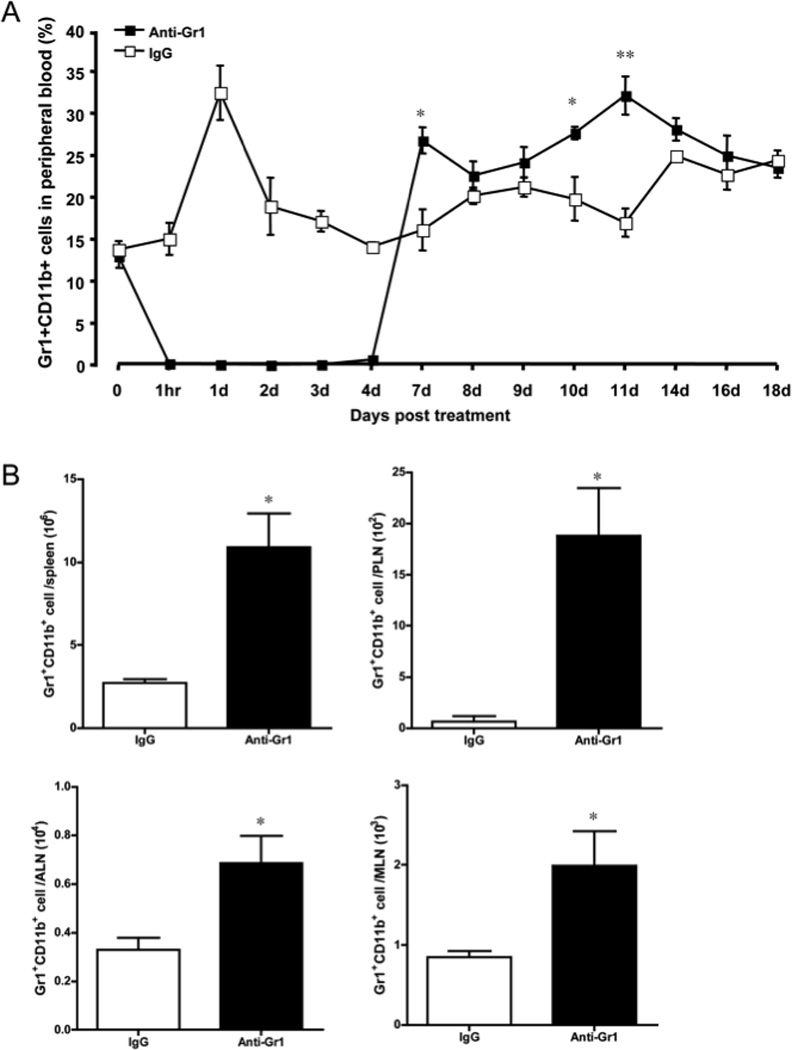
Gr1+CD11b+ cell depletion dynamics. NOD mice were treated with 0.25mg anti-Gr1 or control IgG antibody (n=5 each group). A) The percentage of Gr1+CD11b+ cells in peripheral blood was analyzed by flow cytometry over time. B) Gr1+CD11b+ cell number in peripheral lymphoid organs (spleen, PLN, ALN and MLN) was analyzed by flow cytometry 7 days after anti-Gr1 treatment (n=4 each group). The error bars represent standard error of the mean. P values were calculated by Student’s t test. * P<0.05, ** P<0.001.
To confirm that the expansion of Gr1+CD11b+ cells was responsible for diabetes protection by a single anti-Gr1 antibody injection, we tried to deplete Gr1+CD11b+ cells by multiple anti-Gr1 injections. We treated 9 week old female NOD mice with 4 weekly injections of anti-Gr1 antibody (0.25mg/mouse i.p.), such that reconstitution of the Gr1+CD11b+ cells was prevented during a critical period of diabetes protection. It is interesting that the continued depletion of Gr1+CD11b+ cells and more importantly, inhibition of the expansion of these cells over a longer period of time, abrogated diabetes protection (Fig. 6). The data further support the notion that diabetes protection was mediated by the expansion of Gr1+CD11b+ cells.
Figure 6.
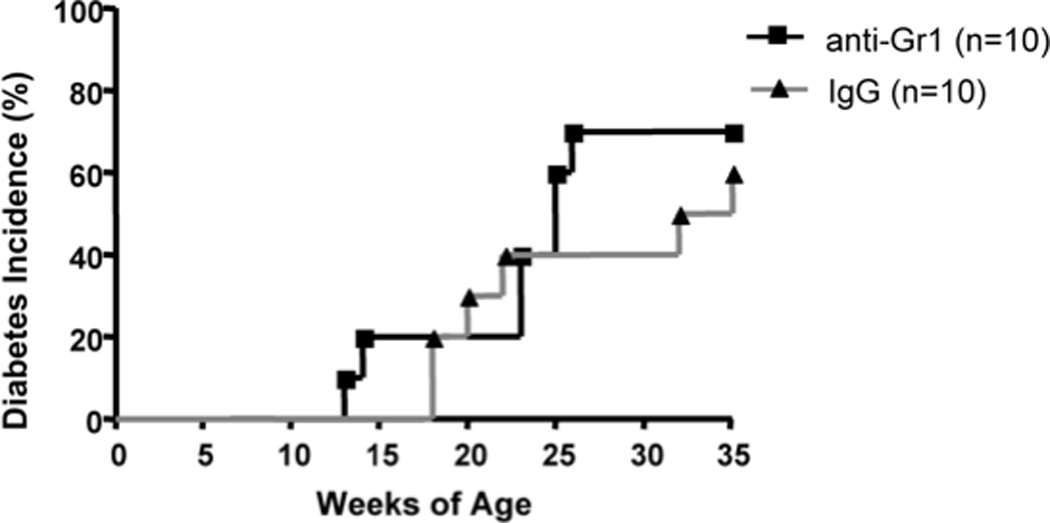
Prolonged depletion of Gr1 cells failed to prevent diabetes development in NOD mice. 9 week-old female NOD mice were treated with 4 injections (i.p.) of 0.25mg anti-Gr1 or control IgG antibody at 7 day intervals. Diabetes development was monitored by testing for glycosuria twice a week. Disease was confirmed by blood glucose level greater than 250 mg/dl. There was no statistically significant difference between control and experimental groups (n=10 each group, Log-rank test P=0.6262).
TGF-β is required for the induction of Foxp3+ regulatory T cells by anti-Gr1 treatment
To test the hypothesis that anti-Gr1 treatment could promote activity of other regulatory cells, including regulatory T cells (Treg), we analyzed FoxP3+ Treg in NOD mice one month after a single anti-Gr1 or rat IgG injection. It is interesting that we found a consistent increase in FoxP3+ Treg cells in the anti-Gr1 antibody treated mice compared to IgG treated control mice, which suggested that anti-Gr1 treatment induced Treg cells (Fig. 7A). The increase of CD4+Foxp3+ Treg cells was more evident at later time points, seen at 35 weeks of age, when the experiments were terminated (Figure 7B). We further tested whether Gr1+CD11b+ cells could induce Foxp3+ cells in vitro. We sorted naïve splenic CD4 T cells and Gr1+CD11b+ cells by FACS and co-cultured them for 5 days. FoxP3+ cells were then analyzed by intracellular staining. We found that a significant number of naïve T cells differentiated into FoxP3+ cells when they were co-cultured in the presence of Gr1+CD11b+ cells (Fig. 7C). The expression of the foxp3 gene was further confirmed at transcriptional level by quantitative PCR (Fig. 7D). It is known that TGF-β is essential for regulatory T cell differentiation. To understand whether TGF-β was involved in Treg induction by Gr1+CD11b+ cells, we measured the TGF-β level in the supernatant from the above co-culture experiments by ELISA and found an increased level of bioactive TGF-β in supernatants of co-cultured T cells and Gr1+CD11b+ cells (Fig. 8A). The upregulation of TGF-β was also confirmed at transcriptional level by qPCR (Fig. 8B). To test the role of TGF-β in the induction of FoxP3 expression, we added neutralizing anti-TGF-β monoclonal antibody (clone 1D11) to the co-culture system. The presence of neutralizing anti-TGF-β monoclonal antibody completely abrogated the induction of FoxP3 expression (Fig. 8C). To investigate the effect on Treg induction in vivo, we treated groups of NOD mice with anti-Gr1 antibody (single injection) with or without neutralizing anti-TGF-β antibody. We found that anti-TGF-β treated mice had fewer Foxp3+ Treg cells in the spleen compared to control mice treated with anti-Gr1 alone (Fig. 8D). Furthermore, the administration of anti-TGF-β antibody in vivo also abolished the protective effect of a single anti-Gr1 injection on diabetes development (Fig. 8E).
Figure 7.
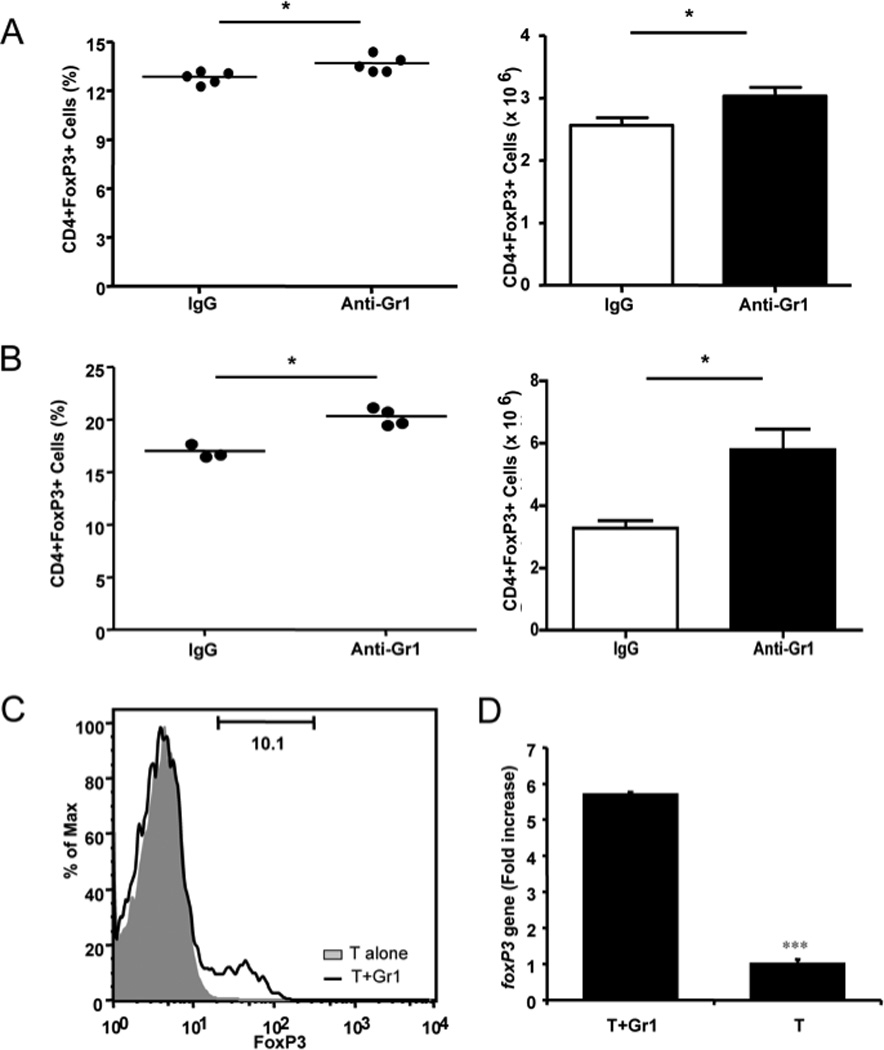
Anti-Gr1 treatment induced regulatory T cells. A) Percentage (left) and absolute numbers ± SEM (right) of splenic FoxP3+CD4 regulatory T cells from mice 1 month after control rat IgG or anti-Gr1 treatment. B) Percentage (left) and absolute numbers ± SEM (right) of regulatory FoxP3+CD4 T cells from splenocytes of the mice 6 months after control rat IgG or anti-Gr1 treatment. P values are shown on the graph (Student’s t test). C) Purified Gr1+CD11b+ cells were co-cultured with purified naïve T cells at 1:5 ratio for 5 days. The differentiation of naïve T cells into FoxP3+ regulatory T cells was determined by flow cytometry. D) FoxP3 mRNA was detected by quantitative PCR in the T cells cocultured with Gr1+CD11b+ cells (T+Gr1) compared with naïve T cells alone (T). Data from one of 4 representative experiments are shown. *p<0.05, ***p<0.0001.
Figure 8.

Gr1+CD11b+ cells induce regulatory T cell differentiation in a TGF-beta dependent manner. A) Secreted TGF-beta from culture supernatants (Fig. 7C) was tested by ELISA (BD Bioscience). Purified naïve T cells were co-cultured with purified Gr1+CD11b+ cells (T+Gr1) compared with T cells alone (T). *** p<0.0001. B) TGF-β expression was detected at transcriptional level by qPCR (5’-primer: TGGAGCCTGGACACACAGTA, 3’-primer: GTTGGACAACTGCTCCACCT). qPCR was performed in triplicate in 3 separate experiments. Data from one of 3 experiments are shown. *** p<0.0001. C) Purified Gr1+CD11b+ cells were co-cultured with purified naïve T cells at 1:5 ratio for 5 days in the presence or absence of neutralizing anti-TGF-β (clone 1D11; 10 µg/ml). The differentiation of naïve T cells into regulatory T cells was determined by flow cytometry. Three independent experiments were performed. D) and E) Nine week-old female NOD mice were injected with one dose of 0.25mg anti-Gr1 or control IgG antibody intravenously together with or without neutralizing anti-TGF-β (3 injections of 0.25 mg/mouse, 3 day intervals). FoxP3+CD4+ cells were examined at 35 weeks of age in both antibody and control IgG treated mice (D, percentage on the left and absolute numbers on the right, *p<0.05), together with the development of diabetes (E). Diabetes development was monitored by testing for glycosuria twice a week until 35 weeks of age. Disease was confirmed by a blood glucose level greater than 250 mg/dl. P=0.035 for anti-Gr1 vs anti-Gr1+anti-TGFβ groups; p=0.331 for control IgG vs anti-Gr1+anti-TGFβ groups; p=0.045 for anti-Gr1 vs control IgG groups.
Discussion
In this study, we have shown that B cell depletion expanded an immunosuppressive cell population that are Gr1+CD11b+ and can produce IL-10. We further showed that these Gr1+CD11b+ cells could suppress T cell function in an IL-10, nitric oxide, and cell contact dependent manner. Single anti-Gr1 treatment could also transiently expand Gr1+CD11b+ cells, which induced Foxp3+ regulatory T cells in a TGF-β dependent manner. The novelty of our study lies in the induction method of Gr1+CD11b+ cells where we found that both B cell depletion and anti-Gr1 treatment can each expand Gr1+CD11b+ cells. This expansion of the Gr1+CD11b+ cells is different from previous reports using tumor bearing mice or bone marrow as the source of Gr1+CD11b+ cells. This builds on our earlier study showing that temporary B cell depletion could prevent and reverse type 1 diabetes in hCD20/NOD mice through several mechanisms including induction of regulatory T and B cells, and suppression of antigen-presenting function of macrophages and dendritic cells (18). This highlights the fact that depleting a major subset of immune cells leads to an altered balance of cell populations as the cells repopulate with an increase in a number of regulatory cell types with immunosuppressive functions. We believe that all of these immune suppressive functions contribute to the prevention of type 1 diabetes development in transiently B cell depleted NOD mice.
Although Gr1+CD11b+ MDSCs are present in tumors and a variety of chronic inflammatory states, (6–8, 11), their presence will have a number of different consequences. In cancer, the immunosuppressive effects prevent an anti-tumor T cell response. In this setting, these cells have a deleterious effect, and reducing or removing them could be of therapeutic value. In autoimmunity, however, increasing the Gr1+CD11b+ cells and activating them could be of considerable importance, given their ability to reduce T cell responses directly and induce the expansion of regulatory T cells. Here, we showed that reconstitution of cells after both B cell depletion and anti-Gr1 treatment could also transiently increase Gr1+CD11b+ cells. One possible explanation for the expansion of Gr1+CD11b+ cells is that antibody treatment leads to Gr1 cell apoptosis. In response to these apoptotic cells, the immune system generates more immature myeloid cells which later become phagocytic cells including neutrophils, macrophages, and dendritic cells to phagocytose the dead cells. This was supported by the fact that when we adoptively transferred apoptotic syngeneic splenocytes, NOD recipients had expansion of Gr1+CD11b+ cells, which led to delayed diabetes development (unpublished data). The detailed mechanism(s) is under further investigation. It is, however, in their immature state that Gr1+CD11b+ cells exert their suppressive function.
Why are these immature myeloid cells present in these chronic inflammatory states? Delano and colleagues using a model of sepsis (23), showed that TLR signaling is important in expansion of this group of cells. Tissue damage induced in chronic inflammation may be important in inducing suppressive cells to reduce the inflammation. Clearly, under normal circumstances in diabetes, their presence in this context is not sufficient to stop the inflammation and beta cell destruction. Maintaining the cells in an immature state, whilst also activating them to carry out immunosuppressive functions over a longer period of time, would be an important therapeutic challenge. This is evidenced by transfer experiments where purified bone marrow-derived Gr1+CD11b+ cells were shown to delay but not completely protect NOD.SCID recipients from diabetes when co-transferred with diabetogenic NOD splenocytes (supplementary Fig. 1).
Interestingly, although the Gr1+CD11b+ cells in this study were induced by B cell depletion or single anti-Gr1 treatment, they exhibit immunosuppressive function through similar mechanisms to those of MDSCs found in tumor environments. Firstly, the suppressive function requires the production of nitric oxide by Gr1+CD11b+ cells and inhibition of nitric oxide synthase partially blocked their function. Secondly, cell-cell contact is essential for the regulatory function as our in vitro experiments showed that the presence of a transwell completely abrogated the immunosuppressive function of Gr1+CD11b+ cells. The upregulation of iNOS and Arginase1 gene expression in Gr1+CD11b+ cells further supported the role of NO for the immunosuppression (supplementary Fig 2). However, Gr1+CD11b+ cells induced by B cell depletion can suppress both CD4 and CD8 T cell proliferation, while MDSCs from tumor environments appeared to mainly affect CD8 T cells (22).
Our previous study showed an increase of Treg cells after B cell depletion (18). It was not clear how the depletion of B cells induced regulatory T cells. Our current study indicates that Treg induction is likely to be mediated by Gr1+CD11b+ cells after B cell depletion. This occurred in a TGF-β dependent manner. We suggest that B cell depletion induces Gr1+CD11b+ cells that not only function by producing NO and reactive oxygen mediators but also produce IL-10 and TGF-β. These cytokines will induce the differentiation of regulatory T cells and could further stimulate both CD4 T cells and Gr1+CD11b+ cells to produce more IL-10 and TGF-β.
Thus, in our study, we have reported the role of Gr1+CD11b+ cells in the development of type 1 diabetes in the NOD mouse model of T1D. This adds another regulatory population, both endogenously as well as by adoptive transfer, that could be therapeutically exploited to increase immune regulation in diabetes. It is striking that a single injection of anti-Gr1 antibody in pre-diabetic NOD mice promoted long-lasting immune regulation leading to diabetes protection. It is clear that immune regulation mediated by Gr1+CD11b+ cells is complex and more than one mechanism is involved in the regulatory function. Further understanding of how Gr1+CD11b+ cells function in situ in the pancreatic islet to modulate autoreactive T cells will provide new insight into pancreatic autoimmunity.
Supplementary Material
Acknowledgments
This study was supported by grants from the Juvenile Diabetes Research Foundation International (Research grant 1-2007-586 to LW, advanced postdoctoral fellowship 10-2010-478 to CYH). This study was also supported in part by the Diabetes Mouse Core of DERC (NIDDK) to LW.
Abbreviations
- PLN
pancreatic draining lymph node
- MLN
mesenteric lymph node
- ALN
auxiliary lymph node
- DC
dendritic cell
- T1D
type 1 diabetes
Footnotes
There are no conflicts of interest relevant to this article.
CYH, LW and FSW designed the experiments. CYH performed most of the experiments; WD and LZ performed some of the experiments; LW supervised the project and CYH, FSW and LW wrote the manuscript.
References
- 1.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, Taylor PA, Panoskaltsis-Mortari A, Serody JS, Munn DH, Tolar J, Ochoa AC, Blazar BR. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010;116:5738–5747. doi: 10.1182/blood-2010-06-287839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med. 2011;17:281–292. doi: 10.2119/molmed.2010.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, Khoury SJ. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228–5237. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- 7.Marhaba R, Vitacolonna M, Hildebrand D, Baniyash M, Freyschmidt-Paul P, Zoller M. The importance of myeloid-derived suppressor cells in the regulation of autoimmune effector cells by a chronic contact eczema. J Immunol. 2007;179:5071–5081. doi: 10.4049/jimmunol.179.8.5071. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson LB, Raveney BJ, Munder M. Monocyte dependent regulation of autoimmune inflammation. Curr Mol Med. 2009;9:23–29. doi: 10.2174/156652409787314499. [DOI] [PubMed] [Google Scholar]
- 9.Raveney BJ, Copland DA, Dick AD, Nicholson LB. TNFR1-dependent regulation of myeloid cell function in experimental autoimmune uveoretinitis. J Immunol. 2009;183:2321–2329. doi: 10.4049/jimmunol.0901340. [DOI] [PubMed] [Google Scholar]
- 10.Iwata Y, Furuichi K, Kitagawa K, Hara A, Okumura T, Kokubo S, Shimizu K, Sakai N, Sagara A, Kurokawa Y, Ueha S, Matsushima K, Kaneko S, Wada T. Involvement of CD11b+ GR-1 low cells in autoimmune disorder in MRL-Fas lpr mouse. Clin Exp Nephrol. 2010;14:411–417. doi: 10.1007/s10157-010-0309-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, Liang S, Wu J, Horuzsko A. Human inhibitory receptor immunoglobulin-like transcript 2 amplifies CD11b+Gr1+ myeloid-derived suppressor cells that promote long-term survival of allografts. Transplantation. 2008;86:1125–1134. doi: 10.1097/TP.0b013e318186fccd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adeegbe D, Serafini P, Bronte V, Zoso A, Ricordi C, Inverardi L. In vivo induction of myeloid suppressor cells and CD4+Foxp3+ T regulatory cells prolongs skin allograft survival in mice. Cell Transplant. 2010 doi: 10.3727/096368910X540621. [DOI] [PubMed] [Google Scholar]
- 13.Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, Vuillefroy de Silly R, Usal C, Smit H, Martinet B, Thebault P, Renaudin K, Vanhove B. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–7906. doi: 10.4049/jimmunol.180.12.7898. [DOI] [PubMed] [Google Scholar]
- 14.Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, Ma G, Hashimoto D, Li Y, Boros P, Grisotto M, van Rooijen N, Matesanz R, Tacke F, Ginhoux F, Ding Y, Chen SH, Randolph G, Merad M, Bromberg JS, Ochando JC. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, Casares S, Chen SH, Yang WC, Pan PY. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol. 2010;185:5828–5834. doi: 10.4049/jimmunol.0903636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steptoe RJ, Ritchie JM, Jones LK, Harrison LC. Autoimmune diabetes is suppressed by transfer of proinsulin-encoding Gr-1+ myeloid progenitor cells that differentiate in vivo into resting dendritic cells. Diabetes. 2005;54:434–442. doi: 10.2337/diabetes.54.2.434. [DOI] [PubMed] [Google Scholar]
- 18.Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Judkowski V, Pinilla C, Schroder K, Tucker L, Sarvetnick N, Wilson DB. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J Immunol. 2001;166:908–917. doi: 10.4049/jimmunol.166.2.908. [DOI] [PubMed] [Google Scholar]
- 20.Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, Serreze DV, Shabanowitz J, Hunt DF, Nathenson SG, Santamaria P, DiLorenzo TP. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2003;100:8384–8388. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong FS, Karttunen J, Dumont C, Wen L, Visintin I, Pilip IM, Shastri N, Pamer EG, Janeway CA., Jr Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat Med. 1999;5:1026–1031. doi: 10.1038/12465. [DOI] [PubMed] [Google Scholar]
- 22.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, O'Malley KA, Wynn JL, Antonenko S, Al-Quran SZ, Swan R, Chung CS, Atkinson MA, Ramphal R, Gabrilovich DI, Reeves WH, Ayala A, Phillips J, Laface D, Heyworth PG, Clare-Salzler M, Moldawer LL. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–1474. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.