

Abstract
Background and aims
The disintegrin and metalloproteinase ADAM17, also known as tumor necrosis factor alpha converting enzyme, is expressed in adipocytes. Importantly, elevated levels of ADAM17 expression have been linked to obesity and insulin resistance. Therefore, the aim of this study was to evaluate the association of six ADAM17 single nucleotide polymorphisms (SNPs) (m1254A>G, i14121C>A, i33708A>G, i48827A>C, i53440C>T, and i62781G>T) with insulin-resistance phenotypes and obesity risk, and their potential interactions with dietary polyunsaturated fatty acids (PUFA).
Methods and results
ADAM17 SNPs were genotyped in 936 subjects (448 men/488 women) who participated in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study. Anthropometrical and biochemical measurements were determined by standard procedures. PUFA intake was estimated using a validated questionnaire. G allele carriers at the ADAM17_m1254A>G polymorphism exhibited significantly higher risk of obesity (P=0.003), were shorter (P=0.017), had higher insulin (P=0.016), and lower HDL-C concentrations (P=0.027) than AA subjects. For the ADAM17_i33708A>G SNP, homozygotes for the A allele displayed higher risk of obesity (P=0.001), were heavier (P=0.011), had higher BMI (P=0.005), and higher waist measurements (P=0.023) than GG subjects. A significant gene-diet interaction was found (P=0.030), in which the deleterious association of the i33708A allele with obesity was observed in subjects with low intakes from (n-6) PUFA (P<0.001), whereas no differences in obesity risk were seen among subjects with high (n-6) PUFA intake (P>0.5)
Conclusion
These findings support that ADAM17 (m1254A>G and i33708A>G) SNPs may contribute to obesity risk. For the ADAM17_i33708A>G SNP, this risk may be further modulated by (n-6) PUFA intake.
Keywords: gene-diet interaction, obesity, BMI, HDL-cholesterol, insulin concentrations
INTRODUCTION
Prevalence of obesity is steadily increasing among Americans, leading to increased morbidity and mortality due to type 2 diabetes and cardiovascular disease (CVD) (1). An environment characterized by an excessive energy intake and lower physical activity leads to obesity. However, not all people in societies with sedentary lifestyles and abundant food become obese, reinforcing the possibility that variation in genetic susceptibility may influence the risk of obesity. Several family and twin studies have demonstrated that genetic factors contribute 40–70% to the susceptibility variation in obesity (2). Therefore, obesity is a multifactorial disease which occurs as a result of the joint action of genetic and environmental factors, as well as their interaction (3).
Recently, several genome wide association analyses (GWAs) have discovered novel genes for common diseases, including obesity (4,5). Particularly, a disintegrin and metalloproteinase gene (ADAM17), also known as tumor necrosis factor alpha converting enzyme (TACE), has been associated with diabetes and CVD (6). Cleavage by TACE releases the soluble form of tumor necrosis factor, transforming growth factor-α, and other proteins such as the pre-adipocyte factor 1 (Pref-1) from their membrane-bound precursors (a phenomenon named ‘shedding’) (7). TNF-α is overexpressed in the adipose tissue of obese animals and humans, and its secretion from explanted adipose tissue is higher in obese as compared to lean samples (8). Likewise, ADAM17 is overexpressed in adipose tissue of obese mice and mRNA levels show modulation upon development of obesity and during differentiation of pre-adipocytes (9). Furthermore, ADAM17 is responsible for the cleavage that generates the large soluble form of Pref-1, leading to the inhibition of adipocyte differentiation (10).
In humans, only one study has investigated the effects of several ADAM17 genetic variants on phenotypic traits. Morange et al (11) identified two novel polymorphisms, C-154A and Ser747Leu, associated with TNF-α plasma levels and risk of cardiovascular death, respectively. Likewise, several TNF genetic variants have been associated with triglycerides (TG), as well as obesity-related traits, pointing out as an obesity-susceptibility candidate gene (12–15). Particularly, two TNF genetic variants at positions -308 (G>A) and -238 (G>A) in the 5′ regulatory region of the gene have been related to body fat, insulin resistance, as well as arterial blood pressure (13–15). However, prior studies failed to find an association between these TNF variants and obesity-related traits (16,17).
Because ADAM17 is a pivotal element in TNF-α and Pref-1 shedding, it is likely that this metalloproteinase contributes to obesity and its complications. Although the importance of TNF in obesity has been revealed, only one recent study (11) has assessed the effect of five ADAM17 polymorphisms in patients with coronary artery disease. Moreover, that study did not assess the contribution of dietary factors which may modulate the risk for obesity. Hence, the aims of the present study were first to assess the association of novel polymorphisms in the ADAM17 gene with anthropometric variables, lipid concentrations and obesity-related phenotypes. Secondly, we investigated whether ADAM17 SNPs interact with dietary fatty acids to modulate the risk of obesity.
METHODS
Subjects
The study population consisted of 936 subjects (448 men and 488 women, aged 49±16 years) participating in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study. Participants were recruited from three-generational pedigrees from two National Heart, Lung, and Blood Institute Family Heart Study field centers (Minneapolis, MN, and Salt Lake City, UT) (18). The study population was homogeneous with regard to ethnic background, being all individuals of European origin. The detailed design and methodology of the study has been described previously (19). The protocol was approved by the Institutional Review Boards at the University of Alabama, the University of Minnesota, the University of Utah, and Tufts University. Written informed consent was obtained from each participant.
Data collection
For GOLDN participants, clinical examinations at the baseline visit included anthropometrical and blood pressure (BP) measurements. Weight was measured with a beam balance and height with fixed stadiometer. BMI was calculated as weight, in kilograms, divided by the square of height, in meters. Waist circumference was measured at the umbilicus, whereas hip circumference was taken at the maximum posterior extension of the buttocks, measured in meters. BP was measured twice with an oscillometric device (Dinamap Pro Series 100, GE Medical Systems) while subjects were seated and had rested for five min. Reported systolic and diastolic BP values were the mean of the two measurements. Questionnaires were administered to assess demographic information, medical and medication history.
The habitual dietary food intake was assessed by the Diet History Questionnaire developed by the National Cancer Institute. It consisted of 124 food items and included portion size and dietary supplement questions. Two studies have confirmed its validity (20,21).
Laboratory methods
Blood samples were drawn after fasting overnight. Fasting glucose was measured using the method of a hexokinase-mediated reaction and total cholesterol using a cholesterol esterase cholesterol oxidase reaction on a Hitachi 911 autoanalyzer (Roche Diagnostics). The same reaction was used to measure HDL-C after precipitation of non-HDL cholesterol with magnesium/dextran. Low-density lipoprotein cholesterol (LDL-C) was measured by use of a homogeneous direct method (LDL Direct Liquid Select Cholesterol Reagent; Equal Diagnostics). TGs were measured by glycerol-blanked enzymatic method on the Roche COBAS FARA centrifugal analyzer (Roche Diagnostics). Fasting insulin and total adiponectin values were measured by specific radioimmunoassay kits (Linco Research).
Genetic analyses
DNA was extracted from blood samples and purified using commercial Puregene reagents (Gentra Systems) following the manufacturer’s instructions. Six ADAM17 SNPs (m1254A>G, rs11684747; i14121C>A, rs1880439, i33708A>G, rs10495563; i48827A>C, rs1056204; i53440C>T, rs34367192; i62781G>T, rs4622692) were genotyped. SNPs were selected using two criteria: bioinformatics functional assessment and linkage disequilibrium (LD) structure. Assessing LD structure at the ADAM17 locus facilitated the selection of tag SNPs representing different LD blocks. Although ADAM17_i62781G>T SNP maps to intron 16, just 20 bp upstream of the splice acceptor site of exon 17, this SNP did not map within a poly-pyrimidine tract normally found 20–50 bp upstream of splice acceptors. Intronic SNPs were also analyzed with MAPPER to uncover potential allele-specific transcription factor binding sites (22) and manually checked for altered mRNA splice donor and acceptor sites and transversions affecting the poly-pyrimidine tract near splice acceptors. Genotyping was performed using a TaqMan® assay with allele-specific probes on the ABIPrism 7900 HT Sequence Detection System (Applied Biosystems) according to routine laboratory protocols (19). The description of ADAM17 SNPs, as well as ABI assay-on-demand ID is available upon request. The pairwise LD between SNPs was estimated as correlation coefficient (r2) in unrelated subjects using the Helixtree software package (Golden Helix).
Statistical analyses
SPSS software (version 16.0) was used for statistical analyses. A logarithmic transformation was applied to measures of plasma insulin and adiponectin to normalize the distribution of the data. Data are presented as means±SE for continuous variables and as frequencies or percentages for categorical variables. Differences in mean values were assessed by using analysis of variance and unpaired t-tests. Categorical variables were compared by using the Pearson chi-square or the Fisher’s exact tests. Potential confounding factors were age, sex, physical activity, smoking habit (current vs. never and past smokers), alcohol consumption (current vs. never and past drinkers), prior coronary heart disease (CHD), diabetes, and family relationships. Obesity was defined as BMI ≥30 kg/m2. We fitted logistic regression models to estimate the odds ratio (OR) and 95% CI of obesity across ADAM17 genotypes stratified by dietary polyunsaturated fatty acid (PUFA) intakes (as dichotomous variables based on less than and equal or greater than median intakes) and to control for the effect of covariates and total energy intake (in kcal). Two-sided P values <0.05 were considered statistically significant.
RESULTS
Characteristics of the GOLDN participants and genotype frequencies by sex are shown in Table 1. Although BMI did not differ by sex, average weight was higher in men compared to women. BP, glucose and TG concentrations were higher in men, whereas hip measurements and HDL-C and adiponectin concentrations were higher in women. No significant differences in other variables were observed. For all ADAM17 polymorphisms, there was no departure from the Hardy-Weinberg equilibrium (P >0.05). The pairwise LD for all the six SNPs is presented as correlation coefficients in Supplemental Table 1. Given that two ADAM17 SNPs (i53440C>T and i33708A>G) were in strong LD (>0.8) with ADAM17_ i62781G>T, the latter was excluded from further analyses. Because homozygous minor allele carriers were few, we analyzed two ADAM17 SNPs (m1254A>G and i14121C>A) using two genotype categories (AA vs. AG+GG and AA+AC vs. CC, respectively). Other SNPs were analysed using three genotype categories (additive model). Considering the lack of a significant ADAM17 gene-gender interaction for all variables examined, men and women were pooled together for subsequent analyses.
Table 1.
General characteristics of the GOLDN population.
| Men (n=448) | Women (n=488) | P | |
|---|---|---|---|
| Age, years | 49±16.3 | 49±16.3 | 0.559 |
| Weight, kg | 91±16.0 | 76±17.7 | <0.001 |
| Height, m | 1.78±0.73 | 1.65±0.70 | <0.001 |
| BMI, kg/m2 | 28.7±4.7 | 28.2±6.4 | 0.159 |
| Waist circumference, m | 1.01±0.13 | 0.93±0.19 | <0.001 |
| Hip circumference, m | 1.06±0.09 | 1.09±0.15 | 0.001 |
| Current smokers, n (%) | 36(8) | 38(8) | 0.904 |
| Current alcohol drinkers, n (%) | 224(50) | 255(52) | 0.513 |
| Systolic blood pressure, mmHg | 119±14.7 | 113±17.3 | <0.001 |
| Diastolic blood pressure, mmHg | 71±8.7 | 66±9.1 | <0.001 |
| Glucose, mmol/L | 5.77±1.05 | 5.39±0.91 | <0.001 |
| Log (Insulin, mU/L) | 1.10±0.21 | 1.07±0.21 | 0.040 |
| Total cholesterol, mmol/L | 4.94±0.95 | 4.99±1.07 | 0.449 |
| LDL cholesterol, mmol/L | 3.22±0.76 | 3.12±0.85 | 0.051 |
| HDL cholesterol, mmol/L | 1.07±0.25 | 1.35±0.36 | <0.001 |
| Triglycerides, mmol/L | 1.68±1.22 | 1.40±0.93 | <0.001 |
| Log (adiponectin, μg/L) | 3.73±0.24 | 3.96±0.23 | <0.001 |
| Obesity, n (%) | 149(33) | 169(35) | 0.679 |
| ADAM17 SNPs, n (%) | |||
| m1254A>G | |||
| AA | 287(64) | 328(67) | 0.335 |
| AG+GG | 161(36) | 160(33) | |
| i14121C>A | |||
| AA+AC | 131(29) | 128(26) | 0.307 |
| CC | 317(71) | 360(74) | |
| i33708A>G | |||
| AA | 179(40) | 206(42) | 0.732 |
| AG | 217(48) | 224(46) | |
| GG | 52(12) | 58(12) | |
| i48827A>C | |||
| AA | 224(50) | 222(46) | 0.300 |
| AC | 181(40) | 208(43) | |
| CC | 43(10) | 58(12) | |
| i53440C>T | |||
| CC | 115(26) | 115(24) | 0.699 |
| CT | 227(51) | 249(51) | |
| TT | 106(24) | 124(25) | |
| i62781G>T | |||
| GG | 134(30) | 148(30) | 0.369 |
| GT | 225(50) | 260(53) | |
| TT | 89(20) | 80(16) | |
All values are means ± SD.
We examined associations between ADAM17 SNPs and anthropometric measurements and lipids (Table 2). For the ADAM17_m1254A>G SNP, G allele carriers were shorter (1.70±0.01 vs 1.72±0.01 in m; P=0.017), and displayed higher insulin concentrations (1.11±0.01 vs. 1.07±0.02 in mU/L; P=0.016), and lower HDL-C concentrations (1.19±0.01 vs. 1.24±0.01 in mmol/L; P=0.027) than AA subjects (Table 2). As a result, G allele carriers exhibited higher risk of obesity compared with AA subjects (P=0.003) (Table 3). For the ADAM17_ i14121C>A, minor A allele carriers were taller (1.72±0.01 vs. 1.71±0.01 in m; P=0.023) and displayed higher glucose (5.68±0.05 vs. 5.54±0.03 in mmol/L; P=0.023), and lower adiponectin concentrations (3.83±0.01 vs. 3.86±0.01 in μg/L; P=0.041) than CC subjects. However, no differences in obesity risk were observed between genotypes (P>0.8) (Table 3). For the ADAM17_i33708A>G SNP, homozygotes for the AA allele were heavier (84±0.8 vs. 79±1.6 in kg; P=0.011), and displayed higher BMI (28.7±0.3 vs. 26.9±0.5 in kg/m2; P=0.005), and higher waist measurements (0.97±0.01 vs. 0.93±0.02 in m; P=0.023) than GG subjects. Likewise, individuals homozygous for the A allele displayed higher risk of obesity than GG subjects (P=0.001) (Table 3). We did not observe any significant association between ADAM17_i48827A>C SNP and anthropometric variables or fasting lipids. For ADAM17_i53440C>T SNP, homozygotes for the major allele (C) were taller than TT subjects (P=0.040) (Table 2).
Table 2.
Association between ADAM17 genetic variants and anthropometric variables and lipids.
| Variable | ADAM17_m1254A>G | ADAM17_i14121C>A | ADAM17_i33708A>G | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA (n=615) | AG+GG (n=321) | P* | CC (n=677) | CA+AA (n=259) | P* | AA (n=385) | AG (n=441) | GG (n=110) | P | |
| Weight, kg | 83±0.7 | 84±0.9 | 0.285 | 83±0.6 | 84±1.0 | 0.300 | 84±0.8 | 84±0.8 | 79±1.6 | 0.011 |
| Height, m | 1.72±0.01 | 1.70±0.01 | 0.017 | 1.71±0.01 | 1.72±0.01 | 0.023 | 1.71±0.01 | 1.71±0.01 | 1.71±0.01 | 0.734 |
| BMI, kg/m2 | 28.2±0.2 | 28.9±0.3 | 0.051 | 28.4±0.2 | 28.5±0.3 | 0.865 | 28.7±0.3 | 28.6±0.3 | 26.9±0.5 | 0.005 |
| Waist, m | 0.96±0.01 | 0.98±0.01 | 0.107 | 0.97±0.01 | 0.97±0.01 | 0.592 | 0.97±0.01 | 0.98±0.01 | 0.93±0.02 | 0.023 |
| Hip, m | 1.07±0.02 | 1.08±0.02 | 0.197 | 1.08±0.02 | 1.07±0.02 | 0.828 | 1.08±0.01 | 1.08±0.01 | 1.05±0.01 | 0.147 |
| Glucose, mmol/L | 5.56±0.03 | 5.61±0.04 | 0.342 | 5.54±0.03 | 5.68±0.05 | 0.023 | 5.61±0.04 | 5.61±0.03 | 5.44±0.07 | 0.141 |
| Log (Insulin, mU/L) | 1.07±0.01 | 1.11±0.01 | 0.016 | 1.08±0.01 | 1.09±0.01 | 0.617 | 1.08±0.01 | 1.10±0.01 | 1.06±0.02 | 0.240 |
| Total cholesterol, mmol/L | 4.98±0.03 | 4.94±0.05 | 0.483 | 4.99±0.03 | 4.94±0.05 | 0.603 | 4.96±0.04 | 4.96±0.04 | 5.04±0.08 | 0.620 |
| LDL cholesterol, mmol/L | 3.15±0.03 | 3.19±0.04 | 0.520 | 3.17±0.02 | 3.14±0.04 | 0.415 | 3.17±0.03 | 3.14±0.03 | 3.25±0.07 | 0.487 |
| HDL cholesterol, mmol/L | 1.24±0.01 | 1.19±0.01 | 0.027 | 1.22±0.01 | 1.22±0.01 | 0.796 | 1.22±0.01 | 1.22±0.01 | 1.24±0.02 | 0.627 |
| Triglycerides, mmol/L | 1.56±0.05 | 1.49±0.06 | 0.401 | 1.57±0.07 | 1.52±0.04 | 0.550 | 1.52±0.06 | 1.57±0.05 | 1.44±0.11 | 0.711 |
| Log (adiponectin, μg/L) | 3.86±0.01 | 3.85±0.01 | 0.565 | 3.86±0.01 | 3.83±0.01 | 0.041 | 3.86±0.01 | 3.85±0.01 | 3.87±0.02 | 0.612 |
| Variable | ADAM17_i48827A>C | ADAM17_i53440C>T | ||||||
|---|---|---|---|---|---|---|---|---|
| AA (n=446) | AC (n=389) | CC (n=101) | P | CC (n=230) | CT (n=476) | TT (n=230) | P | |
| Weight, Kg | 84±0.8 | 83±0.8 | 83±1.7 | 0.909 | 83±1.1 | 83±0.8 | 84±1.1 | 0.797 |
| Height, m | 1.71±0.01 | 1.71±0.01 | 1.71±0.01 | 0.533 | 1.72±0.01 | 1.71±0.01 | 1.70±0.01 | 0.040 |
| BMI, Kg/m2 | 28.5±0.3 | 28.5±0.3 | 28.4±0.5 | 0.974 | 28.1±0.4 | 28.5±0.3 | 28.9±0.4 | 0.292 |
| Waist, m | 0.97±0.01 | 0.96±0.01 | 0.95±0.02 | 0.512 | 0.96±0.01 | 0.97±0.01 | 0.97±0.02 | 0.903 |
| Hip, m | 1.08±0.01 | 1.07±0.01 | 1.07±0.02 | 0.661 | 1.08±0.01 | 1.07±0.01 | 1.08±0.02 | 0.657 |
| Glucose, mmol/L | 5.61±0.03 | 5.54±0.04 | 5.60±0.08 | 0.386 | 5.57±0.05 | 5.59±0.03 | 5.56±0.01 | 0.857 |
| Log (Insulin, mU/L) | 1.10±0.01 | 1.08±0.01 | 1.06±0.02 | 0.099 | 1.09±0.01 | 1.09±0.01 | 1.08±0.01 | 0.796 |
| Total cholesterol, mmol/L | 4.94±0.04 | 5.01±0.04 | 4.94±0.09 | 0.534 | 4.96±0.06 | 4.96±0.04 | 4.96±0.06 | 0.967 |
| LDL cholesterol, mmol/L | 3.17±0.03 | 3.19±0.03 | 3.09±0.07 | 0.552 | 3.16±0.05 | 3.17±0.03 | 3.17±0.05 | 0.980 |
| HDL cholesterol, mmol/L | 1.22±0.01 | 1.22±0.01 | 1.24±0.03 | 0.530 | 1.22±0.02 | 1.22±0.01 | 1.19±0.02 | 0.691 |
| Triglycerides, mmol/L | 1.51±0.05 | 1.56±0.06 | 1.54±0.12 | 0.810 | 1.56±0.08 | 1.52±0.05 | 1.53±0.08 | 0.916 |
| Log (adiponectin, μg/L) | 3.84±0.01 | 3.87±0.01 | 3.87±0.02 | 0.111 | 3.84±0.02 | 3.85±0.01 | 3.87±0.02 | 0.438 |
Values are mean±SE.
P values are adjusted for age, sex, smoking habit, alcohol consumption, diabetes, coronary heart disease, and family relationships.
Table 3.
Odds ratio for obesity depending on ADAM17 genotypes.
| Genotype | OR (95% confidence interval) | P* | P* for trend |
|---|---|---|---|
| ADAM17_m1254A>G | |||
| AA (n=615) | 1 (Reference) | 0.003 | 0.003 |
| AG+GG (n=321) | 1.56 (1.16–2.08) | ||
| ADAM17_i14121C>A | |||
| CC (n=677) | 1 (Reference) | 0.878 | 0.878 |
| AA+AC (n=259) | 0.98 (0.71–1.34) | ||
| ADAM17_i33708A>G | |||
| AA (n=385) | 1 (Reference) | <0.001 | 0.001 |
| AG (n=441) | 0.94 (0.55–1.61) | 0.001 | |
| GG (n=110) | 0.37 (0.28–1.72) | ||
| ADAM17_i488274A>C | |||
| AA (n=446) | 1 (Reference) | 0.598 | 0.426 |
| AC (n=389) | 1.16 (0.71–1.89) | 0.261 | |
| CC (n=101) | 0.88 (0.61–1.61) | ||
| ADAM17_i53440C>T | |||
| CC (n=230) | 1 (Reference) | 0.018 | 0.057 |
| CT (n=476) | 1.34 (0.95–1.87) | 0.237 | |
| TT (n=230) | 1.64 (0.67–1.51) | ||
P values were adjusted for age, sex, smoking habit, alcohol consumption, diabetes, coronary heart disease, and family relationships.
We next examined whether associations between ADAM17 SNPs and obesity risk were modified by PUFA intake in this population (Supplemental Table 2). Because there were no significant differences in dietary intake according to genotype groups (data not shown), we investigated whether ADAM17 gene-PUFA interactions could modulate the observed association with obesity risk. We dichotomized dietary intakes according to the median value for total PUFA, (n-3) PUFA, and (n-6) PUFA. A statistically significant gene-diet interaction was found (P=0.026), in which subjects homozygous for the i33708A allele consuming diets containing low PUFA were associated with a higher risk of obesity than subjects homozygous for the i33708G allele (P>0.001), whereas no differences in obesity risk was observed among subjects consuming diets with high PUFA independent of the genotype (P>0.3) (Supplemental Table 2). We further tested if different types of PUFA would have different effects on the association between ADAM17 SNPs and obesity. A significant gene-diet interaction was observed for (n-6) PUFA (P=0.030) in which the deleterious association of the i33708A allele with obesity was observed in subjects with low dietary intake from (n-6) PUFA (P>0.001), whereas no effect of this SNP on obesity was seen among subjects with high (n-6) PUFA intake (P>0.5) (Figure). No significant gene-diet interactions were found for (n-3) PUFA intake (P>0.3) nor other dietary fatty acids (data not shown). No significant gene-PUFA interactions were found for other analyzed ADAM17 SNPs, regardless of the PUFA family investigated: (n-6) or (n-3) (Supplemental Table 2).
Figure.
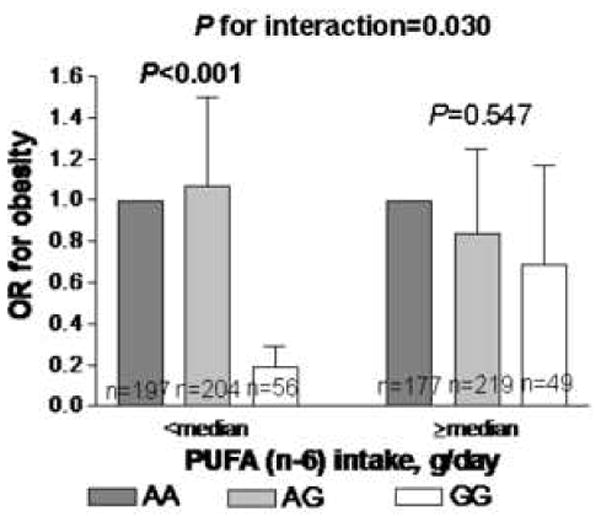
OR and 95% CI obesity depending on ADAM17_i33708A>G genotypes and (n-6) PUFA intakes.
DISCUSSION
In the present study, we observed that two ADAM17 genetic variants (m1254A>G and i33708A>G) were associated with obesity and insulin resistance-related phenotypes (higher insulin and lower HDL-C concentrations). Interestingly, a significant gene-diet interaction was found in which the deleterious association between the i33708A allele and obesity risk was only seen in those subjects with low intakes from PUFA, particularly from (n-6) PUFA. Overall, these findings suggest that dietary habits may modulate the contributions of ADAM17 polymorphisms on the genetic susceptibility towards developing obesity.
The current study is the first to examine in a relatively large population the effects of several ADAM17 genetic variants on insulin-resistance phenotypes and obesity in the context of dietary intake of PUFA. We found that two ADAM17 genetic variants were significantly (m1254A>G, i33708A>G) associated with obesity, in which carriers of the m1254G allele and homozygotes for the i33708A allele had higher BMI. To date, only one prior study (11) has investigated the effect of five ADAM17 SNPs in a prospective cohort of patients with coronary artery disease. The authors found that those patients carrying the 747Leu allele exhibited higher risk of cardiovascular death. Moreover, it has been recently described that HDL activates ADAM17-mediated substrate shedding and stimulates TNF-α release (23). This explains the association observed between ADAM17_m1254A>G and HDL-C concentrations. Because ADAM17 releases TNF-α from adipocytes (7) and prior data support their role in obesity-related phenotypes (9,11), we hypothesize that ADAM17 is an obesity-susceptibility candidate gene. Prior evidence supported an association between two TNF polymorphisms at positions -308(G>A) and -238 (G>A) with increased anthropometric measurements, insulin resistance, and body fat in several populations (13–15). By contrast, no association between both polymorphisms and obesity was seen in 424 subjects self-referred to the Johns Hopkins Weight Management Center (16) and in 380 healthy subjects (17). Inconsistencies regarding the magnitude and direction of the association between these polymorphisms and obesity-related traits may be due to population-specific differences.
The mechanism by which these ADAM17 polymorphisms may contribute to the observed associations is unknown. Given that five of the analyzed SNPs map to non-coding regions, the likelihood that these SNPs represent a functional mutation is low. However, the presence of transcriptional enhancers and other regulatory elements, observed frequently in intronic regions, could explain our findings. In this regard, for the m1254A>G polymorphism, computational analysis by MAPPER indicated a potential allele-specific binding site for the hepatocyte nuclear factor-1 alpha (HNF1A/B) transcription factor, whose motif appears enriched in certain genes involved in insulin sensitivity and development of diabetes (24). Moreover, ADAM17 _i33708A>G polymorphism has a potential allele-specific binding sites for the hepatocyte nuclear factor-2 (FOXA2) that appears to play a pivotal role as a physiological regulator of adipocyte differentiation and metabolism (25).
Interestingly, Dixon et al (5) reported that the polymorphism ADAM17_i62781G>T regulates the expression of ADAM17 gene. Particularly, the major allele G was associated with a higher expression of ADAM17. As mentioned in the results section, the aforementioned SNP was in strong LD with SNP i33708A>G. Therefore, we can speculate that subjects homozygous for the major allele (A) may exhibit higher expression of ADAM17 and accordingly, an increased risk of obesity. In addition to genetic susceptibility, environmental factors such as dietary habits may contribute to the risk of obesity. A growing body of evidence supports the pivotal role that PUFAs play as important regulators of gene expression, particularly of genes involved in lipogenesis, triglyceride synthesis and fatty acid oxidation (26). Moreover, diets lower in PUFA (27) have been associated with an increased risk of obesity through mechanisms involving dyslipidemia, vascular dysfunction and insulin resistance. Although epidemiological evidence supports a protective role against obesity for dietary (n-3) PUFA (27), no significant interactions between obesity and intake of (n-3) PUFA were seen in this study. It has been reported that dietary (n-6) PUFA decreases white adipose tissue mass in rodents through the up-regulation of genes involved in energy-demanding processes like urea synthesis and gluconeogenesis (28). Moreover, Fontaine-Bisson et al (29) found that lean subjects with the -308A allele and the -238GG genotype at the TNF gene had higher risk of obesity with lower intakes of (n-6) PUFA. By contrast, a case-control study from the EPIC cohort (30) showed that the risk of being obese was higher for carriers of the -308A allele with large intakes of (n-6) PUFA. The described anti- or pro-adipogenic effect for (n-6) PUFA (31) may explain differences between studies. However, both findings are supportive of a synergistic relationship between genes and diet by which tailored dietary recommendations targeted at reducing the risk of obesity may modulate the genetic expression of several ADAM17 variants.
Despite the evidence, we should be cautious in the interpretation of our data. The cross-sectional design of this study may weaken or distort any true relationship between ADAM17 and PUFA intakes. Therefore, large prospective studies with a long period of follow-up are required. Another limitation is that dietary intake was measured within a limited period of time and may not be representative of previous or habitual exposures. Thus, replication and extension of research efforts to other populations and ethnic groups is clearly warranted.
In conclusion, the present study carried out in a White population supports the hypothesis that two ADAM17 polymorphisms (m1254A>G and i33708A>G) play a pivotal role in obesity. The latest association was particularly evident in subjects with low dietary intake of (n-6) PUFA. Interestingly, this gene-diet interaction offers the potential to identify dietary and other lifestyle changes which, when implemented, may obviate the onset of obesity in individuals with a specific genetic background. Therefore, our findings have wide-ranging implications for health initiatives targeted at reducing obesity risk.
Supplementary Material
Acknowledgments
This work was supported by NIH Heart, Lung and Blood Institute grant U 01 HL72524, Genetic and Environmental Determinants of Triglycerides, grants HL-54776 and DK075030, and by contracts 53-K06-5-10 and 58-1950-9-001 from the US Department of Agriculture Research Service. M. Junyent is supported by a grant from the Fulbright-Spanish Ministry of Education and Science (reference 2007-1086). C. E. Smith is supported by the grant T32 DK007651-19.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sullivan PW, Ghushchyan VH, Ben-Joseph R. The impact of obesity on diabetes, hyperlipidemia and hypertension in the United States. Qual Life Res. 2008;17:1063–71. doi: 10.1007/s11136-008-9385-7. [DOI] [PubMed] [Google Scholar]
- 2.Clément K. Genetics of human obesity. Proc Nutr Soc. 2005;64:133–42. doi: 10.1079/pns2005416. [DOI] [PubMed] [Google Scholar]
- 3.Ordovas JM, Shen J. Gene-environment interactions and susceptibility to metabolic syndrome and other chronic diseases. J Periodontol. 2008;79:1508–13. doi: 10.1902/jop.2008.080232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KC. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–7. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 6.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29:258–89. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 9.Voros G, Maquoi E, Collen D, Lijnen HR. Differential expression of plasminogen activator inhibitor-1, tumor necrosis factor-α, TNF-α converting enzyme and ADAMS family members in murine fat territories. Biochim Biophys Acta. 2003;1625:36–42. doi: 10.1016/s0167-4781(02)00589-4. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Kim KA, Kim JH, Sul HS. Pref-1, a preadipocyte secreted factor that inhibits adipogenesis. J Nutr. 2006;136:2953–6. doi: 10.1093/jn/136.12.2953. [DOI] [PubMed] [Google Scholar]
- 11.Morange PE, Tregouet DA, Godefroy T, Saut N, Bickel C, Rupprecht HK, et al. Polymorphisms of the tumor necrosis factor-alpha (TNF) and the TNF-alpha converting enzyme (TACE/ADAM17) genes in relation to cardiovascular mortality: the AtheroGene study. J Mol Med. 2008;86:1153–61. doi: 10.1007/s00109-008-0375-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhao LJ, Xiong DH, Pan F, Liu X-G, Recker RR, Deng HW. Polymorphisms of the tumor necrosis factor-alpha receptor 2 gene are associated with obesity phenotypes among 405 Caucasian nuclear families. Hum Genet. 2008;124:171–7. doi: 10.1007/s00439-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brand E, Schorr U, Kunz I, Kertmen E, Ringel J, Distler A, et al. Tumor necrosis factor-alpha—-308G/A polymorphism in obese Caucasians. Int J Obes Relat Metab Disord. 2001;25:581–5. doi: 10.1038/sj.ijo.0801576. [DOI] [PubMed] [Google Scholar]
- 14.Corbalán MS, Marti A, Forga L, Patiño A, Martınez-Gonzalez MA, Martınez JA. Influence of two polymorphisms of the tumoral necrosis factor-alpha gene on the obesity phenotype. Diabetes Nutr Metab. 2004;17:17–22. [PubMed] [Google Scholar]
- 15.Sookoian SC, Gonzalez C, Pirola CJ. Meta-analysis on the G-308A tumor necrosis factor α gene variant and phenotypes associated with the metabolic syndrome. Obes Res. 2005;13:2122–31. doi: 10.1038/oby.2005.263. [DOI] [PubMed] [Google Scholar]
- 16.Walston J, Seibert M, Yen C-J, Cheskin LJ, Andersen RE. Tumor necrosis factor-α--238 and -308 polymorphisms do not associate with traits related to obesity and insulin resistance. Diabetes. 1999;48:2096–8. doi: 10.2337/diabetes.48.10.2096. [DOI] [PubMed] [Google Scholar]
- 17.Rasmussen SK, Urhammer SA, Jensen JN, Hansen T, Borch-Johnsen K, Pedersen O. The -238 and -308 G>A polymorphisms of the tumor necrosis factor α gene promoter are not associated with features of the insulin resistance syndrome or altered birth weight in Danish Caucasians. J Clin Endocrinol Metab. 2000;85:1731–4. doi: 10.1210/jcem.85.4.6563. [DOI] [PubMed] [Google Scholar]
- 18.Higgins M, Province M, Heiss G, Eckfeldt J, Ellison RC, Folsom AR, et al. NHLBI Family Heart Study: objectives and design. Am J Epidemiol. 1996;143:1219–28. doi: 10.1093/oxfordjournals.aje.a008709. [DOI] [PubMed] [Google Scholar]
- 19.Junyent M, Lee Y, Smith CE, Arnett DK, Tsai MY, Kabagambe EK, et al. The effect of a novel intergenic polymorphism (rs11774572) on HDL cholesterol concentrations depends on TaqIB polymorphism in the cholesterol ester transfer protein gene. Nutr Metab Cardiovasc Dis. 2009 doi: 10.1016/j.numecd.2009.02.010. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson FE, Subar AF, Brown CC, Smith AF, Sharbaugh CO, Jobe JB, et al. Cognitive research enhances accuracy of food frequency questionnaire reports: results of an experimental validation study. J Am Diet Assoc. 2002;102:212–25. doi: 10.1016/s0002-8223(02)90050-7. [DOI] [PubMed] [Google Scholar]
- 21.Subar AF, Thompson FE, Kipnis V, Midthune D, Hurwitz P, McNutt S, et al. Comparative validation of the Block, Willett, and National Cancer Institute food frequency questionnaires: the Eating at America’s Table Study. Am J Epidemiol. 2001;154:1089–99. doi: 10.1093/aje/154.12.1089. [DOI] [PubMed] [Google Scholar]
- 22.Marinescu VD, Kohane IS, Riva A. MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinformatics. 2005;6:79. doi: 10.1186/1471-2105-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tellier E, Canault M, Poggi M, Bonardo B, Nicolay A, Alessi M, et al. HDLs activate ADAM17-dependent shedding. J Cell Physiol. 2008;214:687–93. doi: 10.1002/jcp.21265. [DOI] [PubMed] [Google Scholar]
- 24.Stride A, Ellard S, Clark P, Shakespeare L, Salzmann M, Shepherd M, et al. Beta-cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes care. 2005;28:1751–6. doi: 10.2337/diacare.28.7.1751. [DOI] [PubMed] [Google Scholar]
- 25.Wolfrum C, Shih DQ, Kuwajima S, Norris AW, Kahn CR, Stoffel M. Role of FOXA2 in adipocyte metabolism and differentiation. J Clin Invest. 2003;112:345–56. doi: 10.1172/JCI18698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampath H, Ntambi JM. Polyunsaturated fatty acid regulation of gene expression. Annu Rev Nutr. 2005;25:317–40. doi: 10.1146/annurev.nutr.25.051804.101917. [DOI] [PubMed] [Google Scholar]
- 27.Ramel A, Martinez A, Kiely M, Morais G, Bandarra NM, Throsdottir I. Beneficial effects of long-chain n-3 fatty acids included in an energy-restricted diet on insulin resistance in overeweight and obese European young adults. Diabetologia. 2008;51:1261–8. doi: 10.1007/s00125-008-1035-7. [DOI] [PubMed] [Google Scholar]
- 28.Madsen L, Pedersen LM, Liaset B, Ma T, Petersen RK, van den Berg S, et al. cAMP-dependent signaling regulates the adipogenic effect of n-6 polyunsaturated fatty acids. J Biol Chem. 2008;283:7196–205. doi: 10.1074/jbc.M707775200. [DOI] [PubMed] [Google Scholar]
- 29.Fontaine-Bisson B, El-Sohemy A. Genetic polymorphisms of tumor necrosis factor-alpha modify the association between dietary polyunsaturated fatty acids and plasma high-density lipoprotein-cholesterol concentrations in a population of young adults. J Nutrigenet Nutrigenomics. 2008;1:215–23. doi: 10.1159/000149825. [DOI] [PubMed] [Google Scholar]
- 30.Nieters A, Becker N, Linseisen J. Polymorphisms in candidate obesity genes and their interaction with dietary intake of n-6 polyunsaturated fatty acids affect obesity risk in a sub-sample of the EPIC-Heidelberg cohort. Eur J Nutr. 2002;41:210–21. doi: 10.1007/s00394-002-0378-y. [DOI] [PubMed] [Google Scholar]
- 31.Madsen L, Petersen RK, Kristiansen K. Regulation of adipocyte differentiation and function by polyunsaturated fatty acids. Biochim Biophys Acta. 2005;1740:266–86. doi: 10.1016/j.bbadis.2005.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.