

Abstract
BACKGROUND
Lipopolysaccharide (LPS) has a deleterious effect on several organs including the liver and eventually leads to endotoxic shock and death. LPS-induced hepatotoxicity is characterized by disturbed intracellular redox balance and excessive reactive oxygen species (ROS) accumulation, leading to liver injury. We have shown that treatment with suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor (HDACI), improves survival in a murine model of LPS-induced shock, but the protective effect of SAHA against liver damage remains unknown. The goal of this study was to investigate the mechanism underlying SAHA action in murine livers.
METHOD
Male C57BL/6J mice (6-8 weeks) weighing 20-25 g were randomly divided into three groups: (A) a sham group was given isotonic sodium chloride solution (10 μL/g body weight, intraperitoneal, i.p.) with DMSO (1 μl/g body weight, i.p.); (B) a LPS group was challenged with LPS (20 mg/kg, i.p.) dissolved in isotonic sodium chloride solution with DMSO; (C) a LPS plus SAHA group was treated with SAHA (50 mg/kg, i.p.) dissolved in DMSO immediately after injection of LPS (20 mg/kg, i.p.). Mice were anesthetized, and their livers were harvested 6 or 24 hours after injection to analyze whether SAHA affected production of reactive oxygen species (ROS) and activation of apoptotic proteins in the liver cells of challenged mice.
RESULTS
SAHA counteracted LPS-induced production of ROS (thiobarbituric acid reactive substances (TBARS) and nitrite) and reversed an LPS-induced decrease in antioxidant enzyme, glutathione (GSH). SAHA also attenuated LPS-induced hepatic apoptosis. Moreover, SAHA inhibited activation of the redox-sensitive kinase, apoptosis signal-regulating kinase-1 (ASK1), and the mitogen-activated protein kinases (MAPKs) p38 and Jun N-terminal kinase (JNK).
CONCLUSION
Our data indicates, for the first time, that SAHA is capable of alleviating LPS-induced hepatotoxicity and suggests that a blockade of the upstream events required for ASK1 action may serve as a new therapeutic option in the treatment of LPS-induced inflammatory conditions.
Keywords: Histone deacetylase inhibitor, LPS, Liver, Inflammation, Apoptosis
INTRODUCTION
Sepsis, a systemic inflammatory response syndrome capable of inducing endotoxic shock, is a leading cause of mortality in the intensive care unit and has proven to be an exceedingly difficult condition to treat [1]. Infection from Gram-negative bacteria is currently the most common cause of sepsis, and lipopolysaccharide (LPS) found on such bacteria are an endotoxin that has been implicated in the pathogenesis of infection and ensuing septic shock [2]. The pro-oxidant action of LPS induces excessive accumulation of reactive oxygen species (ROS), leading to cellular injury through the impairment of vital macromolecules as well as subsequent altered membrane fluidity and mitochondrial function [3-4]. Growing evidence has demonstrated that ROS are important mediators in caspase-9-dependent apoptotic processes, which have been implicated in a variety of inflammatory and stress signaling pathways [5].
Apoptosis signal-regulating kinase-1 (ASK1) is a key ROS-regulated kinase that controls mitogen-activated protein kinase (MAPK) pathway activation [6]. The inactive form of ASK1 is bound to the reduced form of thioredoxin and 14-3-3 proteins, while thioredoxin oxidation and the release of 14-3-3 result in the activation of both ASK1 and p38 kinase [7]. Additionally, LPS-mediated ROS production has previously been reported to activate ASK1, further suggesting that the ROS-dependent ASK1-p38 axis plays a crucial role in LPS-mediated mammalian innate immunity [8-9].
Chromatin structure plays a central role in regulating gene expression and cellular activity. The acetylation of histones is an essential epigenetic mechanism controlling chromatin structure, DNA accessibility for transcription factors, and gene expression. Such protein acetylation is regulated by the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) and affects diverse biological functions, including cell survival [10]. Suberoylanilide hydroxamic acid (SAHA, vorinostat), a potent histone deacetylase inhibitor (HDACI), has recently emerged as a possible therapeutic intervention for hyper-inflammation, as we and others have shown that SAHA reduces the lethality of hemorrhagic shock, suppresses pro-inflammatory cytokine expression, and improves survival of mice in models of endotoxic shock [10, 11]. Recently we have reported that LPS injection reduces acetylation of proteins, including histone H3K9, H2AK5, and H2BK5, in a murine model of LPS-induced shock. Treatment of mice with SAHA inhibits HDACs, restores the protein acetylation, suppresses pro-inflammatory cytokine expression, and improves survival [12]. The precise mechanism underlying SAHA-mediated HDAC inhibition, however, remains largely unknown. The aim of the present study was to investigate whether SAHA affects (1) expression of iNOS and COX-2, (2) oxidative stress, (3) activation of caspase-3 and -9, (4) liver injury, and (5) phosphorylation of ASK1, p38 and JNK in a murine model of LPS-induced shock.
MATERIALS AND METHODS
Materials
LPS from Salmonella typhosa and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). SAHA was purchased from Biomol International (Plymouth Meeting, PA). Trizol, SuperScript™ II Reverse Transcriptase, and Platinum PCR SuperMix were purchased from Life Technologies (Grand Island, NY).
Mouse protocols
Research was conducted in compliance with the Animal Welfare Act and was approved by the Institutional Animal Care and Use Committee. Male C57BL/6J mice (6-8 weeks) weighing 20-25 g were purchased from Jackson Labs (Bar Harbor, ME). The mice were randomly divided into three groups: (A) a sham group was given isotonic sodium chloride solution (10 μL/g body weight, intraperitoneal, i.p.) with DMSO (1 μl/g body weight, i.p.); (B) a LPS group was challenged with LPS (20 mg/kg, i.p.) dissolved in isotonic sodium chloride solution with DMSO; (C) a LPS plus SAHA group was treated with SAHA (50 mg/kg, i.p.) dissolved in DMSO immediately after injection of LPS (20 mg/kg, i.p.). Mice were anesthetized and their livers were harvested 6 or 24 hours after injection and frozen with liquid nitrogen until further use.
Determination of TBARS, nitrite, and GSH
Thiobarbituric acid reactive substances (TBARS), nitrite, and glutathione (GSH) assay kits were purchased from Cayman Chemical (Ann Arbor, MI). Assays were performed according to manufacturer instructions.
Western blot analysis and antibodies
Antibodies for caspase-3, caspase-9, phospho-p38, phospho-JNK, phospho-ASK1 and ASK1, and β-actin were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for JNK, iNOS and COX-2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The protein content of liver homogenates was measured using a bicinchoninic acid (BCA) kit (Thermo Scientific, Rockford, IL). Protein aliquots (10-40 μg) were electrophoresed on 4%–20% gradient SDS-PAGE gels (Life Technologies, Grand Island, NY) and then transferred to PVDF membranes (Millipore, Billerica, MA). The blocked membranes were incubated with primary antibody in tris-buffered saline tween 20 (TBST) containing 5% nonfat milk or BSA at 4°C overnight, followed by incubation with the appropriate secondary antibodies (Cell Signaling Technology) according to the manufacturer's instructions. Membranes were developed with super-signal west femto maximum sensitivity substrate (Thermo Scientific). Protein loading was routinely confirmed with an antibody against β-actin. Densitometric analysis was conducted using Image Lab Software (Bio-Rad, Hercules, CA). Quantitative analysis was performed by calculating the densitometry ratios versus β-actin.
Quantitative real time PCR
RNA was extracted from livers using TRIzol Reagent (Life Technologies, Grand Island, NY) according to manufacturer's instructions. RNA was reverse-transcribed using SuperScript II RNase H– Reverse Transcriptase (Life Technologies, Grand Island, NY). qRT-PCR assays using iTaqTM SYBR Green Supermix with ROX (Bio-Rad) were performed on an Applied Biosystems 7300 thermal cycler (Foster City, CA). Quantification was performed using the relative standard curve method. All measurements were performed in triplicate. PCR was performed with primers of cyclooxygenase-2 (COX2), GCTGTACAAGCAGTGGCAAA, GCTCGGCTTCCAGTATTGAG; inducible nitric oxide synthase (iNOS), GAGGCCGCATGAGCTTGGTGTTT, GGGGGTTGCATTTCGCTGTCTCC; and β-actin, GTGGGCCGCTCTAGGCACCA, TGGCCTTAGGGTTCAGGGGG.
Statistics
All data are expressed as mean ± SEM. Data for all experiments were analyzed by unpaired Student's t test (2 groups) or ANOVA (>2 groups) using the Statview 6.0 software program (SAS Institute, Cary, NC). Comparisons between groups and tests of interactions were made assuming a two-factor analysis. The interaction term tested each main effect, and the residual error tested the interaction. All comparisons were made using Fisher's least significant difference procedure so that multiple comparisons were made at the 0.05 level only if the overall F test from the ANOVA was significant at p < 0.05.
RESULTS
SAHA inhibits LPS-induced iNOS and COX-2 production in liver
While many genes are altered during LPS treatment, iNOS and Cox-2 were primarily considered on the basis that such genes are closely associated with apoptosis and play central roles in ROS production [13]. iNOS and COX-2 proteins as well as mRNA were examined by Western blot analysis and qRT-PCR analysis, respectively. iNOS and COX-2 protein expression levels were very low in control livers but markedly increased after 6 hours of LPS treatment. In LPS-stimulated livers, however, SAHA treatment significantly decreased iNOS and COX-2 protein expression (Fig. 1A). PCR analysis showed that expression of iNOS and COX-2 and mRNA correlated with corresponding protein levels, indicating that LPS exposure increased the level of iNOS and COX-2 proteins and mRNA (Fig. 1B, C). As expected, treatment with SAHA significantly suppressed LPS-stimulated mediators through transcriptional inhibition.
Figure 1.
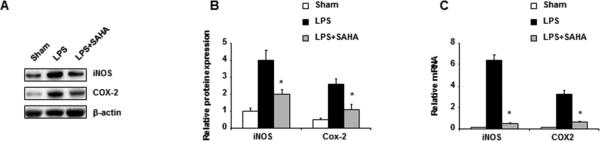
SAHA inhibits LPS-induced expression of iNOS and COX-2. Mice were given SAHA (50 mg/kg, i.p.) immediately after injection of LPS (20 mg/kg, i.p.). Livers were harvested 6 h after injection. (A) A representative image of a Western blot for iNOS and Cox-2 in liver homogenate from each group. (B) Graphic representation of relative abundance protein expression of iNOS and Cox-2 normalized to β-actin. (C) qRT-PCR analysis of relative mRNA levels of iNOS and Cox-2 in liver homogenate from each group. Data represent mean ± SEM from three independent experiments. Asterisks indicate a significant difference (*p<0.05) relative to livers treated with LPS in the absence of SAHA.
SAHA attenuates LPS-induced oxidative stress
The potential effects of SAHA on LPS-induced oxidative stress were then determined via TBARS assay, which effectively screens and monitors lipid peroxidation. Nitric oxide (NO), synthesized by iNOS from L-arginine, is a key mediator of inflammation [14]. NO production was determined by the quantification of nitrite, a stable metabolite of NO. SAHA treatment significantly reduced LPS-induced TBARS and nitrite production in livers 6 hours after LPS challenge (Fig. 2A, B). Additionally, GSH levels were measured to assess the antioxidant capacity of SAHA on LPS-induced oxidative stress. SAHA counteracted the LPS-induced decreases in liver GSH levels (Fig. 2C).
Figure 2.
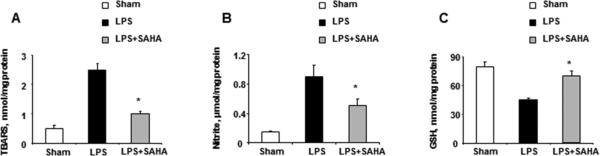
SAHA reduces LPS-induced oxidative stress in livers. Mice were given SAHA (50 mg/kg, i.p.) immediately after injection of LPS (20 mg/kg, i.p.). Livers were harvested 6 h after injection. (A) TBARS, (B) nitrite and (C) GSH in the liver homogenates were analyzed by ELISA. Data represent mean ± SEM from three independent experiments with each experiment performed in triplicate. Asterisks denoted a response that was significantly different from LPS group (*p<0.05).
SAHA suppresses LPS-induced activity of caspase-9 and -3 in LPS-treated mouse liver
ROS, such as NO, are mitochondrial apoptotic factors that result in the activation of procaspase-9 [15]. Thus, the activities of caspase-9 and caspase-3 in the liver of mice treated with or without SAHA were examined to determine the effect of SAHA on LPS-induced liver apoptosis. Liver tissue was harvested from sham, LPS, and LPS plus SAHA groups 6 hours after LPS injection, and resultant caspase activity was detected by Western blot analysis. Livers from mice challenged with LPS exhibited notably greater caspase-9 and -3 activity than those from sham mice. Moreover, livers treated with SAHA after LPS injection expressed significantly suppressed caspase-9 and -3 activity, indicating that the hepatoprotective effect of SAHA involves the inhibition of pro-appoptotic enzyme activation (Fig. 3A, B).
Figure 3.
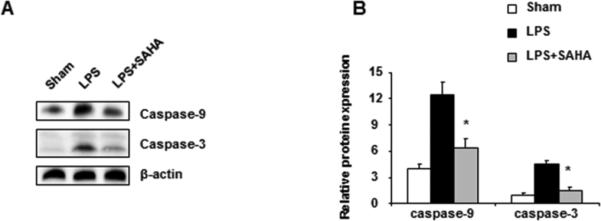
Effect of SAHA on LPS-induced activation of caspase-9 and -3 in mouse liver homogenates. Mice were given SAHA (50 mg/kg, i.p.) immediately after injection of LPS (20 mg/kg, i.p.). Livers were harvested 6 h after LPS injection. (A) Liver homogenates were then prepared and subjected to Western blotting with antibodies specific to cleaved forms of caspase-9 and -3. Shown here is one representative result from three independent experiments. (B) The bar graph represents densitometric measurements of caspase-9 and -3 bands relative to the corresponding β-actin bands and normalized to caspase-9 and -3/β-actin ratio. *p<0.05 compared with LPS group.
SAHA reduces LPS-induced liver damage by histology
Histopathological analysis was performed to assess the SAHA-mediated protection on LPS-induced liver injury. As expected, indications of injury were absent from liver samples of sham animals (Fig. 4A). The inflammatory response to 24-hour LPS challenge in murine liver was considerably enhanced, as evidenced by increasingly intense infiltration of inflammatory cells within the parenchyma, where necrotic and apoptotic cell death of hepatocytes was also observed (Fig. 4B). The architecture of the tissue in mice treated with SAHA after LPS injection was better preserved relative to the group injected with LPS alone (Fig. 4C).
Figure 4.
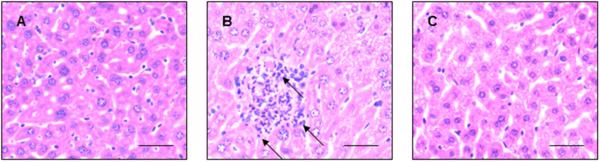
SAHA reduces LPS-induced liver injury. Mice were given SAHA (50 mg/kg, i.p.) immediately after injection of LPS (20 mg/kg, i.p.). Livers of mice 24h after LPS administration were observed pathologically (H&E stain). (A) A non-injured mouse shows normal architecture and very minimal inflammation. (B) The liver of a mouse that received LPS shows markedly increased immune cell infiltration. Arrows show examples of increased parenchymal immune cells. (C) The pathological changes caused by LPS were significantly alleviated in SAHA-treated mice. The results are representative of three independent experiments. Scale bars: 100 μm.
SAHA inhibits LPS-induced activation of ASK1 in livers
ASK1 is a key ROS-regulated kinase that controls the activation of MAPK pathways [7]. Thus, we determined whether LPS-induced activation of ASK1 was inhibited by SAHA. LPS treatment increased phosphorylation of ASK1 in liver, while such activation of ASK1 was prevented by SAHA pretreatment (Fig. 5A, B). Our data also indicate that SAHA reversed LPS-triggered p38 and JNK pathway activation (Fig. 5A, B).
Figure 5.
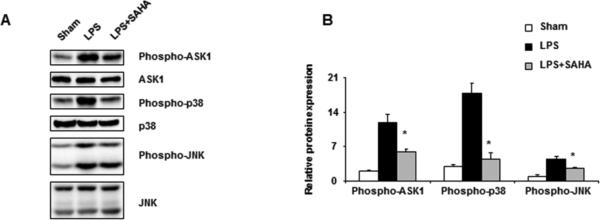
SAHA inhibits LPS-induced activation of phosphorylation of ASK1, p38, and JNK in livers. Mice were given SAHA (50 mg/kg, i.p.) immediately after injection of LPS (20 mg/kg, i.p.). Livers were harvested 6 h after LPS injection. (A) Liver homogenates were prepared and levels of phospho-ASK1, phospho-p38 and phospho-JNK were analyzed by Western blot. Results from a representative experiment of three independent experiments are shown. (B) Densitometric analyses of bands. Phospho-ASK1, phospho-p38 and phospho-JNK data were normalized with total ASK1, p38 and JNK, respectively. *p <0.05 compared with LPS group.
DISCUSSION
Over the past quarter century, attempts at designing drugs to reduce the severity of septic hyper-inflammation have proven ineffective, as the underlying mechanisms are largely unknown [16]. Given that septic shock results from complex patho-physiological processes of numerous molecules, strategies that target a specific biomarker are typically unsuccessful in managing the overall problem. The multi-faceted nature of systemic septic inflammation requires a therapeutic agent with diverse physiological and pharmacological functions [17].
Liver is a vital organ for survival during bacterial infection. In the 1970's, Ferluga and Allison demonstrated that even a small dose of LPS caused lethal hepatitis to mice that were pre-injected with Corynebacterium parvum in the liver lobules [18]. We recently found that inhibition of HDAC6 can prevent liver damage in a lethal septic model (manuscript in preparation). In the present study, we focused on the mechanism of action of SAHA and investigated how this HDAC inhibitor protects against liver damage.
Both excessive formation and release of ROS as well as diminished antioxidant enzyme activities are principal components of oxidative stres [19]. Furthermore, NO is a free radical that is converted to nitrate and nitrite after exposure to endotoxins or stimulation of cytokines in inflamed tissues and vessel walls [20, 21]. NO-induced oxidative stress (nitric oxidative stress) contributes to septic shock, as well as many other conditions, and overproduction of NO has been linked with an LPS-dependent expression of inducible (iNOS) and mitochondrial (mtNOS) isoforms of NO synthase [21-24]. NO primarily reacts with superoxide anion, leading to the formation of a potent cytotoxic oxidant peroxynitrite anion (ONOO), which oxidizes sulfhydryl groups and generates hydroxyl radicals (•OH) [25, 26]. Both ONOO and •OH lead to cellular lipid peroxidation, protein oxidation, and impairment of mitochondrial function, eventually resulting in oxidative damage to many tissues including the liver [27].
In contrast, antioxidant agents, such as GSH, decrease levels of free radicals. GSH is known as a highly effective extra-and intra-cellular antioxidant compound, as its scavenging and antioxidant properties allow it to neutralize hydrogen peroxide and hydroperoxides. A consistent and sufficient glutathione level can prevent LPS-induced damage [28]. Here, LPS-induced liver damage was accompanied by increased TBARS and nitrite production in addition to decreased content of major antioxidant GSH. These findings suggest that oxidative stress may enhance LPS hepatotoxicity and consequently, that SAHA may protect hepatocytes against oxidants by directly scavenging intracellular ROS. The mechanism of hepatoprotection by SAHA against LPS may involve the restoration of GSH concentration in liver.
It has been reported that the activation of ROS-sensitive ASK1 is required for LPS induction of p38 but is not required for induction of the NF-κB pathway [10]. LPS activated ASK1, p38, and JNK, while SAHA suppressed ASK1 and JNK expression, which may account for the selective inhibition of the LPS-induced p38 pathway. Previous studies have shown that ASK1 is required for the activation of JNK signaling in cells stimulated by tumor necrosis factor-α (TNF-α) [29]. LPS-induced TNF-α was inhibited by SAHA in mouse liver (data not shown). Thus, SAHA may inhibit JNK signaling through the inhibition of ASK1, which is activated by LPS-induced TNF-α signaling activation.
ROS is a significant regulator of apoptosis and can induce apoptotic processes [30]. The origin of LPS-induced ROS production is not limited to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase – interestingly the involvement of mitochondria has also been proposed [14, 31, 32]. Mitochondrial damage leads to excessive ROS release, which may exacerbate damage and induce the loss of mitochondrial membrane potentials that initiate caspase-9 activity [33]. In the present study, SAHA decreased LPS-induced ROS levels and inhibited caspase-9 activity in LPS-activated liver. In light of previous results, such findings suggest that SAHA inhibits LPS-induced hepatocyte apoptosis that is caused by oxidative stress through ROS.
This study has certain limitations which should be addressed. Intraperitoneal administration of drugs is in routine in mouse models, and we have previously administered SAHA intraperitoneally with good systemic effects, but this approach is not clinically very relevant. So far, we have only examined the effects of SAHA on acute lung injury [34] and acute liver injury (the present study). Further research investigating the impact of SAHA on the other vital organs should be conducted. Moreover, this was essentially a proof-of-concept study. LPS is endotoxin, a component of the Gram-negative bacterial wall. Although it does not completely replicate all aspects of septic shock, the LPS model is nevertheless well established. An ongoing study is being prepared to examine the impact of SAHA in a CLP model.
Previous evidence suggest that apoptosis is the primary cause of the septic inflammatory response [1]. SAHA therapy attenuated apoptotic effects by demonstrating the novel capacity to powerfully suppress LPS-induced apoptosis in mouse liver. SAHA has been shown to have low toxicity, and therefore administration of SAHA has promise as a new therapy that may be capable of decreasing sepsis-mediated mortality without inducing deleterious side effects [35].
Highlights for Review.
Histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA, vorinostat) has only recently emerged as a possible therapeutic intervention for hyper-inflammation, as we and others shown that SAHA reduces the lethality of hemorrhagic shock, suppresses pro-inflammatory cytokines expression, and improves survival of mice in models of endotoxic shock. While it has been reported that SAHA reduces inflammation in a LPS-induced endotoxic shock model, the present study demonstrates for the first time antioxidant, anti-apoptotic and anti-inflammatory effects of SAHA on hepatotoxicity induced by LPS as well as the mechanisms involved in such hepatoprotection.
Acknowledgements
This work was supported by NIHRO1 GM084127 to H.B.A.
ABBREVIATIONS USED
- ASK1
apoptosis signal-regulating kinase 1
- COX-2
cyclooxygenase-2
- GSH
glutathione
- NO
nitric oxide
- JNK
Jun N-terminus kinase
- ROS
reactive oxygen species
- SAHA
suberoylanilide hydroxamic acid
- TBARS
Thiobarbituric acid reactive substances
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ contribution: Y.L. and H.B.A. designed this study, for which H.B.A. secured funding. Y.Z. performed experiments, collected and analyzed data. B.L. provided experimental support. Y.Z. and Y.L. wrote the manuscript, which was critically revised by Y.L., P.Z, T.B., R.M., and H.B.A. All authors read and approved the final manuscript.
Author Disclosure Statement
Ralph Mazitschek has financial interests in SHAPE Pharmaceuticals and Acetylon Pharmaceuticals. He is also the inventor on IP licensed to these two entities. His interests were reviewed and are managed by Massachusetts General Hospital and Partners Health Care in accordance with their conflict of interest policies. The other authors declare no conflict of interest.
REFERENCES
- 1.Oberholzer C, Oberholzer A, Clare-Salzler M, Moldawer LL. 2001. Apoptosis in sepsis: a new target for therapeutic exploration. FASEB J. 2001;15:879. doi: 10.1096/fj.00-058rev. [DOI] [PubMed] [Google Scholar]
- 2.Westphal M, Stubbe H, Sielenkämper AW, Borgulya R, Van Aken H, Ball C, Bone HG. Terlipressin dose response in healthy and endotoxemic sheep: impact on cardiopulmonary performance and global oxygen transport. Intensive Care Med. 2003;29:301. doi: 10.1007/s00134-002-1546-5. [DOI] [PubMed] [Google Scholar]
- 3.Cadenas S, Cadenas AM. Fighting the stranger-antioxidant protection against endotoxin toxicity. Toxicology. 2002;180:45–63. doi: 10.1016/s0300-483x(02)00381-5. [DOI] [PubMed] [Google Scholar]
- 4.Mallis RJ, Buss JE, Thomas JA. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J. 2001;355:145. doi: 10.1042/0264-6021:3550145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuo Y, Xiang B, Yang J, Sun X, Wang Y, Cang H, Yi J. Oxidative modification of caspase-9 facilitates its activation via disulfide-mediated interaction with Apaf-1. Cell Res. 2009;19:449. doi: 10.1038/cr.2009.19. [DOI] [PubMed] [Google Scholar]
- 6.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 7.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J Immunol. 2006;176:5720. doi: 10.4049/jimmunol.176.10.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1–p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Alam HB. Modulation of acetylation: creating a pro-survival and anti-inflammatory phenotype in lethal hemorrhagic and septic shock. J Biomed Biotechnol. 2011;2011:523481. doi: 10.1155/2011/523481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, Schrenzel J, François P, Calandra T. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117(4):1205–1217. doi: 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, deMoya M, Hales CA, Alam HB. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32(5):517–523. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 13.Payne CM, Bernstein C, Bernstein H. Apoptosis overview emphasizing the role of oxidative stress, DNA damage and signal-transduction pathways. Leuk Lymphoma. 1995;19:43. doi: 10.3109/10428199509059662. [DOI] [PubMed] [Google Scholar]
- 14.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 15.Herrera B, Alvarez AM, Sánchez A, Fernández M, Roncero C, Benito M, Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor β in fetal hepatocytes. FASEB J. 2001;15:741. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- 16.Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol. 2006;6:813. doi: 10.1038/nri1943. [DOI] [PubMed] [Google Scholar]
- 17.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferluga J, Allison AC. Role of mononuclear infiltrating cells in pathogenesis of hepatitis. Lancet. 1978;2(8090):610–611. doi: 10.1016/s0140-6736(78)92828-3. [DOI] [PubMed] [Google Scholar]
- 19.Davies KJ. Protein damage and degradation by oxygen radicals. I. General aspects. J Biol Chem. 1987;262:9895. [PubMed] [Google Scholar]
- 20.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 21.Thiemermann C. The role of the l-arginine: nitric oxide pathway in circulatory shock. Adv Pharmacol. 1994;28:45. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- 22.Titheradge MA. Nitric oxide in septic shock. Biochim Biophys Acta. 1995;1411:437. doi: 10.1016/s0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]
- 23.Szabo C. Alterations in nitric oxide production in various forms of circulatory shock. New Horiz. 1995;3:2. [PubMed] [Google Scholar]
- 24.Boveris A, Alvarez S, Navarro A. The role of mitochondrial nitric oxide synthase in inflammation and septic shock. Free Radic Biol Med. 2002;33:1186. doi: 10.1016/s0891-5849(02)01009-2. [DOI] [PubMed] [Google Scholar]
- 25.Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 26.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sewerynek E, Melchiorri D, Chen L, Reiter RJ. Melatonin reduces both basal and bacterial lipopolysaccharide-induced lipid peroxidation in vitro. Free Radic Biol Med. 1995;19:903. doi: 10.1016/0891-5849(95)00101-3. [DOI] [PubMed] [Google Scholar]
- 28.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 29.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 30.England K, Cotter TG. Direct oxidative modifications of signaling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005;10:237. doi: 10.1179/135100005X70224. [DOI] [PubMed] [Google Scholar]
- 31.Cakir Y, Ballinger SW. Reactive species-mediated regulation of cell signaling and the cell cycle: the role of MAPK. Antioxid Redox Signal. 2005;7:726. doi: 10.1089/ars.2005.7.726. [DOI] [PubMed] [Google Scholar]
- 32.Woo CH, Lim JH, Kim JH. Lipopolysaccharide induces matrix metalloproteinase-9 expression via a mitochondrial reactive oxygen species-p38 kinase-activator protein-1 pathway in Raw 264.7 cells. J Immunol. 2004;173:6973. doi: 10.4049/jimmunol.173.11.6973. [DOI] [PubMed] [Google Scholar]
- 33.Ricci JE, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160:65. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, Jin G, Lu J, deMoya MA, Velmahos GC, Alam HB. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;148(2):246–254. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]