

Abstract
Diabetic cardiomyopathy is a significant contributor to the morbidity and mortality associated with diabetes and metabolic syndrome. However, the underlying molecular mechanisms that lead to its development have not been fully elucidated. Hydrogen sulfide (H2S) is an endogenously produced signaling molecule that is critical for the regulation of cardiovascular homeostasis. Recently, therapeutic strategies aimed at increasing its levels have proven cardioprotective in models of acute myocardial ischemia-reperfusion injury and heart failure. The precise role of H2S in the pathogenesis of diabetic cardiomyopathy has not yet been established. Therefore, the goal of the present study was to evaluate circulating and cardiac H2S levels in a murine model of high fat diet (HFD)-induced cardiomyopathy. Diabetic cardiomyopathy was produced by feeding mice HFD (60% fat) chow for 24 weeks. HFD feeding reduced both circulating and cardiac H2S and induced hallmark features of type-2 diabetes. We also observed marked cardiac dysfunction, evidence of cardiac enlargement, cardiac hypertrophy, and fibrosis. H2S therapy (SG-1002, an orally active H2S donor) restored sulfide levels, improved some of the metabolic perturbations stemming from HFD feeding, and attenuated HFD-induced cardiac dysfunction. Additional analysis revealed that H2S therapy restored adiponectin levels and suppressed cardiac ER stress stemming from HFD feeding. These results suggest that diminished circulating and cardiac H2S levels play a role in the pathophysiology of HFD-induced cardiomyopathy. Additionally, these results suggest that H2S therapy may be of clinical importance in the treatment of cardiovascular complications stemming from diabetes.
Keywords: diabetes, diabetic cardiomyopathy, hydrogen sulfide, ER stress
1.1 Introduction
Cardiovascular disease refers to any disease that affects the cardiovascular system, including cardiac disease, vascular diseases of the brain and kidney, and peripheral artery disease. Worldwide, it is on the rise and continues to be the leading cause of morbidity and mortality in both men and women [1]. A major contributor to the rise in cardiovascular disease is the increased prevalence of diabetes mellitus [2]. According to the World Health Organization, diabetes affects 347 million people worldwide (http://www.who.int/mediacentre/factsheets/fs312/en/). Diabetes is responsible for diverse cardiovascular complications, such as hypertension, atherosclerosis, and heart failure [3]. Additionally, diabetes and insulin resistance are powerful predictors of cardiovascular morbidity and mortality, as well as independent risk factors for death in patients with established heart failure [4; 5]. Diabetes can also affect cardiac structure and function in the absence of changes in blood pressure and coronary artery disease [3], a condition termed “diabetic cardiomyopathy” [6]. The pathophysiology of diabetic cardiomyopathy is multifactorial with evidence that autonomic dysfunction, metabolic derangements, and the development of interstitial fibrosis all contribute to myocardial stiffness and impaired contractility [3; 7; 8]. However, the molecular mechanisms that underlie the development of diabetic cardiomyopathy require further investigation.
The endoplasmic reticulum (ER) is the central organelle for secretory/transmembrane protein folding, calcium storage, and lipid synthesis [2]. Pathological stimuli such as oxidative stress, the accumulation of lipids, and the accumulation of misfolded proteins, interfere with the function of the ER [9]. As a result, ER stress develops and the unfolded protein response (UPR) is triggered [10]. In an effort to ameliorate ER stress, the UPR activates three pathways to antagonize the cellular stress. As an initial step, the chaperone protein BiP (GRP78) senses unfolded or damaged proteins. It then subsequently activates PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). Engagement of the ER stress/UPR response acutely reduces protein synthesis in the ER, enhances protein degradation of damaged or misfolded proteins, and selectively induces expression of protective proteins [11; 12]. While the acute activation of the ER stress/UPR is protective, prolonged activation triggers an apoptotic-signaling cascade resulting in cell death [9]. Mounting evidence suggests that ER stress/UPR plays a critical role in the heart during the development of diabetic cardiomyopathy [2; 9]. For instance, markers of ER stress are elevated in the hearts of diabetic animals [13; 14; 15] and therapeutic strategies that manipulate components of the ER stress response improve cardiac function in the setting of diabetes [16; 17]. This suggests that targeting ER stress may be a viable treatment option to prevent or attenuate the development of diabetic cardiomyopathy.
Hydrogen sulfide (H2S) is an endogenously produced gaseous signaling molecule that is critical for the regulation of cardiovascular homeostasis [18; 19]. Recently, therapeutic strategies aimed at increasing the levels of H2S have been shown to be cardioprotective in models of acute myocardial ischemia-reperfusion (MI/R) injury and heart failure [20; 21; 22; 23]. These cytoprotective effects are attributed to the ability of H2S to upregulate antioxidant defenses and to reduce apoptosis, inflammation, and mitochondrial injury [24]. In brief, cardiomyopathic oxidative stress states cause an increase in reactive oxygen species (ROS) production, leading to mitochondrial damage, lipid peroxidation, and DNA strand breaks. These and related maladaptations are mitigated by antioxidants, which scavenge and neutralize the ROS [25]. H2S has been found to upregulate antioxidant properties, attenuating the adverse effects of oxidative stress [26]. Increasing evidence suggests an association between oxidative stress and the progression of diabetic complications in various tissues, including the heart [27; 28; 29]. Thus, the reported antioxidant effects of H2S may be of critical importance in the diabetic myocardium. However, circulating levels of H2S are decreased in animal models of diabetes [30; 31; 32; 33] and in T2DM patients [30; 34]. Previous work from our lab found that lower levels of H2S are not confined to the circulation in the setting of diabetes, as evidenced by the findings that cardiac levels of H2S were also decreased in db/db diabetic mice [35]. Furthermore, we found that treatment with exogenous H2S for 7 days increased cardiac H2S levels and reduced myocardial injury following I/R. This latter finding supports the idea that decreased H2S levels in the setting of diabetes contribute to the pathophysiology of the disease [36], especially in relation to cardiovascular complications.
To our knowledge, the precise role of H2S in the pathogenesis of diabetic cardiomyopathy has not yet been established. Therefore, the goal of the present study was to evaluate circulating and cardiac H2S levels in a murine model of high fat diet (HFD)-induced cardiomyopathy. In addition, we investigated if H2S therapy ameliorated the development of diabetic cardiomyopathy by decreasing ER stress.
2.1 Materials and Methods
2.1.1 Animals
Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) at 6 weeks of age. Starting at 8 weeks of age, different groups of mice were maintained on one of the following diets for 24 weeks: (1) Control Diet (10% fat diet, Open Source Diets D12450Bi), (2) High Fat Diet (HFD, 60% fat; Open Source Diets D12492i), or (3) HFD supplemented with SG-1002, an orally active H2S donor. SG-1002 (provided by Sulfagenix, Cleveland, OH) was added to the HFD mice to achieve a dosage of 20 mg/kg/day [37]. All diets were purchased from Research Diets, Inc. (New Brunswick, NJ). All experimental protocols were approved by the Institute for Animal Care and Use Committee at Emory University School of Medicine and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 86-23, revised 1996), and with federal and state regulations.
2.1.2 Blood and Tissue Biochemistry
Blood obtained via a tail snip following 24 weeks of HFD feeding was screened using an Xtra glucose-monitoring system (Precision). Serum insulin (Millipore), adiponectin (Millipore) and cholesterol (Sigma) levels were determined using standard assay kits according to the manufacturers' recommendations. Cardiac lipid content was determined using a standard assay kit (Sigma) according to the manufacturer's recommendations.
2.1.3 Glucose Tolerance Test
Intraperitoneal glucose tolerance test was performed as follows. A glucose solution (2.0g/kg body weight in a 10% solution) was intraperitoneally administered into mice that were fasted for 8 hours. Both before and after injection (at 0,15,30,45,60,90,120 minutes), blood samples were obtained by tail vein clipping to determine blood glucose using an Xtra glucose-monitoring system (Precision).
2.1.4 Hydrogen Sulfide Measurements
Hydrogen sulfide and sulfane sulfur levels were measured in the heart and blood according to previously described methods [38]. For heart tissue, the amount of H2S is reported as nmol/mg wet weight. For the blood, the amount of H2S is reported as μM.
2.1.5 Histology
For histological analysis, hearts were collected, fixed in 10% buffered formalin, and embedded in paraffin. Serial 5-μm heart sections from each group were stained with Picrosirius Red (to detect fibrosis). Digital images of the slides were then captured and analyzed using ImageJ. For each heart, we analyzed multiple sections taken from the mid-ventricle and then averaged these numbers to obtain a single % fibrosis/LV measurement for each animal.
2.1.6 Wheat Germ Agglutinin Staining
Cell surface area (μm2) and cross-sectional area (μm2) were analyzed by staining cardiac cryosections with wheat germ agglutinin (WGA)-Texas Red-X conjugate (Life Technologies) as described previously to show myocyte membranes in histological sections [38]. Paraffin fixed slides were deparaffinized, washed in 1XPBS and then incubated in 10μg WGA-Texas Red-X conjugate for 1 hour at room temperature followed by additional washes in 1XPBS. Slides were mounted with Vectashield mounting medium (Vector Labs) and sealed. Digital images were captured and cell surface area was assessed with NIS Elements Imaging Software (version 3.22.11) in 5 animals per group with at least 5 randomly taken sections per heart and at least 100 myocytes counted per animal.
2.1.7 Western Blot Analysis
Western blot analysis was performed as described previously [38]. Protein concentrations were measured with the DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein were loaded into lanes of polyacrylamide-SDS gels. The gels were electrophoresed, followed by transfer of the protein to a PVDF membrane. The membranes were then blocked and probed with primary antibodies overnight at 4°C. Immunoblots were next processed with secondary antibodies (Cell Signaling) for 1 hour at room temperature. Immunoblots were then probed with a Super Signal West Dura kit (Thermo) to visualize signal, followed by exposure to X-ray film (Denville Scientific). Immunoblots were subsequently stained with Coomassie blue to detect total protein load. The film was scanned to make a digital copy and densitometric analysis was performed to calculate relative intensity with ImageJ software from the National Institutes of Health (version 1.40g) using the Rodbard function. Results were presented as the ratio of the expression of the protein of interest to the total protein load. We used total protein load as our control because an initial analysis revealed that the expression of GAPDH changed significantly in response to HFD feeding (Supplemental Fig. 1).
2.1.8 Echocardiograph Analysis
Baseline echocardiography images were obtained prior to administration of diets. The mice were lightly anesthetized with isoflourane (1-5% in 100% oxygen) and in vivo transthoracic echocardiography of the left ventricle (LV) using a 38-MHz linear array scanhead interfaced with a Vevo 2100 (Visualsonics) was used to obtain high-resolution M-mode images. From these images LV end-diastolic diameter (LVEDD), LV end-systolic diameter (LVESD), fractional shortening (FS), and ejection fraction (EF) were calculated. Echocardiography images were obtained and analyzed again every 4 weeks for 24 weeks.
2.1.9 Hemodynamic Analysis
Following the last echocardiography session, mice were anesthetized with isoflourane (1-5% in 100% oxygen). LV hemodynamics were assessed by passing a 1.2F pressure catheter (Scisense) into the LV lumen via the right common carotid artery. The catheter was connected to a computer and data was collected with LabScribe2 software (Version 2.334, iWorx Systems, Inc). Circumferential stress was calculated as previously described [38].
2.1.10 Statistical Analysis
All the data are expressed as mean ± standard error (SEM). Means were compared using Prism 4 (GraphPad Software, Inc) with oneway analysis of variance (ANOVA), or two-way ANOVA where indicated. For the ANOVA, if a significant result was found, the Tukey (one-way ANOVA) or Bonferroni (two-way ANOVA) test was used as the post hoc analysis. For all data, a p value less than 0.05 was considered significant.
3.1 Results
3.1.1 Oral H2S therapy ameliorates HFD-induced cardiac dysfunction
Our initial experiments examined the effects of HFD feeding on the myocardial expression of the three known H2S-producing enzymes, as well as the levels of circulating and myocardial sulfide levels. As expected, HFD feeding for 24 weeks induced increases in body weight, serum glucose levels, glucose intolerance, serum insulin levels, and serum cholesterol levels, recapitulating hallmark features of type-2 diabetes (Table and Supplemental Fig. 2). Immunoblot analysis of whole cell extracts from control and HFD-fed mouse hearts revealed that the expression of cystathionine-γ-lyase (CSE), cystathionine-β-synthase (CBS), and 3-mercaptopyruvate sulfutransferase (3-MST) were unaltered under HFD conditions (Fig. 1A-B). However, free H2S and sulfane sulfur levels were significantly lower in the blood and hearts of HFD-fed mice when compared to control mice (Fig. 1C-F), indicating that HFD feeding decreases circulating and cardiac sulfide levels.
Table 1. Body weight and blood analysis of mice fed a HFD for 24 weeks.
| Control | HFD | HFD-S | HFD-D | |
|---|---|---|---|---|
| Body Weight (grams) | 32.3 ± 0.8 | 53.3 ± 1.6*** | 46.5 ± 1.9*** | 51.1 ± 0.9*** |
| Blood Glucose (mg/dL) | 137.5 ± 4.9 | 206.0 ± 5.2*** | 184.1 ± 5.9ψ | 183.7 ± 6.8ψ |
| Insulin (ng/mL) | 0.8 ± 0.1 | 22.61 ± 0.7*** | 5.8 ± 1.6φ | 13.4 ± 1.8Φ |
| Total Cholesterol (mg/dL) | 39.3 ± 1.5 | 80.1 ± 3.4*** | 80.1 ± 3.2*** | 85.1 ± 3.3*** |
Values are means ± SEM for n = 8-15 per group.
p<0.001vs. Control.
p<0.001 vs. Control and p<0.05 vs. HFD.
p<0.05 vs. Control, p<0.001 vs. HFD, and p<0.01 vs. HFD-D.
Fig. 1.
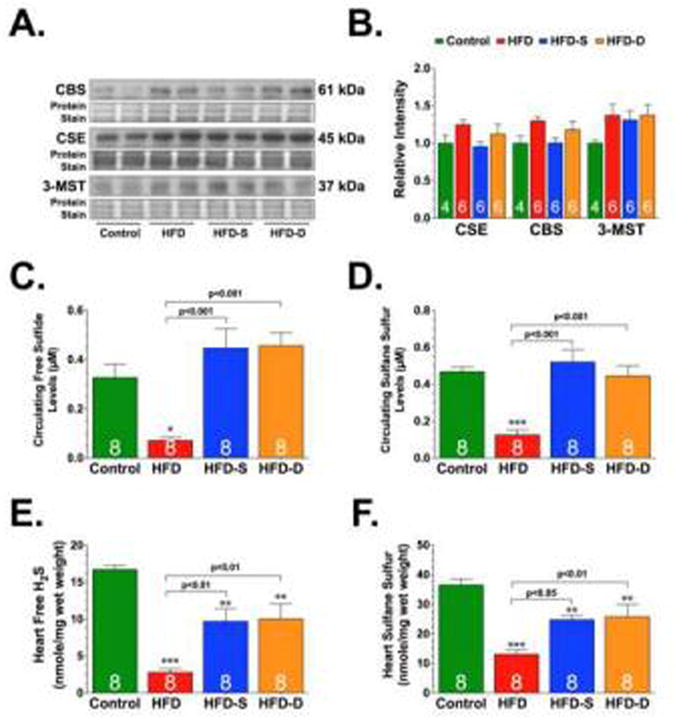
Representative immunoblots and densitometric analysis of cystathionine beta synthase (CBS), cystathionine gamma lyase (CSE), and 3-mercaptopyruvate sulfutransferase (3-MST) (A-B). Circulating (C-D) and cardiac (E-F) levels of free hydrogen sulfide (H2S) and sulfane sulfure. All samples (blood and hearts) were collected from control, high-fat diet (HFD), and HFD-fed mice supplemented with SG-1002 (20 mg/kg/day). One group of mice received the SG-1002 supplemented HFD chow for the entire 24 weeks (HFD-S). We delayed the treatment in another group mice (HFD-D). These mice received the HFD chow for 12 weeks before being switched to the SG-1002 supplemented HFD chow for the final 12 weeks of the study. Results are expressed as mean ± SEM. Numbers in bars represent the sample size. *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
Next, we examined the effects of oral H2S therapy on the development of HFD-induced cardiomyopathy. For these experiments, we supplemented SG-1002 (20 mg/kg/day) in the HFD chow. One group of mice received the SG-1002 supplemented HFD chow for the entire 24 weeks (HFD-S). We delayed the treatment in another group of mice (HFD-D). These mice received the HFD chow for 12 weeks before being switched to the SG-1002 supplemented HFD chow for the final 12 weeks of the study. Our initial studies found that SG-1002 supplementation restored circulating sulfide levels and partially restored cardiac sulfide levels under HFD conditions without providing any alterations in the expression of the H2S-producing enzymes (Fig. 1).
In association with the metabolic perturbations stemming from HFD, we also observed marked cardiac dysfunction beginning at 4 weeks of HFD feeding, as exemplified by increased LVESD, decreased LV ejection fraction, and decreased LV fractional shortening (Fig. 2A-C). Additionally, analysis of LV hemodynamics at 24 weeks of HFD feeding revealed decreased LV dP/dt max, decreased LV dP/dt min, increased relaxation constant Tau, and increased circumferential stress (Fig. 2D-F). Both groups of H2S treated mice displayed improvements in blood glucose levels, glucose tolerance and serum insulin levels when compared to mice in the HFD group (Table). However, H2S treatment did not alter the HFD-induced increase in body weight or serum cholesterol levels. The mice in the HFD-S group developed less severe changes in cardiac dilatation and dysfunction when compared to the HFD group. The mice in the HFD-D group were indistinguishable from the HFD mice up until 12 weeks. However, by 16 weeks (4 weeks on the SG-1002 supplemented HFD chow) these mice displayed improvements in cardiac dilatation and dysfunction that were maintained until the conclusion of the 24 weeks. Importantly, the HFD-D mice were indistinguishable from the HFD-S mice after the addition of SG-1002 in the diet.
Fig 2.
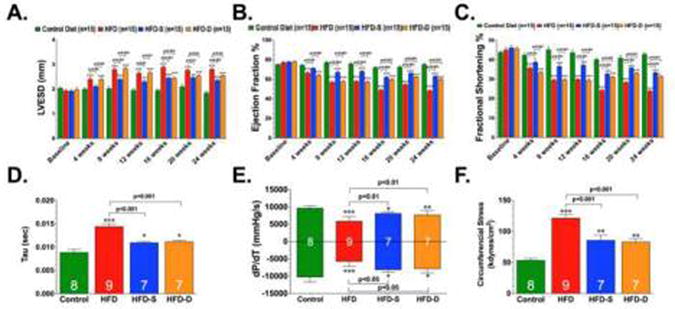
Left Ventricular End Systolic Diameter (LVESD) (A), LV Ejection Fraction (B), LV Fractional Shortening (C) relaxation constant Tau (D), Max dP/dT and Min dP/dT, (E) and Circumferential Stress (F) were evaluated in control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Results are expressed as mean ± SEM.In panels A-C, ***p<0.001 vs. Baseline. In panels D-F, *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
3.1.2 Oral H2S therapy ameliorates HFD-induced cardiac hypertrophy and fibrosis
Hypertrophy and fibrosis are pathological responses of cardiomyocytes to the stress of the HFD condition [39; 40]. Therefore, we examined the effects of HFD and H2S therapy on cardiac remodeling in terms of morphological changes at 24 weeks of HFD feeding. Consistent with the echocardiography and hemodynamic findings, our morphological and histological analysis revealed that HFD induced increases in heart weight to tibia length ratios (cardiac enlargement), increases in the cell surface area and cross sectional areas of cardiomyocytes (cardiac hypertrophy) and increases in Picrosirius Red staining (fibrosis) (Fig. 3). Although evidence of cardiac enlargement, cardiac hypertrophy, and fibrosis were present in the HFD-S and HFD-D groups, these morphological and histological changes were significantly less than the changes observed in the HFD group.
Fig 3.
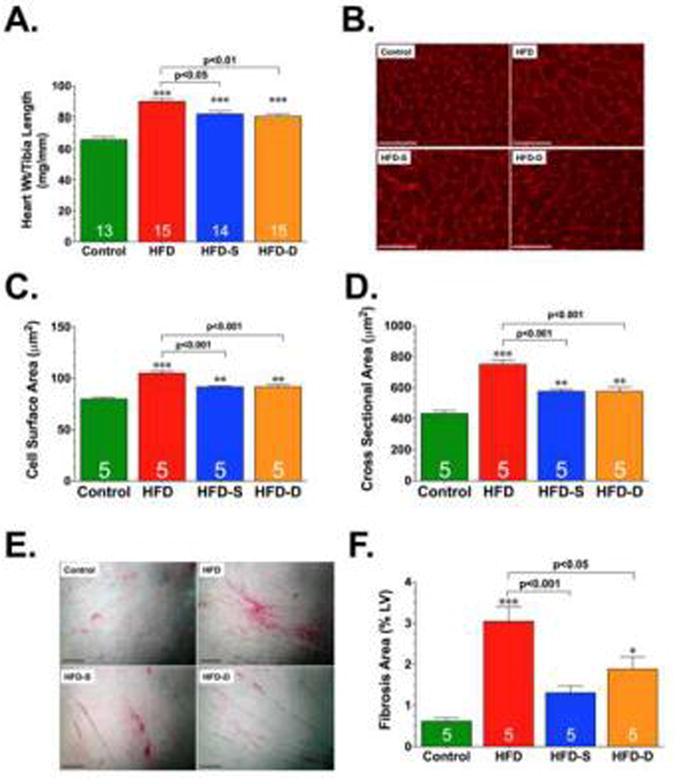
Heart weight to body weight ratios (A), Representative photomicrographs of wheat germ agglutinin stained hearts (B), Summary of myocyte cell surface area (C) and cross sectional area measurements (D) of wheat germ agglutinin stained hearts, Representative photomicrographs of Picrosirius Red stained hearts (E), and summary of fibrosis area as % of the LV calculated from the Picrosirius Red sections (F), in control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Scale bar equals 100 μm in panels B and D, respectively. Results are expressed as mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
3.1.3 Oral H2S therapy induces Adiponectin-AMPK signaling in the setting of HFD feeding
Adiponectin (APN), an adipokine secreted from adipose tissue, plays an important role in maintaining cardiovascular health [41; 42; 43]. Specifically, there is a correlation between reduced APN levels and increase cardiovascular risk [43]. Emerging evidence indicates that APN deficiency and resistance occurs in the early stages of metabolic syndrome and contributes to the development of insulin resistance and lipid accumulation [42; 44]. We, therefore, examined the effects of HFD and H2S therapy on circulating APN levels. Our analysis revealed lower circulating levels of APN in response to HFD feeding (Fig 4A). Although the levels of APN were lower in the HFD-S and HFD-D groups when compared to the Control group, the levels were significantly higher than those observed in the HFD group. APN delivers much of its metabolic-regulatory effects via the induction of AMP-activated protein kinase (AMPK) [45]. Our analysis revealed that the expression of phosphorylated AMPK was significantly decreased in the hearts of HFD-fed mice compared to control mice (Fig. 4B-C). In contrast, this HFD-induced decrease was attenuated in the hearts of mice in the HFD-S and HFD-D groups. No differences in the expression of total AMPK were noted in the hearts of any of the groups. We next turned our attention to downstream targets of AMPK. The activation of AMPK coordinates the regulation of several steps in substrate transport and metabolism, which are critical for energy generation and conservation under times of stress [46]. For instance, AMPK stimulates glucose transport by increasing the expression of glucose transporter 4 (GLUT4). Immunoblot analysis revealed a significant decrease in the expression of GLUT4 under HFD conditions (Fig. 4B and D). However, this HFD-induced decrease was attenuated in the hearts of mice in the HFD-S and HFD-D groups. AMPK also modulates cardiac fatty acid oxidation and subsequently lipid accumulation through the phosphorylation and inhibition of acetyl-coenzyme A carboxylase (ACC) [47]. As with AMPK, the phosphorylation of ACC was decreased under HFD conditions (Fig. 4B and E). In contrast, oral H2S therapy increased the phosphorylation of ACC when compared to the HFD group. Finally, to examine the effects of H2S therapy on HFD-induced lipid accumulation, we evaluated triglyceride levels in hearts collected from Control, HFD, HFD-S, and HFD-D mice. Consistent with our findings related to ACC, direct measurement of triglyceride content revealed that oral H2S therapy decreased HFD-induced lipid accumulation (Fig. 4F).
Fig. 4.
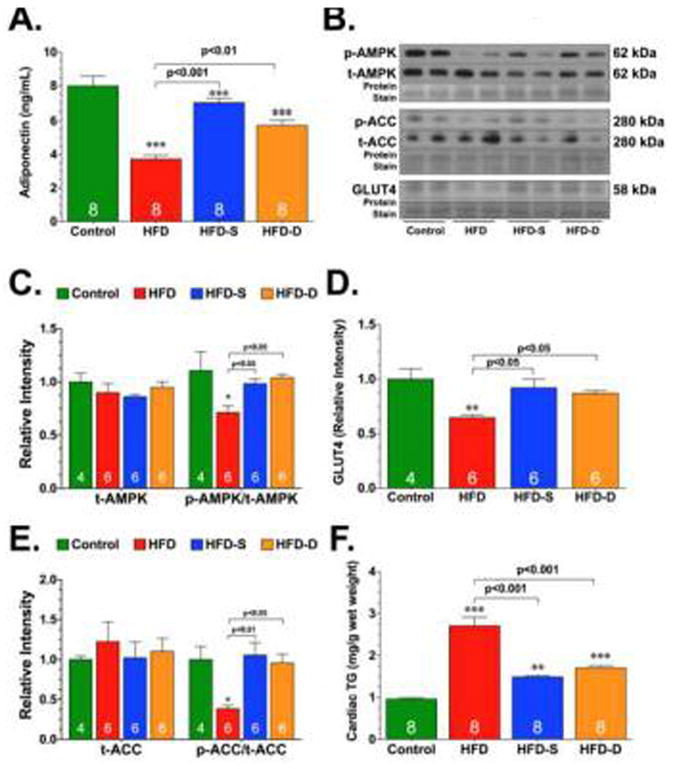
Circulating adiponectin levels (A), representative immunoblots (B) and densitometric analysis of total AMP-activated protein kinase (t-AMPK) and the ratio of phosphorylated AMPK (p-AMPK) to t-AMPK (C), glucose transporter 4 (GLUT4) (D), total acetyl-coenzyme A carboxylase (t-ACC) and the ratio of phosphorylated ACC (p-ACC) to t-ACC (E), and cardiac triglyceride levels (F) in control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Results are expressed as mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
3.1.4 Oral H2S therapy attenuates HFD-induced ER stress in the heart
Lipotoxicity is thought to contribute to the development of diabetic cardiomyopathy in part via the induction of ER stress [9]. We next examined changes in ER stress markers in heart tissue collected from the experimental groups. First, we focused on the PERK arm of the ER stress/UPR. The activation of PERK leads to an early and transient attenuation of protein biosynthesis [9]. This in turn leads to an increase in the expression of ATF4, which in turn induces the expression of numerous genes involved in the resolution of ER stress [9]. Immunoblot analysis of heart homogenates collected from Control, HFD, HFD-S, and HFD-D mice revealed that the expression of ATF4 remained unchanged (Fig. 5). Next, we focused on the ATF6 arm of the ER stress/UPR. ATF6 is a transmembrane basic leucine zipper transcription factor that normally exists in the ER as a dimer linked by intermolecular disulfide bonds in the luminal domain [9; 48]. In response to ER stress, ATF6 translocates to the Golgi where it undergoes cleavage by site-1 and site-2 proteases [9]. This yields cleaved forms of ATF6, which translocate to the nucleus and induce target ER genes. Immunoblot analysis of heart homogenates revealed significant increases in the expression of the cleaved form of ATF6 appearing at 60 kDa under HFD conditions (Fig. 5). However, the HFD-induced cleavage of ATF6 was dampened in the hearts of mice in the HFD-S and HFD-D groups.
Fig. 5.
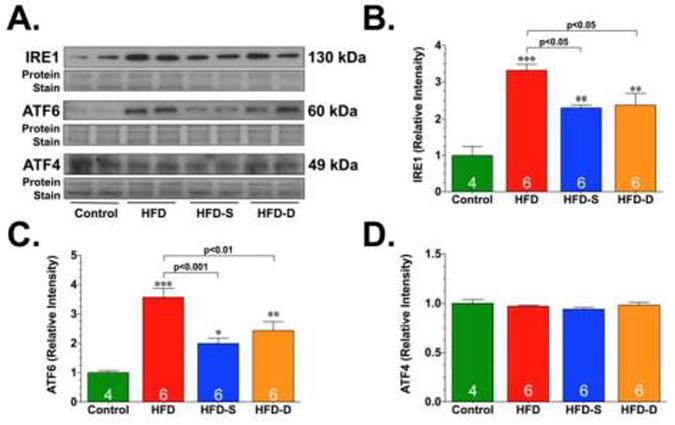
Representative immunoblots (A) and densitometric analysis of inositol-requiring enzyme 1 (IRE1) (B), activating transcription factor 6 (ATF6) (C), and ATF4 (D). All samples were collected from the hearts of control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Results are expressed as mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
Next, experiments evaluated the effects of HFD and H2S therapy on the IRE arm of the ER stress/UPR. IRE1 is a type 1 ER transmembrane protein that functions as a kinase and as an endoribonuclease [12]. In response to ER stress, the endoribonuclease activity of IRE1 is induced. In mammalian cells, the substrate for the IRE1 endoribonuclease is the mRNA for X-box–binding protein-1 (XBP1) [12]. Immunoblot analysis revealed a significant increase in the expression of IRE1 (Fig. 5), as well as an increase in the spliced form of XBP1 (Fig. 6) under HFD conditions. Again, as observed for the cleavage of ATF6, the HFD-induced changes in the expression of IRE1 and spliced XBP1 were attenuated in the hearts of mice in the HFD-S and HFD-D groups. In addition to its ability to target XBP1, IRE1 can also induce a c-Jun N-terminal kinase (JNK) signaling pathway [9]. This is important in the context of diabetic cardiomyopathy because JNK can influence cardiac remodeling in response to stress via the activation of proapoptotic or pro-hypertrophic signaling cascades [49]. Our analysis revealed that the expression of total JNK was significantly elevated in the hearts of HFD-fed mice compared to control mice (Fig. 6). In contrast, the expression of total JNK (t-JNK) was decreased in the hearts of HFD-S and HFD-D mice when compared to both control and HFD-fed mice. We also found that there was no difference in the phosphorylation of JNK (p-JNK) in HFD, HFD-S, or HFD-D mice when the expression was compared to the expression of total JNK. However, there was a significant increase in the expression of p-JNK when the expression was compared to the total protein load, suggesting that there was more p-JNK under HFD conditions. Importantly, the HFD-induced increase in p-JNK was attenuated in the hearts of mice in the HFD-S and HFD-D groups.
Fig. 6.
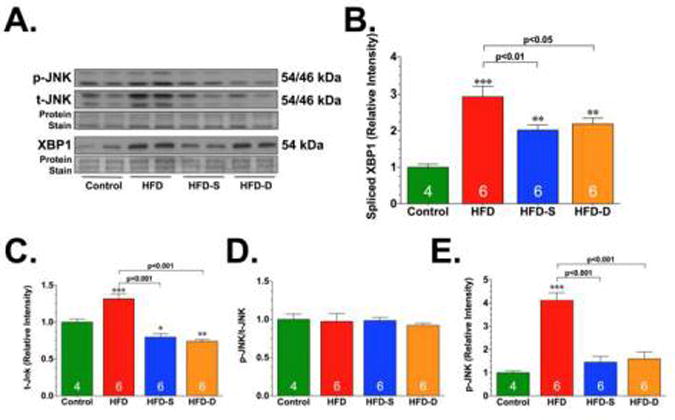
Representative immunoblots (A) and densitometric analysis of X-box– binding protein-1 (XBP1) (B), total JNK (t-JNK) (C), ratio of phosphorylated JNK (p-JNK) to t-JNK (D), and ratio of p-JNK to total protein load (E). All samples were collected from the hearts of control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Results are expressed as mean ± SEM. *p<0.05, **p<0.01 and ***p<0.001 vs. Control.
As noted above, while the acute activation of the ER stress/UPR is protective, prolonged activation triggers cell death [9]. We therefore evaluated the expression of ER chaperone proteins and the expression of pro-cell death proteins induced by ER stress (Fig. 7). The ER chaperone proteins, glucose-regulated protein 78 (BiP or GRP78) and 94 (GRP94) act to promote protein folding by reducing general protein synthesis and enhancing the degradation of misfolded proteins [50]. Immunoblot analysis revealed a significant increase in the expression of GRP94 under HFD conditions. However, this HFD-induced increase was attenuated in the hearts of mice in the HFD-S and HFD-D groups. In contrast, the expression of GRP78 was not altered in any of the experimental groups. Further analysis revealed that the HFD-induced cleavage of capase-12 (active) was attenuated in the hearts of mice in the HFD-S and HFD-D groups. Together, these results suggest that oral H2S therapy mitigates HFD-induced ER stress.
Fig. 7.
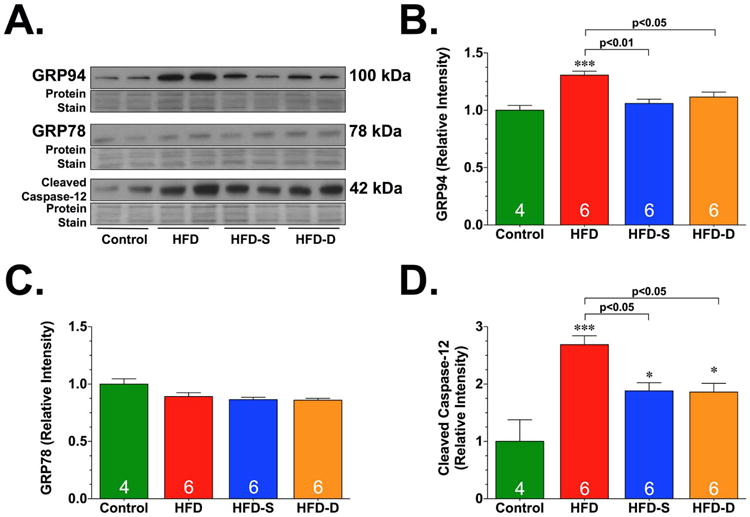
Representative immunoblots (A) and densitometric analysis of glucose-regulated protein 94 (GRP94) (B), GRP78 (C), and cleaved caspase-12 (D). All samples were collected from the hearts of control, HFD, HFD-S, and HFD-D mice following 24 weeks of HFD feeding. Results are expressed as mean ± SEM. *p<0.05 and ***p<0.001 vs. Control.
4.1 Discussion
Diabetic cardiomyopathy is a significant contributor to the morbidity and mortality associated with diabetes and metabolic syndrome [4]. However, the underlying molecular mechanisms leading to the development of diabetic cardiomyopathy have not been fully elucidated. The results of the current study suggest that diminished circulating and cardiac H2S levels play a role in the pathophysiology of HFD-induced cardiomyopathy. Our findings support the following conclusions: (1) HFD feeding decreases circulating and cardiac H2S levels; (2) restoring H2S levels via the oral administration of SG-1002 improves some of the metabolic perturbations stemming from HFD feeding; (3) Oral H2S therapy attenuates and reverses HFD-induced cardiac dysfunction; (4) Oral H2S therapy attenuates HFD-induced cardiac hypertrophy and fibrosis; (5) Oral H2S therapy induces APN-AMPK signaling in the setting of HFD feeding; and (6) Oral H2S therapy suppresses cardiac ER stress stemming from HFD feeding.
Experimental and clinical evidence is now emerging to indicate that alterations in H2S bioavailability play a prominent role in not only the pathophysiology of diabetes but also in the pathophysiology of heart failure. More specifically, levels of H2S are negatively related to diabetes and the severity of heart failure. In the case of diabetes, circulating and cardiac H2S levels are diminished in animal models of diabetes (type-1, type-2, and non-obese models). Clinically, the negative association between diabetes and H2S also exists, as evidenced by the findings that lower circulating H2S levels are detected in plasma samples taken from patients with type-2 diabetes [30; 34]. In the case of heart failure, circulating and cardiac H2S levels are reduced in experimental models of ischemic-induced and pressure-induced heart failure [37; 38]. Additionally, recent clinical evidence suggests that total plasma sulfide is negatively related to severity of congestive heart failure and low plasma sulfide predicts a higher mortality [51]. The findings of the current study now suggest that H2S levels are diminished in the setting of HFD-induced diabetic cardiomyopathy. Specifically, we provide evidence that both circulating and cardiac levels of free H2S and sulfane sulfur are significantly reduced following 24 weeks of HFD feeding. More importantly, we report that chronic oral administration of SG-1002 augments H2S levels and provides protection against HFD-induced cardiac dysfunction, hypertrophy, and fibrosis. These findings strongly suggest that a deficiency of H2S contributes to the pathophysiology and progression of HFD-induced diabetic cardiomyopathy. It is important to note that we used two strategies to investigate the therapeutic potential of H2S therapy in this model. In one group of mice, we provided H2S therapy at the beginning of the HFD feeding. This treatment strategy demonstrated that H2S therapy prevents the development of HFD-induced diabetic cardiomyopathy. In the second group, we provided H2S therapy after the mice displayed signs of cardiac dysfunction (12 weeks of HFD feeding). This treatment strategy demonstrated that H2S therapy is able to reverse the development of HFD-induced diabetic cardiomyopathy. Together, we feel that these results suggest that H2S treatment could potentially be used to treat patients with diabetic cardiomyopathy to achieve a long-term improvement in cardiac function and to decrease the morbidity and mortality resulting from heart failure.
Diabetes is a complex disease of metabolic dysregulation characterized by abnormal glucose metabolism [52]. For instance, endogenous glucose production is accelerated in patients with diabetes. The resulting hyperglycemia triggers glucotoxicity, which in turn contributes to cardiac injury via a multitude of mechanisms such as glucose auto-oxidation, the formation of advanced glycation end products, NADPH oxidases, increased hexosamine pathway flux, xanthine oxidase, and the mitochondrial respiratory chain [53]. The hyperglycemia associated with diabetes stems from absolute or relative insulin deficiency. In the majority of instances, it is associated with insulin resistance. Insulin regulates intermediary metabolism through its ability to orchestrate substrate utilization (glucose and lipids) for storage or oxidation in all cells. As such, insulin is important for protein, carbohydrate, and lipid metabolism throughout the body. Therefore, impairments in insulin signaling have devastating effects in numerous tissues, including the cardiovascular system. For instance, elevated circulating lipids and hyperinsulinemia together increase lipid delivery to cardiac cells, rapidly promoting lipid utilization. However, if the delivery of the lipids to the tissue exceeds the oxidative capacity of the cell, the lipids accumulate and lipotoxicity ensues. This can then increase oxygen demand, provoke mitochondrial uncoupling and ROS generation, decrease ATP synthesis, induce mitochondrial dysfunction and trigger cell death. Together all of these events play an important role in the pathogenesis of diabetes-associated heart disease. The findings of the current study indicate that H2S therapy improved many of the metabolic derangements associated with HFD-induced type-2 diabetes. Specifically, we found that H2S therapy decreased blood glucose levels, improved glucose tolerance, decreased serum insulin levels, and decreased cardiac lipotoxicity. While the mechanisms responsible for the improvements in the diabetic status of the H2S treated animals are not fully known, our findings suggest that APN-AMPK signaling may play a role. As noted above, APN is an adipokine that induces cardioprotective effects and regulates metabolism via the activation of AMPK [41; 42; 45]. A recent study provided novel evidence that in the setting of obesity and insulin resistance, AMPK reduced lipid accumulation through its ability to inhibit ACC, which in turn restored insulin sensitivity and lowered blood glucose levels [47]. Given that H2S therapy partially restored circulating APN levels and increased AMPK signaling, one could argue that it is possible that it improves the metabolic derangements induced by HFD feeding via APN-AMPK signaling. Future studies are definitely warranted to investigate these mechanisms because these findings suggest that chronic H2S therapy has the potential to improve the diabetic state.
As noted, recent studies provide evidence that ER stress/UPR contributes to the development of diabetic cardiomyopathy. Specifically, the cardiac expression of GRP78, GRP94, CHOP, and caspase-12 were found to be elevated as early as 2 weeks following the onset of STZ-induced diabetes [50]. Similarly, markers of ER stress have been reported to be present in the hearts of OLETF rats [13] and ob/ob mice [54]. There are several pathological factors present in the diabetic heart that are thought to induce ER stress. These include hyperglycemia and hyperlipidemia [9]. Both can induce the production of ROS, which in turn induces the accumulation of unfolded or misfolded proteins [48]. The action of the ER stress/UPR is initially an adaptive process, as an increase in ER chaperone proteins will attempt to reestablish homeostasis by restoring proper protein folding. However, the excessive and unremitting activation of the UPR becomes maladaptive and induces apoptotic pathways for cell destruction [2]. Therefore, the presence of ER stress markers, especially Caspase-12, in the diabetic heart is considered to be a pathological marker. More support for this notion stems from the evidence that manipulating components of the UPR with pharmacological agents improves heart function in the setting of diabetes. For instance, the chemical chaperone, tauroursodeoxycholic acid, ablated obesity-associated cardiac contractile dysfunction by reducing ER stress [54]. The findings of the present study suggest that H2S therapy targets ER stress under HFD conditions. Specifically, we found that H2S therapy decreased the HFD-induced activation of the ATF6 and IRE1 arms of the UPR. The findings related to a decrease in the IRE1 arm are of particular importance to the development of diabetic cardiomyopathy for several reasons. First, the chronic activation of IRE1 leads to the cleavage and activation of caspase-12. It then translocates from the ER to the cytosol, where it directly cleaves procaspase-9, which, in turn, activates the downstream effector caspase, caspase-3 [55]. This occurs without the need for mitochondrial amplification indicating that caspase-12-mediated apoptosis is a specific apoptosis pathway of ER, independent of the mitochondria or death receptor activation [56]. Second, chronic activation of IRE1 leads to the activation of Jnk, which can induce apoptosis by activating Bax and Bad[57] and inhibiting Bcl2 and Bcl-xL[58; 59], as well as induce pathological cardiac hypertrophy [49]. Together, these findings suggest that H2S therapy attenuates the development of HFD-induced hypertrophy and cardiac dysfunction in part by suppressing ER stress.
Although the current study demonstrates that H2S therapy attenuates the development of HFD-induced diabetic cardiomyopathy, there are a few caveats that need to be noted. First, our findings related to the ability of H2S to improve the metabolic derangements associated with HFD feeding are in contrast to several studies, including a recent study from our lab [35]. Specifically, the previous studies reported that the protective effects of H2S therapy in models of diabetes were observed in the absence of changes in the metabolic derangements of the animal. The difference more than likely lies in the duration of the treatment and/or the model system. For instance our previous study evaluated the effects of 7 days of H2S therapy, whereas the current study evaluated the effects of 12 weeks or 24 weeks of H2S therapy. Second, the H2S-producing enzymes were unaltered with HFD, while the H2S levels were reduced. This finding is similar to our previous study in which db/db diabetic mice exhibited reduced H2S levels with no correlating change in the protein expression of the enzymes [35]. However, in our previous study we found that the enzymatic activity of the H2S-producing enzymes was decreased in the setting of diabetes. The activity of 3-MST is inhibited by oxidative stress [60]. Therefore, it is possible that the oxidative environment induced by HFD feeding contributes to the reduced levels of sulfide by reducing the activity of the H2S-producing enzymes. Alternatively, Suzuki et al [32] suggests that the elevated levels of glucose seen in diabetes induce the mitochondrial formation of ROS, which in turn causes an increased consumption of H2S. Either or both of the mechanisms could lead to the reduced levels of H2S seen in the setting of HFD feeding. Future studies are definitely warranted to determine the exact mechanism(s). Third, the precise mechanism by which H2S therapy reduces ER stress is currently not known. Given the antioxidant properties of H2S, it is possible that H2S reduces oxidative stress, which in turn reduces the pathological stimulus that induces ER stress. On the other hand, the ability of H2S to improve the diabetic state of the animals may also contribute to the reduction in ER stress. Furthermore, the unaltered GRP78 results did not fit the standard UPR mechanism, as numerous studies have shown GRP78 upregulation after ER stress [61; 62; 63]. Few studies have examined such a chronic high fat diet phenotype, and we believe that GRP78 may be regulated differently than the UPR arms, as it is a molecular chaperone that is omnipresent. Our findings warrant a closer temporal look into the activity of this protein. Regardless of the mechanism, our data suggests that the improvements in cardiac structure and function in response to H2S therapy are associated with improvements in ER stress. Therefore future studies are warranted to investigate if the mechanism is direct or indirect.
In summary, the findings of the current study suggest that a deficiency of H2S contributes to the metabolic derangements induced by HFD feeding. Additionally, our findings indicate that oral H2S therapy prevents and reverses the development of HFD-induced diabetic cardiomyopathy. Finally, we found that the protective effects of H2S therapy were associated with the activation of APNAMPK signaling and suppression of HFD-induced ER stress. Therefore, these findings further support the emergent concept that H2S therapy may be of clinical importance in the treatment of cardiovascular complication stemming from diabetes.
Supplementary Material
Highlights.
High fat diet (HFD) feeding decreases hydrogen sulfide (H2S) levels
Oral H2S therapy attenuates and reverses HFD-induced cardiac dysfunction
Oral H2S therapy attenuates HFD-induced cardiac hypertrophy and fibrosis
Oral H2S therapy induces APN-AMPK signaling in the setting of HFD feeding
Oral H2S therapy suppresses cardiac ER stress stemming from HFD feeding
Acknowledgments
Supported by a grant the National Institutes of Health National Heart Lung and Blood Institute (5R01HL098481-05) to J.W.C. This work was also supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown.
Footnotes
Disclosures: SG-1002 was provided by Sulfagenix, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Battiprolu PK, Lopez-Crisosto C, Wang ZV, Nemchenko A, Lavandero S, Hill JA. Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci. 2013;92:609–15. doi: 10.1016/j.lfs.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 4.Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic Cardiomyopathy: Mechanisms and Therapeutic Targets. Drug Discov Today Dis Mech. 2010;7:e135–e143. doi: 10.1016/j.ddmec.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- 6.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 7.Avendano GF, Agarwal RK, Bashey RI, Lyons MM, Soni BJ, Jyothirmayi GN, Regan TJ. Effects of glucose intolerance on myocardial function and collagen-linked glycation. Diabetes. 1999;48:1443–7. doi: 10.2337/diabetes.48.7.1443. [DOI] [PubMed] [Google Scholar]
- 8.Capasso JM, Robinson TF, Anversa P. Alterations in collagen cross-linking impair myocardial contractility in the mouse heart. Circ Res. 1989;65:1657–64. doi: 10.1161/01.res.65.6.1657. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Zhou Q, Xu W, Cai L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Experimental diabetes research. 2012;2012:827971. doi: 10.1155/2012/827971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–93. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–68. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–84. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 13.Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, Terashima Y, Takada A, Ishikawa S, Shimamoto K. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. 2009;58:2863–72. doi: 10.2337/db09-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, Zhang Y, Ge Z. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. 2007;41:58–67. doi: 10.3164/jcbn.2007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. 2010;59:2033–42. doi: 10.2337/db09-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sukumaran V, Watanabe K, Veeraveedu PT, Gurusamy N, Ma M, Thandavarayan RA, Lakshmanan AP, Yamaguchi K, Suzuki K, Kodama M. Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int J Biol Sci. 2011;7:154–67. doi: 10.7150/ijbs.7.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu T, Dong Z, Geng J, Sun Y, Liu G, Kang W, Zhang Y, Ge Z. Valsartan protects against ER stress-induced myocardial apoptosis via CHOP/Puma signaling pathway in streptozotocin-induced diabetic rats. Eur J Pharm Sci. 2011;42:496–502. doi: 10.1016/j.ejps.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Doeller JE, Isbell TS, Benavides G, Koenitzer J, Patel H, Patel RP, Lancaster JR, Jr, Darley-Usmar VM, Kraus DW. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem. 2005;341:40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 19.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–35. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 20.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–5. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–74. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122:11–9. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra PK, Tyagi N, Sen U, Givvimani S, Tyagi SC. H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H451–6. doi: 10.1152/ajpheart.00682.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicholson CK, Calvert JW. Hydrogen sulfide and ischemia-reperfusion injury. Pharmacol Res. 2010;62:289–97. doi: 10.1016/j.phrs.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barr LA, Calvert JW. Discoveries of hydrogen sulfide as a novel cardiovascular therapeutic. Circ J. 2014;78:2111–8. doi: 10.1253/circj.cj-14-0728. [DOI] [PubMed] [Google Scholar]
- 26.Su YW, Liang C, Jin HF, Tang XY, Han W, Chai LJ, Zhang CY, Geng B, Tang CS, Du JB. Hydrogen sulfide regulates cardiac function and structure in adriamycin-induced cardiomyopathy. Circ J. 2009;73:741–9. doi: 10.1253/circj.cj-08-0636. [DOI] [PubMed] [Google Scholar]
- 27.Marra G, Cotroneo P, Pitocco D, Manto A, Di Leo MA, Ruotolo V, Caputo S, Giardina B, Ghirlanda G, Santini SA. Early increase of oxidative stress and reduced antioxidant defenses in patients with uncomplicated type 1 diabetes: a case for gender difference. Diabetes Care. 2002;25:370–5. doi: 10.2337/diacare.25.2.370. [DOI] [PubMed] [Google Scholar]
- 28.Rosen P, Nawroth PP, King G, Moller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- 29.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 30.Jain SK, Bull R, Rains JL, Bass PF, Levine SN, Reddy S, McVie R, Bocchini JA. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010;12:1333–7. doi: 10.1089/ars.2009.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brancaleone V, Roviezzo F, Vellecco V, De Gruttola L, Bucci M, Cirino G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br J Pharmacol. 2008;155:673–80. doi: 10.1038/bjp.2008.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero D, Szoleczky P, Chang T, Zhou Z, Wu L, Wang R, Papapetropoulos A, Szabo C. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–34. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yusuf M, Kwong Huat BT, Hsu A, Whiteman M, Bhatia M, Moore PK. Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis. Biochem Biophys Res Commun. 2005;333:1146–52. doi: 10.1016/j.bbrc.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 34.Whiteman M, Gooding KM, Whatmore JL, Ball CI, Mawson D, Skinner K, Tooke JE, Shore AC. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia. 2010;53:1722–6. doi: 10.1007/s00125-010-1761-5. [DOI] [PubMed] [Google Scholar]
- 35.Peake BF, Nicholson CK, Lambert JP, Hood RL, Amin H, Amin S, Calvert JW. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner, American journal of physiology. Heart and circulatory physiology. 2013;304:H1215–24. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szabo C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2012;17:68–80. doi: 10.1089/ars.2011.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Sr, Gojon G, Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–27. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholson CK, Lambert JP, Molkentin JD, Sadoshima J, Calvert JW. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler Thromb Vasc Biol. 2013;33:744–51. doi: 10.1161/ATVBAHA.112.300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–18. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monji A, Mitsui T, Bando YK, Aoyama M, Shigeta T, Murohara T. Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2013;305:H295–304. doi: 10.1152/ajpheart.00990.2012. [DOI] [PubMed] [Google Scholar]
- 41.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng Q, Wang Y, Yuan Y, Wang X, Tao L, Li R, Koch W, Ma XL. Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid Redox Signal. 2011;15:1779–88. doi: 10.1089/ars.2010.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouchi N, Shibata R, Walsh K. Cardioprotection by adiponectin. Trends Cardiovasc Med. 2006;16:141–6. doi: 10.1016/j.tcm.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullen KL, Pritchard J, Ritchie I, Snook LA, Chabowski A, Bonen A, Wright D, Dyck DJ. Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R243–51. doi: 10.1152/ajpregu.90774.2008. [DOI] [PubMed] [Google Scholar]
- 45.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 46.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circulation research. 2012;111:800–14. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O'Neill HM, Ford RJ, Palanivel R, O'Brien M, Hardie DG, Macaulay SL, Schertzer JD, Dyck JR, van Denderen BJ, Kemp BE, Steinberg GR. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–54. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–82. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S, Takeda T, Watanabe T, Asahi M, Taniike M, Matsumura Y, Tsujimoto I, Hongo K, Kusakari Y, Kurihara S, Nishida K, Ichijo H, Hori M, Otsu K. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci U S A. 2003;100:15883–8. doi: 10.1073/pnas.2136717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, Cai L. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. 2009;13:1499–512. doi: 10.1111/j.1582-4934.2009.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovacic D, Glavnik N, Marinsek M, Zagozen P, Rovan K, Goslar T, Mars T, Podbregar M. Total plasma sulfide in congestive heart failure. J Card Fail. 2012;18:541–8. doi: 10.1016/j.cardfail.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Nathan DM. Long-term complications of diabetes mellitus. N Engl J Med. 1993;328:1676–85. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- 53.Jay D, Hitomi H, Griendling KK. Oxidative stress and diabetic cardiovascular complications. Free Radic Biol Med. 2006;40:183–92. doi: 10.1016/j.freeradbiomed.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 54.Ceylan-Isik AF, Sreejayan N, Ren J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol. 2011;50:107–16. doi: 10.1016/j.yjmcc.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–94. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 56.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 57.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. Embo J. 2004;23:1889–99. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–78. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–7. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- 60.Nagahara N, Katayama A. Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteinesulfenate in the maintenance of redox homeostasis. J Biol Chem. 2005;280:34569–76. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- 61.Bousette N, Abbasi C, Chis R, Gramolini AO. Calnexin silencing in mouse neonatal cardiomyocytes induces Ca2+ cycling defects, ER stress, and apoptosis. J Cell Physiol. 2014;229:374–83. doi: 10.1002/jcp.24459. [DOI] [PubMed] [Google Scholar]
- 62.Vekich JA, Belmont PJ, Thuerauf DJ, Glembotski CC. Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death. J Mol Cell Cardiol. 2012;53:259–67. doi: 10.1016/j.yjmcc.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.