

Abstract
Hypoxia-induced arginase elevation plays an essential role in several vascular diseases but influence of arginase on hypoxia-mediated angiogenesis is completely unknown. In this study, in-vitro network formation in bovine aortic endothelial cells (BAEC) was examined after exposure to hypoxia for 24 hours with or without arginase inhibition. Arginase activity, protein levels of the two arginase isoforms, eNOS, and VEGF as well as production of NO and ROS were examined to determine the involvement of arginase in hypoxia-mediated angiogenesis. Hypoxia elevated arginase activity and arginase 2 expression but reduced active p-eNOSSer1177 and NO levels in BAEC. Additionally, both VEGF protein levels and endothelial elongation and network formation were reduced with continued hypoxia, whereas ROS levels increased and NO levels decreased. Arginase inhibition limited ROS, restored NO formation and VEGF expression and prevented the reduction of angiogenesis. These results suggest a fundamental role of arginase activity in regulating angiogenic function.
Keywords: Arginase, reactive oxygen species (ROS), endothelial nitric oxide synthase (eNOS), angiogenesis, endothelial cell
1. Introduction
Arginase, an ureohydrolase enzyme, has been shown to be involved in the several vascular diseases such as hypertension, atherosclerosis, and diabetic coronary vascular dysfunction (Yang & Ming, 2006; Sasaki et al., 2007; Romero et al., 2008). The detrimental effects of elevated arginase activity in these conditions are mostly attributed to its capability to inhibit nitric oxide (NO) synthesis by competing with endothelial NO synthase (eNOS) for their common substrate, L-arginine (Morris, 2002, 2009). When the L-arginine supply needed for eNOS activity is insufficient, NOS can become uncoupled, producing less NO and forming superoxide (O2.-). Further, remaining NO can react with O2.- to form the toxic oxidant peroxynitrite (ONOO-) (Forstermann & Munzel, 2006). NO plays a pivotal role in vasodilation and angiogenesis (Dulak et al., 2000; Jones et al., 2004; Morris et al., 2008; Berchner-Pfannschmidt et al., 2010). Reduced NO bioavailability within the vessel wall leads to vasoconstriction, vascular remodeling and an aberrant neovasculature (Madigan & Zuckerbraun, 2013).
Many factors that trigger endothelial dysfunction also up-regulate arginase expression and activity. Hypoxia is one of these factors. Hypoxia has been reported to increase arginase-2 expression and activity in human umbilical vein and pulmonary microvascular endothelial cells (Krotova et al., 2010; Prieto et al., 2011). Also, oxidative stress due to generation of reactive oxygen species (ROS), including superoxide and peroxynitrite, contributes to elevated arginase expression/activity and eNOS uncoupling (Thengchaisri et al., 2006; Sankaralingam et al., 2010; Chandra et al., 2012). Moreover, hypoxia is a major mediator of angiogenesis. Hypoxia-mediated angiogenesis due to elevated levels of hypoxia-induced factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) has been well demonstrated (Fong, 2009; Takenaga, 2011; Ahluwalia & Tarnawski, 2012). NO also is importantly involved in regulation of hypoxia-mediated angiogenesis through a NO/HIF-1α/VEGF axis (Kimura et al., 2000; Kimura et al., 2001; Kimura & Esumi, 2003). Hence, arginase may alter this process via its reduction of NO. However, no studies on a role of arginase in hypoxia-mediated angiogenesis have been reported. The present study examined the hypothesis that enhanced arginase activity during hypoxia limits the angiogenic process. In-vitro network formation in endothelial cells was examined after exposure to hypoxia (2% O2) for 12 and 24 hours with or without arginase inhibition. Protein levels of the two arginase isoforms, eNOS and VEGF in addition of NO and ROS production were examined to determine the role of arginase in hypoxia-mediated angiogenesis.
2. Materials and Methods
2.1 Cell culture and hypoxia
Bovine aortic endothelial cells (BAECs, Cell Application Inc, CA, USA) were used from passages 4 to 7. Cells were cultured in normoxia (21% O2) incubator to 70% confluence and were maintained for 2 hrs in low-arginine M-199 medium (Invitrogen, Carlsbad, CA, USA) containing 0.2% FBS and 50 µM L-arginine, before treatment. The cells were pretreated with the arginase inhibitor 2(S)-amino-6-boronohexanoic acid (ABH, 100 µm, a gift of Dr. Dan Berkowitz) or vehicle (1xPBS) for 1 hr. Thereafter, cells were maintained with a gas mixture (5% CO2-balanced N2) to obtain 2% O2 in a hypoxia chamber for 12 or 24 hrs. Normoxia control group cells were incubated in 21% O2 conditions. This concentration of ABH, 100 µM inhibits arginase activity by over 90% in endothelial cells for over 24 hrs (Steppan et al., 2013).
2.2 Arginase activity
BAEC were washed in PBS and incubated in lysis buffer (50 mM Tris-HCl) containing protease inhibitors. Cell lysates were centrifuged at 12,000 g for 10 min. The enzyme was activated by heating the lysate at 56°C for 10 min in 25 mM Tris buffer (pH 7.4) containing 5 mM MnCl2. L-arginine hydrolysis was then conducted by incubating 50 μl of the activated lysate with 50 μl of 0.5 M L-arginine (pH 9.7) at 37°C for 60 min. The reaction was stopped in acid medium (H2SO4:H3PO4:H2O=1:3:7 v/v). The concentration of urea was determined after adding 25 μl of 9% α-isonitrosopropiophenone. Protein concentration in the lysates was determined by a BCA assay (Thermo Scientific, IL, USA). Results were normalized to arginase activity in normoxia control group and expressed as arbitrary unit.
2.3 Western-blot
BAEC were homogenized in a RIPA Lysis Buffer (EMD Millipore, MA, USA) supplied with phosphatase inhibitor cocktail (Sigma-Aldrich, MO, USA) and protease inhibitor cocktail (Sigma-Aldrich, MO, USA). 20μg protein samples were subjected to 10% SDS-PAGE. Proteins were transferred onto a nitrocellulose membrane and the membrane was blocked with 5% BSA and incubated with primary polyclonal chicken anti-arginase I (1:10,000, gift of Dr. S.M. Morris, Jr.), rabbit anti-arginase II (1:250, SantaCruz Biotech, Texas, USA), rabbit anti-eNOS(1:4000, Santacruz Biotech, Texas, USA), monoclonal mouse anti-eNOS phosphorylated at serine-1177 or threonine-495 (1:1000, BD Bioscience, CA, USA), human anti-VEGF antibody (LifeSpan BioScience, WA, USA), mouse anti-β-actin (1:5000, Sigma-Aldrich, MO, USA) overnight at 4°C. After incubation with secondary antibodies, proteins were detected using an enhanced chemiluminescence and quantified by densitometry. Results were normalized to β-actin.
2.4 Measurement of NO levels and production
Intracellular NO levels at 24 hr were monitored using the NO-specific fluorescent dye 4,5 -diaminofluorescein diacetate (DAF-2 DA, Calbiochem, USA). Thereafter, cells were stained with oxygenated DAF-2 DA solution (10 mM) for 30min in the darkness at 37C, in a shaking water bath. This experimental procedure in saturated oxygen ensures that NO, rather than O2, is the limiting factor in the reaction and the fluorescence is directly proportional to NO. Negative controls for DAF-2 DA were incubated in NOS inhibitor L-NAME (0.1mM) throughout the experimental period. Fluorescence was monitored using fluorescence microscope (excitation, 488 nm; emission, 610 nm) at 20x magnification. Nine pictures were captured in each well (n=3) and the fluorescent intensity was quantified using Image J software. Results were presented as mean fluorescent intensity per field and expressed as percent of control.
Cumulative production of NO by BAEC was also assessed. After 24 hr exposure to normoxia, hypoxia or hypoxia+ABH, the medium was collected. NO levels in 30μl of media was measured using NO-specific chemiluminescence in a Sievers Nitric Oxide Analyzer as previously described (Shatanawi et al., 2011).
2.5 In-vitro EC network formation assay
Cells were collected for in-vitro tube formation assay. ABH (100 μM) or PBS (as control) was added to the wells of 96-well plate before 30 μl Matrigel (reduced growth factor basement membrane matrix, BD Bioscience, USA) mixed with 60 μl medium containing 1.5 ×104 cells were seeded. After 12 hr and 24 hr hypoxia exposure, the network-like structures were visualized and captured using microscope (Axiovert 25, Carl Zesis) at 10x magnification. Five pictures were taken for each well and five wells for each group. Network length in each field was analyzed and quantified using NIH ImageJ. Results were presented as network length in each field and expressed as percent of control.
2.6 Reactive Oxygen Species (ROS)
Dichlorofluorescein (DCF) is the oxidation product of the reagent 2′,7′-dichlorofluorescein diacetate (H2DCFDA; Molecular Probes, CellRox Oxidative Stress Deep Red Reagents, Life technologies, NY, USA). It is a marker of cellular oxidation by hydrogen peroxide, peroxynitrite, and hydroxy radicals. Cells were incubated with low-arginine M199 medium containing 5 μM H2DCFDA at 37°C for 30mins. Fluorescence was monitored using fluorescence microscope (excitation, 640 nm; emission, 665 nm) at 20x magnification. Nine pictures were captured in each well (n=3) and the fluorescent intensity was quantified using NIH ImageJ software. Results were presented as mean fluorescent intensity in each field and expressed as percent of control.
2.7 Statistical Analysis
All the values are presented as mean ± S.E.M. Student's t-test or one-way analysis of variance (ANOVA) with Bonferroni post-test was used to evaluate differences between the groups as appropriate. P<0.05 was considered significant. All statistical analysis was calculated by using GraphPad Prism (V5.01).
3. Results
3.1 In-vitro tube-like formation and VEGF165 expression
We first determined the effects of hypoxia exposure on angiogenic function as shown by the alignment of ECs in tube-like networks and VEGF165 protein levels as shown by Western blot. At 12 hr of exposure to hypoxia, ECs showed significantly increased VEGF165 levels. The increase in VEGF165 was accompanied by EC elongation and alignment, but network formation was not evident. Both VEGF165 levels and EC elongation/alignment in hypoxia were significantly increased by ABH treatment (Figure 1). In normoxic conditions, EC exhibited only small spiky protrusions. After 24 hrs of hypoxia exposure, well-formed EC networks were observed in the normoxia control cultures. However, the network length per field in hypoxia was less than observed in normoxia control, being 63% of that level. ABH treatment partially prevented this hypoxia-induced reduction in tube-like formation (88% of control level) (Figure 2a-d). VEGF165 protein levels in the hypoxia group were reduced to 53% of control and ABH prevented this decline (Figure 2e-f). We therefore used the 24 hrs hypoxia treatment for further analyses of the role of arginase activity in this process.
Figure 1.
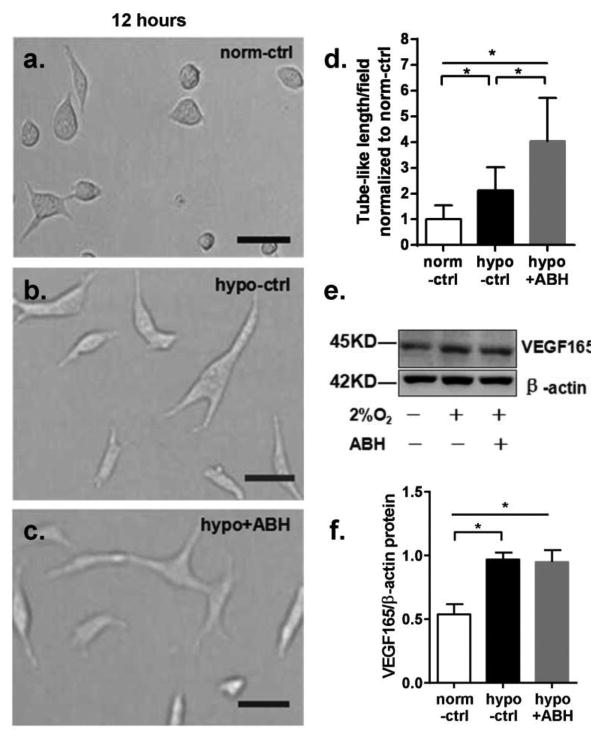
Effect of 12 hr exposure on in-vitro angiogenesis of BAEC cultured in Matrigel and VEGF protein levels in BAEC. Magnification is 40X. Scale bar length is 20 μm. (a) Cells in normoxia control group. (b) Cells in hypoxia group. (c) Cells in hypoxia+ABH group. (d) Mean length of tube-like structures among groups. Values are normalized to normoxia control. (e)Western blot for VEGF165 normalized to β-actin in BAEC exposed to either normoxia(-) or hypoxia (2% O2, +) and with (+) or without ABH (-). (f) VEGF 165/β-actin levels. N=4-6 wells. *P < 0.01.
Figure 2.
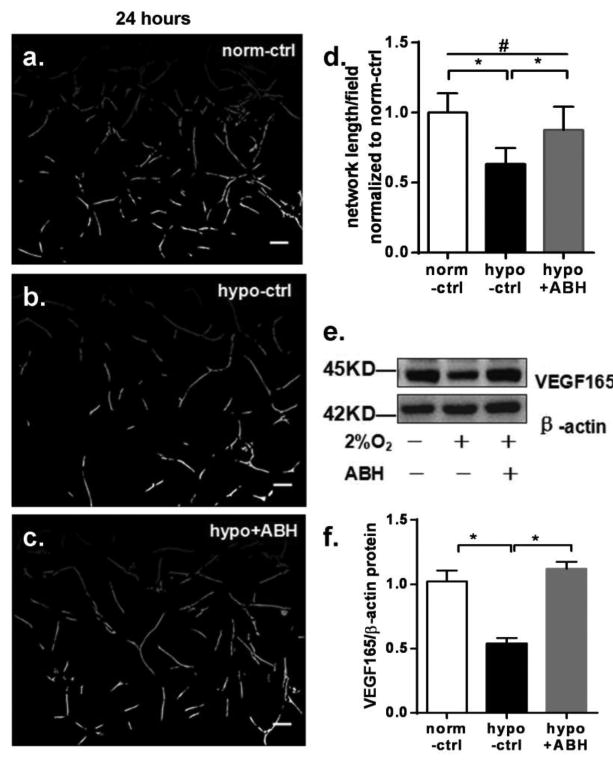
Effect of 24 hr exposure on in-vitro angiogenesis of BAEC cultured in Matrigel and VEGF protein levels in BAEC. Images a-c are normoxia control, hypoxia control and hypoxia+ABH group, respectively (10X). Scale bar length is 50 μm. (d) Mean length of tube-like structures among groups. Values are normalized to normoxia control. (e) Western blot for VEGF165 and β-actin in BAEC exposed to either normoxia (-) or hypoxia (2% O2, +) and with (+) or without ABH (-). (f). VEGF165 /β-actin levels. N=4-6 wells. *P < 0.01,#P < 0.05.
3.2 Arginase expression and activity
Hypoxia increased protein levels of arginase 2 in BAEC but did not change in arginase 1 expression compared with normoxia control. In hypoxia group, arginase 2 protein expression was increased by 87% over normoxia control. Pretreatment with ABH significantly inhibited the hypoxia-induced increase in arginase expression. Hypoxia-induced elevation of arginase activity also was partially inhibited by ABH, but activity still was higher than that of normoxia control (Figure 3).
Figure 3.
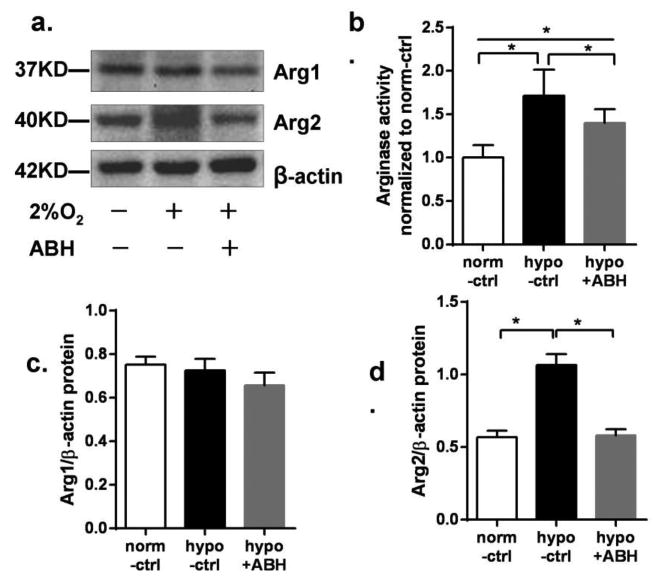
Arginase expression and arginase activity in BAEC exposed to hypoxia for 24 hr. (a) Western blot for arginase 1 (Arg1) and arginase 2 (Arg2) normalized to β-actin in BAEC exposed to either normoxia (-) or hypoxia (2% O2, +) and with (+) or without ABH (-). (b) Arginase activity in BAEC, normalized to normoxia control. (c) Argl/β-actin levels and (d) Arg2/β-actin levels. N=4-6. *P < 0.01.
3.3 eNOS expression
Total eNOS protein was unaltered after 24 hr hypoxia exposure, and was not affected by ABH treatment. However, hypoxia reduced protein levels of eNOS phosphorylated at serine-1177 (p-eNOSSer1177) to 73% of the normoxia control. In contrast, phosphorylation at threonine-495(p-eNOSThr495) was increased by 91% over normoxia control values. ABH pretreatment prevented the changes in p-eNOSSer1177 and p-eNOSThr495 caused by hypoxia (Figure 4). These data indicate that eNOS activity, determined by p-eNOS levels, is affected by arginase.
Figure 4.
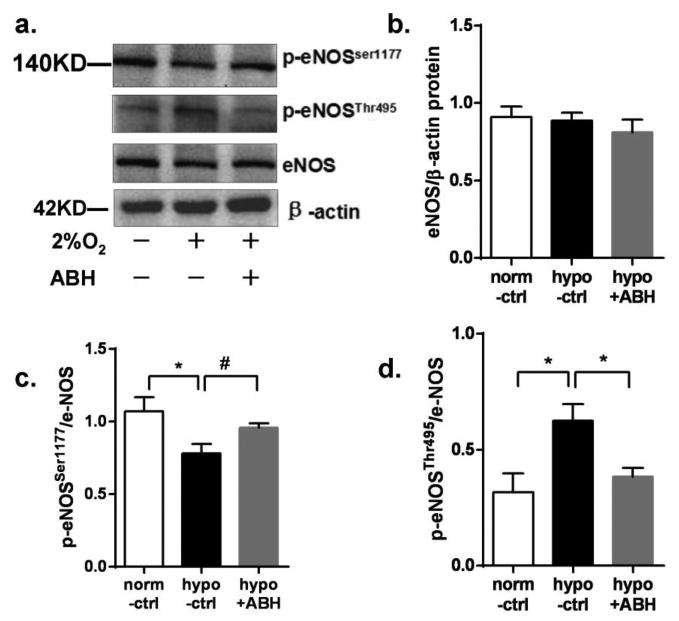
eNOS expression in BAEC exposed to hypoxia or 24 hr. (a) Western blot or p-eNOSSer1177, p-eNOSThr495 and β-actin in BAEC exposed to either normoxia (-) or hypoxia (2% O2, +) and with (+) or without ABH (-). Panels b-d are e-NOS, p-eNOSSer1177, p-eNOSThr495/β-actin levels, respectively N=4-6. *P < 0.01, #P < 0.05.
3.4 NO levels and production
Mean fluorescent intensity of DAF-2 staining for intracellular NO showed that hypoxia markedly decreased intracellular NO levels to 13% of the normoxia control. Extracellular NO accumulation in culture medium, detected by the NO analyzer during hypoxia exposure, declined to 35% of the normoxia control. ABH pretreatment partially prevented the reduction in NO levels and production in response to hypoxia, with values only falling to 44% and 53% of respective controls (Figure 5).
Figure 5.
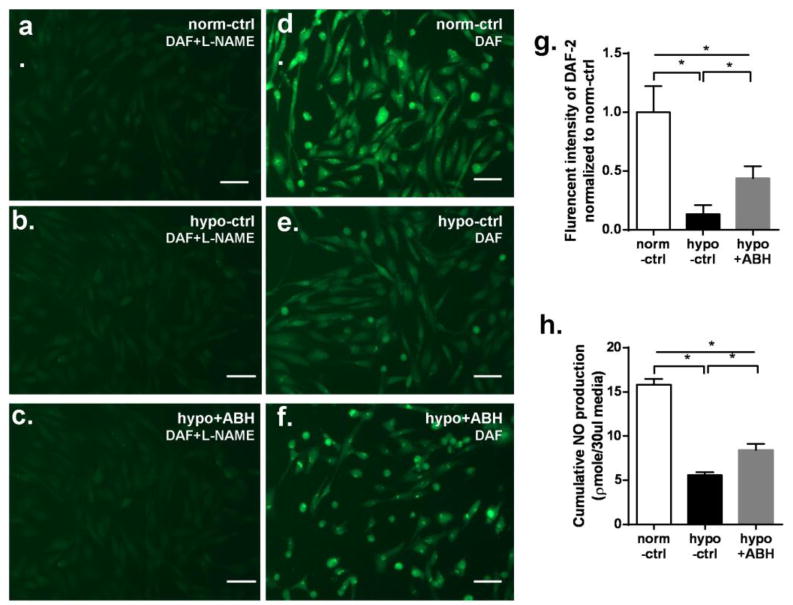
NO production in BAEC exposed to hypoxia for 24 hr. (a-f) NO-specific fluorescence of DAF-2 in BAEC exposed to hypoxia for 24 hr (20x) with L-NAME (a-c) and without L-NAME (d-f). Scale bar length is 50μm. (g) Mean DAF-2 fluorescent intensity levels in EC without L-NAME at 24 hr among groups. Values are normalized to normoxia control. (h) Cumulative NO production measured by chemiluminescence over 24 hr among groups. N=5 wells. *P < 0.01.
3.5 ROS generation
Twenty-four hours of hypoxia exposure significantly increased reactive oxygen species generation in EC cytoplasm to 96% over normoxia control. But ABH pretreatment reduced the hypoxia-induced ROS generation to only 66% above control (Figure 6).
Figure 6.
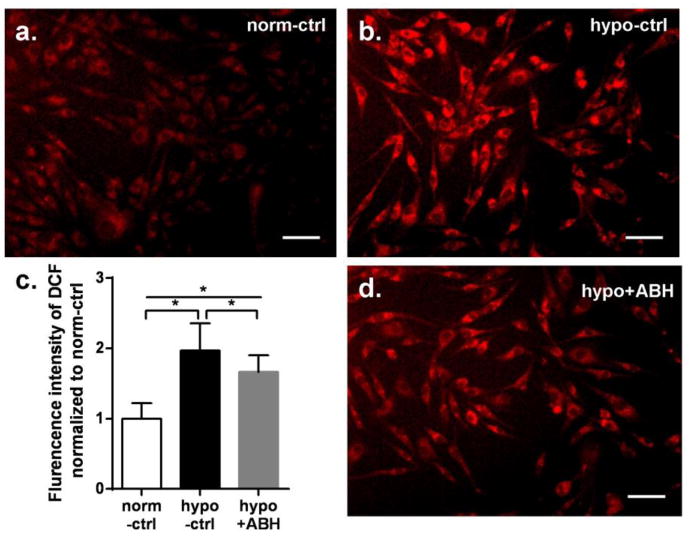
ROS-specific fluorescence of DCF in BAEC exposed to hypoxia for 24 hr. Images a, b, d are for normoxia control, hypoxia control and hypoxia+ABH group, respectively (20x). Scale bar length is 50μm. (c) Mean DCF fluorescent intensity among groups. Values are normalized to normoxia control. N=4-6 wells. *P < 0.01.
4. Discussion
Hypoxia is a major trigger of elevated arginase expression and/or activity in several pathological conditions such as pulmonary arterial hypertension (PAH), cardiovascular dysfunction, and atherosclerosis (Xu et al., 2004; Yang & Ming, 2006; Romero et al., 2008). Furthermore, hypoxia-mediated angiogenesis is a feature of these diseases. For example, hypoxia-mediated neovascularization of pulmonary artery vasa vasorum has been found in PAH and intraplaque hypoxia-induced angiogenesis also has been reported to be present in atherosclerosis (Kuwahara et al., 2002; Gao et al., 2012). However, the role of arginase in hypoxia-mediated angiogenesis has not been determined.
In present study, bovine endothelial cells (BAEC) were grown in normoxic conditions to 70% confluence and then subjected to 2% oxygen, a concentration commonly used for hypoxia studies. After 12 hr of hypoxia exposure, levels of VEGF were significantly increased as compared to the normoxia control cells and the ECs began to elongate and align in primitive networks. Both VEGF and network alignment were significantly increased by ABH treatment. After 24 hr hypoxia exposure, well-formed EC networks were observed. However, compared with normoxia control, there was a significantly lower network length along with a corresponding decrease in VEGF protein levels. Arginase inhibition with ABH partially prevented the suppressive effects of hypoxia.
Hypoxia is a well-known inducer of angiogenesis (Fong, 2009; Takenaga, 2011; Ahluwalia & Tarnawski, 2012). However, sustained hypoxia conditions, such as ischemia, are detrimental for the vasculature through high levels of ROS formation (Webster, 2012). ROS have been noted to be significantly increased in ischemic hindlimbs of wild-type mice, but not in mice deficient in Nox2- NADPH oxidase (NOX2). The NOX2 mice exhibited resistance against ROS production, preserved eNOS expression and improved VEGF-dependent angiogenesis (Haddad et al., 2009). In our study, a significantly higher level of ROS and decreased VEGF levels occurred in hypoxia group. This could explain the impaired network formation after 24 hr of hypoxia exposure.
Importantly, inhibition of arginase with ABH partially prevented the hypoxia-induced impairment of network formation and the hypoxia-related decline in VEGF, which possibly was achieved through its actions to maintain NO production and subsequent VEGF expression (Kimura & Esumi, 2003; Fraisl, 2013). Under the low oxygen tension, NO donors at low concentrations have a synergetic effect on the VEGF gene activation by promoting HIF-1α (Kimura & Esumi, 2003; Abe et al., 2013). Site-directed mutational analysis of the VEGF gene promoter has revealed that a HIF-1 binding site (HBS) and its downstream HIF-1α ancillary sequence (HAS) within the hypoxia response elements (HRE) are required as cis-elements for the transcriptional activation of VEGF by NO (Kimura et al., 2001). Therefore, NO-induced VEGF expression can be partially mediated by activation and subsequent binding of HIF-1α (Kimura et al., 2000). Moreover, increased endogenous NO due to overexpressed eNOS or elevated eNOS activity has been shown to enhance angiogenesis in a human colon tumor cell line and vascular smooth muscle cells (VSMC) (Jenkins et al., 1995; Dulak et al., 2000). Additionally, the protective role of ABH pretreatment in network formation can be partially attributed to its action to reduce ROS levels in hypoxia. Inhibition of arginase has been shown to reduce the ROS production from uncoupled eNOS in previous studies (Reid et al., 2007; Kim et al., 2009; Jung et al., 2010).
More recent studies also report the protective role of arginase inhibition in oxidative stress. Systemic treatment of rat with an arginase inhibitor N-(ω)-hydroxy-nor-L-arginine (nor-NOHA) was shown to significantly prevent both hypoxia-increased tissue level of ROS and lipid peroxides in hypoxic hearts. Nor-NOHA treatment also significantly ameliorated the peroxynitrite formation in hypoxic hearts (Singh et al., 2014). Further, nitroglycerin (NTG)-induced increases of ROS levels in aorta isolated from wild type mice were not observed in aorta pretreated with the arginase inhibitor (s)-(2-boronethyl)-L-cysteine HCl (BEC), nor in aorta isolated from Arg2–/– mice. BEC pretreatment also significantly reduced the ROS levels in NTG-treated HUVEC (Khong et al., 2012).
In parallel with elevating ROS levels, hypoxia significantly elevated arginase activity and arginase 2 protein level, but had no influence on arginase 1 levels. Hypoxia-induced arginase expression appears to involve arginase activity, possibly due to increased levels of ROS which drive arginase expression (Thengchaisri et al., 2006; Sankaralingam et al., 2010; Chandra et al., 2012). Hypoxia-induced arginase 2 expression and activity are also reported in human umbilical vein endothelial cells (HUVEC) (Prieto et al., 2011) and human pulmonary microvascular endothelium (HMVEC) (Krotova et al., 2010). Hypoxia-inducible factor 2α (HIF-2α) likely mediates this induction since the silencing of HIF-2α but not HIF-1α prevented activation of arginase 2 by hypoxia (Krotova et al., 2010). In contrast, hypoxia-induced enhancement of arginase 1 expression and arginase activity was found in rat cardiac and aortic tissue, which was linked to the hypoxia-mediated c-Jun recruitment to AP-1 binding site on ARG1 promoter (Singh et al., 2014). The discrepancy in arginase isoforms induced by hypoxia can be explained by reports that arginase 1 is the predominant isoforms in the rat endothelium while arginase 2 is the major isoforms in human aortic and human umbilical vein endothelial cells (Ming et al., 2004; Ryoo et al., 2006; Gao et al., 2007).
Previous studies have shown that ROS (Zhang et al., 2009), including H2O2 (Thengchaisri et al., 2006) and ONOO- (Sankaralingam et al., 2010) can up-regulate arginase expression. Our recent studies further confirm that this up-regulation of arginase by ROS occurs through the Rho/ROCK pathway and that pretreatment with inhibitors of Rho kinase or protein kinase C (PKC) can prevent this increase (Chandra et al., 2012). Given that arginase inhibition can reduce ROS levels in hypoxic tissue (Takemoto et al., 2002; Gonon et al., 2012), we project that reduced arginase 2 expression in the hypoxia+ABH group is due to decreased ROS production via arginase inhibition.
Hypoxia-induced arginase elevation appears to contribute to the marked reduction in NO production via competition for L-arginine and eNOS uncoupling. Reduced active eNOS (p-eNOSSer1177) levels and increased levels of inactive eNOS (p-eNOSThr495) indicate reduced eNOS activity during hypoxia. Although ABH prevented these changes in p-eNOS levels caused by hypoxia, NO levels and production in hypoxia+ABH groups were still significantly below that of normoxic control. This implies that eNOS function is substantially impaired by hypoxia. However, ABH partially preserved NO synthesis seemingly by attenuating hypoxia-induced arginase expression and activity. Results from in-vitro studies of hypoxia on activation of eNOS has been controversial (Hoffmann et al., 2001; Takemoto et al., 2002; Prieto et al., 2011; Krause et al., 2012). The reasons for the variability of eNOS expression in these studies may reflect differences in cell species and location, oxygen concentration and exposure duration.
Our data shows that pretreatment with the arginase inhibitor ABH is protective in angiogenesis after sustained hypoxia exposure. To our knowledge, ours is the first study to show a detrimental role of arginase in limiting angiogenesis in vitro. Further work is needed to understand the influence of arginase on angiogenic function in vivo. However, our group has shown that deletion of arginase 2 and inhibition of arginase promotes vascular repair in a model of ischemic retinopathy by a mechanism involving normalization of NOS function and reduced superoxide formation (Suwanpradid et al., Manuscript In Review). A protective role of arginase inhibition also has been demonstrated in rodent hearts subjected to ischemic-reperfusion myocardium or to hypobaric hypoxia by restoring eNOS coupling and NO production (Gonon et al., 2012; Singh et al., 2014). In line with these previous studies, our present results suggest that hypoxia elevates arginase activity and arginase 2 expression but reduces active p-eNOSSer1177 and NO levels in BAEC. Furthermore, the impaired network formation and reduced VEGF protein levels with continued hypoxia appear to have occurred because of rising level of ROS and falling levels of NO. However, arginase inhibition prevented this reduction of angiogenesis. These effects appear to be related to a maintained NO availability and sustained VEGF protein levels.
5. Conclusion
In summary, we project that the protective role of arginase inhibition is derived from increased NO production and availability and the subsequent sustained VEGF levels. Additional studies on the role of arginase in hypoxia-mediated angiogenesis and VEGF expression are needed to understand the mechanism or pathways more clearly.
Highlights.
We investigated the role of arginase in hypoxia-mediated angiogenesis.
Endothelial cell (EC) arginase expression and activity are elevated by hypoxia.
Inhibition of arginase enhances EC network formation and NO levels.
Arginase inhibition also limits ROS formation and elevates VEGF levels.
Arginase plays a fundamental role in angiogenesis
Acknowledgments
This work was supported by National Institutes of Health Grants HL70215 (to RWC) and EY11766 (to RBC and RWC).
Footnotes
Contributions of authors: Lin Wang: conception and design, data collection and interpretation, writing paper. Anil Bhatta: support for measurement of nitric oxide, analysis and interpretation of data. Haroldo A. Toque: support for western-blot and arginase activity assay. Modesto Rojas: support for DAF-2 and DCF fluorescence. Lin Yao: statistical analysis and literature search. Zhimin Xu: provision of special reagents. Chintan Patel: support for endothelial cell network formation. Ruth B. Caldwell: guidance in angiogenesis, critical revision of the manuscript and statistical expertise. R. William Caldwell: conception and design, interpretation of data, critical editing, revision, and final approval of article. -- All authors have approved the final version of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe H, Ishikawa W, Kushima T, Nishimura T, Mori C, Onuki A, Suzuki T, Ishii Y, Kansaku N, Miyazaki Y, Tanaka K, Morita H, Takizawa T. Nitric oxide induces vascular endothelial growth factor expression in the rat placenta in vivo and in vitro. Biosci Biotechnol Biochem. 2013;77:971–976. doi: 10.1271/bbb.120923. [DOI] [PubMed] [Google Scholar]
- Ahluwalia A, Tarnawski AS. Critical role of hypoxia sensor--HIF-1alpha in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr Med Chem. 2012;19:90–97. doi: 10.2174/092986712803413944. [DOI] [PubMed] [Google Scholar]
- Berchner-Pfannschmidt U, Tug S, Kirsch M, Fandrey J. Oxygen-sensing under the influence of nitric oxide. Cell Signal. 2010;22:349–356. doi: 10.1016/j.cellsig.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Chandra S, Romero MJ, Shatanawi A, Alkilany AM, Caldwell RB, Caldwell RW. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br J Pharmacol. 2012;165:506–519. doi: 10.1111/j.1476-5381.2011.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wojtowicz A, Szuba A, Cooke JP. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–666. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- Fong GH. Regulation of angiogenesis by oxygen sensing mechanisms. J Mol Med (Berl) 2009;87:549–560. doi: 10.1007/s00109-009-0458-z. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- Fraisl P. Crosstalk between oxygen- and nitric oxide-dependent signaling pathways in angiogenesis. Exp Cell Res. 2013;319:1331–1339. doi: 10.1016/j.yexcr.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Gao L, Chen Q, Zhou X, Fan L. The role of hypoxia-inducible factor 1 in atherosclerosis. J Clin Pathol. 2012;65:872–876. doi: 10.1136/jclinpath-2012-200828. [DOI] [PubMed] [Google Scholar]
- Gao X, Xu X, Belmadani S, Park Y, Tang Z, Feldman AM, Chilian WM, Zhang C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2007;27:1269–1275. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- Gonon AT, Jung C, Katz A, Westerblad H, Shemyakin A, Sjoquist PO, Lundberg JO, Pernow J. Local arginase inhibition during early reperfusion mediates cardioprotection via increased nitric oxide production. PLoS One. 2012;7:e42038. doi: 10.1371/journal.pone.0042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad P, Dussault S, Groleau J, Turgeon J, Michaud SE, Menard C, Perez G, Maingrette F, Rivard A. Nox2-containing NADPH oxidase deficiency confers protection from hindlimb ischemia in conditions of increased oxidative stress. Arterioscler Thromb Vasc Biol. 2009;29:1522–1528. doi: 10.1161/ATVBAHA.109.191437. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Gloe T, Pohl U. Hypoxia-induced upregulation of eNOS gene expression is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. J Cell Physiol. 2001;188:33–44. doi: 10.1002/jcp.1092. [DOI] [PubMed] [Google Scholar]
- Jenkins DC, Charles IG, Thomsen LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S. Roles of nitric oxide in tumor growth. Proc Natl Acad Sci U S A. 1995;92:4392–4396. doi: 10.1073/pnas.92.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, Tsugawa K, Tarnawski AS, Baatar D. Dual actions of nitric oxide on angiogenesis: possible roles of PKC, ERK, and AP-1. Biochem Biophys Res Commun. 2004;318:520–528. doi: 10.1016/j.bbrc.2004.04.055. [DOI] [PubMed] [Google Scholar]
- Jung C, Gonon AT, Sjoquist PO, Lundberg JO, Pernow J. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion. Cardiovasc Res. 2010;85:147–154. doi: 10.1093/cvr/cvp303. [DOI] [PubMed] [Google Scholar]
- Khong SM, Andrews KL, Huynh NN, Venardos K, Aprico A, Michell DL, Zarei M, Moe KT, Dusting GJ, Kaye DM, Chin-Dusting JP. Arginase II inhibition prevents nitrate tolerance. Br J Pharmacol. 2012;166:2015–2023. doi: 10.1111/j.1476-5381.2012.01876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, Santhanam L, Webb A, Camara A, Sikka G, Nyhan D, Shoukas AA, Ilies M, Christianson DW, Champion HC, Berkowitz DE. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol (1985) 2009;107:1249–1257. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Esumi H. Reciprocal regulation between nitric oxide and vascular endothelial growth factor in angiogenesis. Acta Biochim Pol. 2003;50:49–59. [PubMed] [Google Scholar]
- Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D'Acquisto F, Addeo R, Makuuchi M, Esumi H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95:189–197. [PubMed] [Google Scholar]
- Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, Hashimoto K, D'Acquisto F, Makuuchi M, Esumi H. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J Biol Chem. 2001;276:2292–2298. doi: 10.1074/jbc.M008398200. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Prieto CP, Munoz-Urrutia E, San Martin S, Sobrevia L, Casanello P. Role of arginase-2 and eNOS in the differential vascular reactivity and hypoxia-induced endothelial response in umbilical arteries and veins. Placenta. 2012;33:360–366. doi: 10.1016/j.placenta.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Krotova K, Patel JM, Block ER, Zharikov S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am J Physiol Cell Physiol. 2010;299:C1541–1548. doi: 10.1152/ajpcell.00068.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara F, Kai H, Tokuda K, Shibata R, Kusaba K, Tahara N, Niiyama H, Nagata T, Imaizumi T. Hypoxia-Inducible Factor-1 /Vascular Endothelial Growth Factor Pathway for Adventitial Vasa Vasorum Formation in Hypertensive Rat Aorta. Hypertension. 2002;39:46–50. doi: 10.1161/hy1201.097200. [DOI] [PubMed] [Google Scholar]
- Madigan M, Zuckerbraun B. Therapeutic Potential of the Nitrite-Generated NO Pathway in Vascular Dysfunction. Front Immunol. 2013;4:174. doi: 10.3389/fimmu.2013.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- Morris CR, Gladwin MT, Kato GJ. Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders. Curr Mol Med. 2008;8:620–632. doi: 10.2174/156652408786241447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM., Jr Regulation of enzymes of the urea cycle and arginine metabolism. Annu Rev Nutr. 2002;22:87–105. doi: 10.1146/annurev.nutr.22.110801.140547. [DOI] [PubMed] [Google Scholar]
- Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto CP, Krause BJ, Quezada C, San Martin R, Sobrevia L, Casanello P. Hypoxia-reduced nitric oxide synthase activity is partially explained by higher arginase-2 activity and cellular redistribution in human umbilical vein endothelium. Placenta. 2011;32:932–940. doi: 10.1016/j.placenta.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Reid KM, Tsung A, Kaizu T, Jeyabalan G, Ikeda A, Shao L, Wu G, Murase N, Geller DA. Liver I/R injury is improved by the arginase inhibitor, N(omega)-hydroxy-nor-L-arginine (nor-NOHA) Am J Physiol Gastrointest Liver Physiol. 2007;292:G512–517. doi: 10.1152/ajpgi.00227.2006. [DOI] [PubMed] [Google Scholar]
- Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, Caldwell RB, Caldwell RW. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. 2008;102:95–102. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, Shoukas A, Romer LH, Berkowitz DE. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. 2006;99:951–960. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- Sankaralingam S, Xu H, Davidge ST. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc Res. 2010;85:194–203. doi: 10.1093/cvr/cvp277. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Doi S, Mizutani S, Azuma H. Roles of accumulated endogenous nitric oxide synthase inhibitors, enhanced arginase activity, and attenuated nitric oxide synthase activity in endothelial cells for pulmonary hypertension in rats. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1480–1487. doi: 10.1152/ajplung.00360.2006. [DOI] [PubMed] [Google Scholar]
- Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, Caldwell RB, Caldwell RW. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol. 2011;300:C1181–1192. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Padhy G, Vats P, Bhargava K, Sethy NK. Hypobaric hypoxia induced arginase expression limits nitric oxide availability and signaling in rodent heart. Biochim Biophys Acta. 2014;1840:1817–1824. doi: 10.1016/j.bbagen.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Steppan J, Nyhan D, Berkowitz DE. Development of novel arginase inhibitors for therapy of endothelial dysfunction. Frontiers in immunology. 2013;4:278. doi: 10.3389/fimmu.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- Takenaga K. Angiogenic signaling aberrantly induced by tumor hypoxia. Front Biosci (Landmark Ed) 2011;16:31–48. doi: 10.2741/3674. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, Kuo L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8:863–884. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, Goggans T, Thunnissen FB, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. Faseb J. 2004;18:1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- Yang Z, Ming XF. Endothelial arginase: a new target in atherosclerosis. Curr Hypertens Rep. 2006;8:54–59. doi: 10.1007/s11906-006-0041-8. [DOI] [PubMed] [Google Scholar]
- Zhang W, Baban B, Rojas M, Tofigh S, Virmani SK, Patel C, Behzadian MA, Romero MJ, Caldwell RW, Caldwell RB. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am J Pathol. 2009;175:891–902. doi: 10.2353/ajpath.2009.081115. [DOI] [PMC free article] [PubMed] [Google Scholar]