

Abstract
Peptidyl-prolyl cis/trans isomerases (PPIases), a unique family of molecular chaperones, regulate protein folding at proline residues. These residues are abundant within intrinsically disordered proteins, like the microtubule-associated protein tau. Tau has been shown to become hyperphosphorylated and accumulate as one of the two main pathological hallmarks in Alzheimer's disease (AD), the other being amyloid beta (Aβ). PPIases, including Pin1, FK506-binding protein (FKBP) 52, FKBP51, and FKBP12, have been shown to interact with and regulate tau biology. This interaction is particularly important given the numerous proline-directed phosphorylation sites found on tau and the role phosphorylation has been found to play in pathogenesis. This regulation then affects downstream aggregation and oligomerization of tau. However, many PPIases have yet to be explored for their effects on tau biology, despite the high likelihood of interaction based on proline content. Moreover, Pin1, FKBP12, FKBP52, cyclophilin (Cyp) A, CypB, and CypD have been shown to also regulate Aβ production or the toxicity associated with Aβ pathology. Therefore, PPIases directly and indirectly regulate pathogenic protein multimerization in AD and represent a family rich in targets for modulating the accumulation and toxicity.
Keywords: Alzheimer's disease, PPIase, immunophilin, tau, Amyloid β, chaperone
Alzheimer's disease (AD), the most common cause of dementia, is a progressive disease marked by the accumulation of intracellular tangles predominantly comprised of the microtubule-associated protein tau, and extracellular amyloid beta (Aβ) plaques (Hardy & Higgins 1992, Kosik et al. 1984, Wolozin et al. 1986). AD is not the only disease in which tau accumulates, but is only one of over 15 neurological disorders containing tau pathology. These proteinopathies, collectively termed tauopathies, include progressive supranuclear palsy (PSP) and frontotemporal lobar dementia linked to chromosome 17 (FTLD-17) (Mackenzie et al. 2007, Schmidt et al. 1996). The tau protein normally works to regulate microtubule dynamics, but in disease, tau forms oligomers and tangles, a process that correlates with AD progression (Maeda et al. 2006). The Aβ peptide, which is formed by the proteolytic cleavage of the amyloid precursor protein (APP) by β-secretase (BACE) followed by γ-secretase, also accumulates in AD, but as extracellular oligomers and plaques (Kayed et al. 2003). Although the most toxic forms of these proteins are still debated, much is known about their biology and how they are regulated (Bloom 2014, Hardy 2003).
Tau is an intrinsically disordered protein (IDP), due to its proline (Pro)-rich region (Mukrasch et al. 2005). Within this Pro-rich domain lie a number of serine (Ser) and threonine (Thr) residues that produce putative consensus motifs for Pro-directed Ser/Thr kinases, including glycogen synthase kinase 3 beta (GSK3-β), cyclin-dependent kinase (CDK) 2, CDK5 and mitogen-activated protein kinase (MAPK) (Schweers et al. 1994). As a result of this stretch of residues spanning ~100 ααs (residues 151-244), combined with the fact that tau is hyper-phosphorylated in the AD brain, each of these kinases have been implicated in the pathogenesis of tau (Mandelkow & Mandelkow 2012). However, there is an alternative reason for this Pro-rich region that has nothing to do with phosphorylation, but has more to do with tau structure and its flexible nature. Pro/Ser/Thr stretches allow IDPs to undergo major structural changes that convey functional diversity, a process that can be controlled by peptidyl-prolyl cis/trans isomerases (PPIases) (Liou et al. 2011, Lu et al. 2007, Rogers et al. 2014). PPIases, chaperone proteins important for folding and transportation of proteins, rotate peptide bonds directly before Pro residues from cis conformation to trans and vice versa (Edlich & Fischer 2006). A list of PPIs is provided in Table 1. Because the intrinsically unstructured protein tau contains a large number of Pro motifs, adjacent to functionally relevant phosphorylation sties (Bibow et al. 2011, Bulbarelli et al. 2009), PPIases are particularly important for tau biology. In fact, Pro residues have been identified as the single most common amino acid to promote disorder in proteins and are represented almost 1 ½ - 2 times greater than other amino acids in IDPs (Theillet et al. 2013). These Pro residues have been shown to add stability to IDPs and allow regulation of structure (Ge & Pan 2009). While Pro residues are rarely found in secondary structural elements, they are often found adjacent to these structured regions and have been shown to significantly alter the tertiary structure of a protein (MacArthur & Thornton 1991).
Table 1.
List of PPIases.
| Family | Protein | Gene | Primary location |
Secondary locations |
Expression in brain tissue |
Common aliases |
Effects on AD pathology |
|
|---|---|---|---|---|---|---|---|---|
| Parvulins | Pin1 | Pin1 | nucleus | cytosol | ++ | Promotes dephosphoryaltion of tau; Promotes non-amyloidogenic APP processing | ||
| Pin4 | Pin4 | nucleus, mitochondrion, cytoskeleton | cytosol | + | Par14, Par17 | |||
| Immunophilins | FK506-binding protein | FKBP12 | FKBP1A | cytosol | nucleus | ++ | PKC12 | Prevents tau aggregation; Promotes amyloidogenic APP processing |
| FKBP12.6 | FKBP1B | cytosol | ER | ++ | FKBP1L, OTK4 | |||
| FKBP13 | FKBP2 | ER | cytosol | ++ | ||||
| FKBP19 | FKBP11 | extracellular | ER, cytosol | + | ||||
| FKBP22 | FKBP14 | ER | extracellular, cytosol | + | ||||
| FKBP23 | FKBP7 | ER | extracellular | - | ||||
| FKBP25 | FKBP3 | nucleus | cytosol | ++ | ||||
| FKBP36 | FKBP6 | nucleus, cytosol | + | |||||
| FKBP37 | AIP | cytosol | nucleus | ++ | FKBP16, XAP-2, FKBP37.7 | |||
| FKBP38 | FKBP8 | mitochondrion | ER, cytosol | + | ||||
| FKBP51 | FKBP5 | nucleus, extracellular | cytosol | + | FKBP54 | Promotes tau preservation and oligomerization | ||
| FKBP52 | FKBP4 | nucleus, mitochondrion | cytosol, extracellular | ++ | FKBP59 | Promotes tau oligomerization; Protects against Aβ toxicity | ||
| FKBP60 | FKBP9 | ER | mitochondrion | + | FKBP63 | |||
| FKBP65 | FKBP10 | ER, extracellular | + | |||||
| FKBP133 | FKBP15 | Endosome | nucleus, cytoskeleton, cytosol | + | WAFL | |||
| Cyclophilins | Cyclophilin A | PPIA | cytosol, nucleus, extracellular | +++ | CypH, Cyp18 | Protects against Aβ toxicity | ||
| Cyclophilin B | PPIB | ER | nuclear, extracellular | ++ | Cyp22, Cyp-S1 | Protects against Aβ toxicity | ||
| Cyclophilin C | PPIC | ER, cytosol | + | |||||
| Cyclophilin D | PPID | nucleus | cytosol | + | Cyp40 | Protects against Aβ toxicity | ||
| Cyclophilin E | PPIE | nucleus | cytosol | + | Cyp33 | |||
| Cyclophilin F | PPIF | mitochondrion | peroxisome, plasma membrane | ++ | Cyp3, CypD | |||
| Cyclophilin G | PPIG | nucleus | cytosol, extracellular | + | SRCyp, CARS-Cyp | |||
| Cyclophilin H | PPIH | nucleus | cytosol | + | Cyp20 | |||
| Cyclophilin NK | NK-TR | nucleus | cytosol | + | CypNK | |||
| PPlase-like | PPIL1 | PPIL1 | nucleus | cytosol | + | CypL1 | ||
| PPIL2 | PPIL2 | nucleus, golgi | cytosol | + | Cyp60, CYC4 | |||
| PPIL3 | PPIL3 | nucleus | cytosol | ++ | CypJ, CLK1 | |||
| PPIL4 | PPIL4 | nucleus | cytosol | ++ | ||||
| PPIL5 | LRR1 | nucleus | cytosol | + | ||||
| PPIL6 | PPIL6 | cytosol | golgi | + | RSPH12 | |||
| PTPA | PPP2R4 | nucleus | cytoplasm | ++ | PPA | Decreases tau phosphorylation | ||
| PP5 | PPP5C | nucleus, golgi | cytosol | + | PPT | Decreases tau phosphoryaltion; Protects against Aβ toxicity | ||
| RANBP2 | RANBP2 | nucleus | cytosol | + | NUP358, ANE1 | |||
| PPWD1 | PPWD1 | nucleus | cytosol | + | spliceosome-associated Cyp | |||
| CWC27 | CWC27 | nucleus | cytosol | + | SDCCAG-10 | |||
While tau is indeed hyper-phosphorylated in the AD brain, this occurs on specific residues within the Pro-rich region that are not normally phosphorylated outside of disease (Augustinack et al. 2002). This further suggests that there is not a functional use for the phosphorylation of many of these motifs, rather these sites become phosphorylated only after tau has begun to accumulate. In fact, the temporal order of tau phosphorylation in the AD brain would suggest that many of the phospho-tau epitopes that are well-known pathological markers of tau tangles only emerge much later in the life of the tangle (Binder et al. 2005, Grundke-Iqbalet al. 1986). Only a select few are considered “early” phospho-tau sites that could lead to its pathogenic aggregation and only non-Pro-directed motifs within the MBD of tau have any known impact on tau function. Thus, it is likely that the functional use of this Pro-rich region in tau is to convey its flexibility rather than as a hub for kinase activity (Figure 1). However, since PPIases are also known to modulate protein phosphorylation, it is possible that they affect both tau structure and function.
Figure 1. Potential dual role of Proline-directed Ser/Thr motifs in tau.
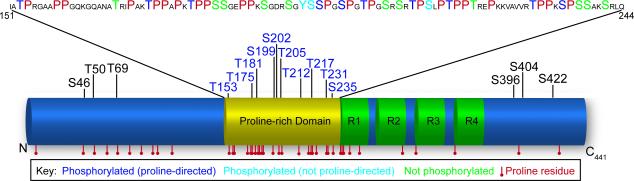
Tau contains 43 Proline residues in the human 441-residue isoform that could be modified by either PPIases or kinases. Many of these Prolines are grouped together in the Proline-rich domain between 151 and 244 αα. Within this domain, only 13 of the 23 Ser/Thr residues are known to be phosphorylated, suggesting that some of these sites may exist to regulate tau structure. PPIases could control both tau phosphorylation and tau structure leading to its oligomerization.
Recent evidence suggests that tau oligomers are the more toxic aggregates in disease rather than the pathological hallmark tangles (Berger et al. 2007, Gomez-Isla et al. 1997, Morsch et al. 1999, Santacruz et al. 2005). Tau oligomerization, which occurs early in AD, has been shown to both require and be accelerated by both the phosphorylation and isomerization of this Pro-rich domain (Blair et al. 2013, Bulbarelli et al. 2009, Chambraud et al. 2010, Lasagna-Reeves et al. 2012, Lu et al. 1999, Giustiniani et al. 2014, Ma et al. 2012). Thus, it is likely that both of these processes are contributing to disease pathogenesis.
In contrast to tau, the Aβ peptide does not contain any Pro residues, so it is not directly regulated by cis/trans isomerization or Pro-directed phosphorylation. Instead, the amyloid precursor protein, APP contains Pro residues that could also be modified by both phosphorylation and isomerization. It is known that APP can be phosphorylated by Pro-directed kinases at Thr668 (Balastik et al. 2007, Pastorino et al. 2006), which can lead to increased Aβ production (Lee et al. 2003). In fact, phosphorylation at this site is increased in AD (Lee et al. 2003). Like tau, Aβ can form oligomers which have been shown to act as seeds to drive tau oligomerization (Lasagna-Reeves et al. 2010). Thus, Pro-directed phosphorylation regulates two major constituents of AD pathology and alters their oligomerization. But the role of isomerization in APP processing and Aβ pathogenesis remains unknown.
There are two major groups of proteins containing PPIase domains, the parvulins and the immunophilins (He et al. 2004, Lu et al. 1996, Sinars et al. 2003, Wiederrecht et al. 1991). The immunophilins, which includes both the FK506-binding proteins (FKBPs) and the cyclophilins (Cyps), are also targets of immunosuppressive drugs. Previous work has shown that Pin1, FKBP52, FKBP51, and possibly FKBP12 can regulate tau oligomerization (Blair et al. 2013, Bulbarelli et al. 2009, Chambraud et al. 2010, Giustiniani et al. 2014, Lu et al. 1999, Ma et al. 2012). As more and more evidence supports tau oligomers being the most toxic species in AD, more effort is being put into identifying molecular targets which regulate this process. The PPIases are a unique family of chaperones which have already been shown to directly affect the oligomerization of tau. This review will summarize this work and provide a basis for investigating other members of the PPIase chaperone family.
Parvulins
Parvulins are a well-conserved family of PPIases vital for mitosis (Mueller & Bayer 2008). This family, unlike the other family of PPIases, uses an N-terminal WW (Trp-Trp) domain to bind clients and a C-terminal PPIase domain for protein isomerization (Sun et al. 2012). An important member of this family, protein interacting with NIMA (never in mitosis) 1 (Pin1), is the only PPIase to recognize phosphorylated Ser/Thr-Pro bonds (Zhou et al. 2000). Pin4 (Par14/ Par17) is also a notable member of this family; however, much less is known about this protein (Mueller et al. 2006).
Pin1
Pin1 is a highly conserved protein which is highly expressed in neurons (Nakamura et al. 2012b) and has been shown to regulate both tau and Aβ biology (Ma et al. 2012, Pastorino et al. 2012, Zhou et al. 2000). In fact, mice lacking Pin1 develop tau pathology and neuron loss (Liou et al. 2003). While this is the case for wild-type tau, it is not the case for mutant P301L tau, which was found to have reduced levels in Pin1 knock-out (KO) mice crossed with P301L tau transgenic mice (Lim et al. 2008). The inverse has also been shown to be true. That is, Pin1 over-expression leads to the degradation of wild-type tau, like that in AD, and the preservation of P301L tau, like that found in fronto-temporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) (Lim et al. 2008). Therefore, Pin1 regulates mutant and wild-type tau differently. Furthermore, Pin1 has been shown to restore misfolded tau in such a way that it is able to bind the microtubules: tau must be phosphorylated at Thr231 in order for Pin1 to bind (Lu et al. 1999), which promotes a cis (“unhealthy” tau) to trans (“healthy” tau) isomerization of tau (Nakamura et al. 2013). Pin1 isomerization restores tau to a conformation which is able to once again bind the microtubules (Lu et al. 1999, Nakamura et al. 2013). This is particularly important to tau oligomerization, since the degree of tau phosphorylation at Thr231 correlates with tau oligomerization (Lasagna-Reeves et al. 2012). To our knowledge, no direct test of tau oligomerization has been performed with Pin1 modulation. However, it was shown that Aβ oligomers affect Pin1 regulation of tau, in part by stimulating the dephosphorylation of tau at Thr231 (Bulbarelli et al. 2009). This is one of the multiple indirect ways that Pin1 regulates tau phosphorylation and folding.
Pin1 was also shown to directly bind to the APP pThr668-Pro motif via its WW domain and catalyze cis to trans isomerization via its PPIase domain (Ma et al. 2012). This isomerization of APP from the cis conformation, which leads to amyloidogenic processing, into the trans conformation, which slows the production of amyloidogenic Aβ, reduced Aβ synthesis (Pastorino et al. 2006). Pin1 also inhibited GSK-3β activity, which increased the rate of APP turnover. Moreover, Pin 1 has been shown to directly modulate GSK-3β by binding the Thr330-Pro motif (Ma et al. 2012), which could lead to additional downstream regulation of tau and Aβ. Since GSK3ß is also hyper-activated in AD brain this provides another mechanism through which Pin1 might impact AD pathogenesis (Forlenza et al. 2011).
Moreover, AD brains have reduced levels of soluble Pin1 and reduced Pin1 activity (Lu et al. 1999, Pastorino et al. 2006). This down-regulation of Pin1 is thought to be the result of the combination of increased oxidation of Pin1 (Yao et al. 2004) which inactivates Pin1 (Butterfield et al. 2006) and inactivation of Pin1 via phosphorylation by death-associated protein kinase 1 (DAPK1), which is not only up-regulated in AD, but has been genetically linked to late-onset AD and shown to regulate tau stability (Kim et al. 2014, Li et al. 2006). Insufficient Pin1 activity may contribute to elevated levels of tau, through decreased degradation, and elevated levels of Aβ, through decreased GSK-3β inhibition. This increase in tau and/or Aβ levels may be sufficient to initiate the AD disease process, as this has been shown to model disease pathologies and phenotypes in animal models of AD (Pastorino et al. 2012, Grundke-Iqbal & Iqbal 1999, Guo et al. 2013). In fact, surviving neurons in the degenerating brain have been shown to have high levels of Pin1 expression (Liou et al. 2003).
Pin4 (Par14)
Pin4, another parvulin PPIase also known as Par14, has not been well characterized. It is elevated in Pin1 KO mice, demonstrating a compensatory effect between PPIases (Zhang et al. 2013); however, Par14 is unable to cis/trans isomerize Ser/Thr-Pro residues (Uchida et al. 1999). Par14 regulates insulin receptor substrate 1 (IRS-1), which affects downstream proteins including phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt); specifically, Par14 over-expression induced IRS-1 phosphorylation leading to Akt phosphorylation (Zhang et al. 2013). In a separate study, phospho-Akt (active form) was shown to increase the amount of p-GSK3-β, which inactivates this kinase (Lee et al. 2009). Taken together, perhaps depletion of Pin1 leads to a compensatory activation of Par14 to provide additional protection against the aberrant folding of proteins; however, more work needs to be done to properly assess the importance of Par14 in AD.
Immunophilins
Immunophilins are made of two families of proteins, the FKBPs and the Cyps. These proteins, in addition to their PPIase activity, bind to immunosuppressive drugs (Barik 2006, Soldin et al. 1993). However, each family binds a unique set of these drugs. FKBPs bind FK506 and rapamycin while Cyps bind cyclosporine, all of which lead to the inhibition of calcineurin and mammalian target of rapamycin (mTOR) (Hamilton & Steiner 1998, Li et al. 2002). Interestingly, studies using Saccharomyces cerevisiae have shown that immunophilins are not necessary for viability in any combination (Dolinski et al. 1997). Several immunophilins including FKBP52, FKBP51, FKBP38, FKBP37 (hepatitis B virus protein X associated protein 2; Xap2), and CyP40 contain tetratricopeptide repeat (TPR) domains (Schulke et al. 2010), which allow for a second mechanism of action in AD biology, through direct binding to major heat shock proteins Hsp70 and Hsp90 (Schweiger et al. 2013). A few well-studied members of this family, like FKBP51, FKBP52, and CypA, have relationships with both tau and Aβ proteins (Cao & Konsolaki 2011, Ge et al. 2009, Sanokawa-Akakura et al. 2010).
FK506-binding proteins (FKBPs)
Members of the highly conserved FKBP family are found throughout the nervous system and have been well studied in the context of steroid hormone complexes and AD biology (Steiner et al. 1992). These chaperones bind to the immunosuppressive drug FK-506, which has been shown to regulate tau and Aβ in vivo (Dineley et al. 2010, Luo et al. 2008, Yoshiyama et al. 2007, Zhao et al. 2006). These studies revealed that FK-506 reduced tau and Aβ pathology in the P301S mouse and Aβ oligomer-treated mice respectively (Dineley et al. 2010, Yoshiyama et al. 2007). However, in wild-type mice another study found an increase in tau phosphorylation under FK-506 treatment (Luo et al. 2008). In particular, FKBP52, FKBP38, FKBP37, FKBP25, FKBP22, and FKBP12 have been shown to regulate tau and/or Aβ pathology (Cao & Konsolaki 2011).
FKBP52
One of the most well studied FKBPs, FKBP52, is a TPR-containing PPIase. FKBP52, encoded by FKBP4, was shown to be highly expressed in neurons and has been well characterized for regulating steroid hormone complexes (Tatro et al. 2009). In particular, it has been shown to play an important role in the translocation of steroid hormones, like glucocorticoid receptors (GR), into the nucleus (Tatro et al. 2009). Glucocorticoids have been shown to up-regulate both tau and Aβ pathology and may represent a secondary mechanism by which FKBP52 is involved in AD (Green et al. 2006). FKBP52, which is a ubiquitously expressed protein with high levels in the hippocampus, cortex and basal ganglia, was shown to have decreased levels in the frontal cortex of AD brains (Giustiniani et al. 2012) and has been shown to regulate both tau and Aβ (Sanokawa-Akakura et al. 2010).
FKBP52 has been shown to directly bind to tau and regulate microtubule dynamics (Chambraud et al. 2007, Chambraud et al. 2010). Moreover, FKBP52 was recently shown to stimulate P301L tau oligomerization, which eventually led to fibril formation. The same study demonstrated that FKBP52 depletion in a P301L zebrafish model of tauopathy rescued synaptic deficits, which was attributed to changes in tau phosphorylation at Thr181 (Chambraud et al. 2010, Giustiniani et al. 2014). These studies also showed that high levels of FKBP52 were associated with decreased neurite growth and decreased tau accumulation (Chambraud et al. 2010). Conversely, FKBP52 expression has been shown to be inversely related to Aβ toxicity in an Aβ transgenic fly model (Sanokawa-Akakura et al. 2010). These inverse effects on tau and Aβ may complicate therapeutically targeting FKBP52 for AD; however, FKBP52 may be a better target for tauopathies, without Aβ pathology.
FKBP51
FKBP51, a homolog of FKBP52, is also highly expressed in neurons and has been primarily studied for its role in regulating GR activity (Grad & Picard 2007, Pratt & Toft 1997). Encoded by FKBP5, FKBP51 is a part of the mature GR complex. FKBP51 is up-regulated by GR activity through multiple glucocorticoid response elements (GREs) found throughout the FKBP5 gene (Binder 2009). However, FKBP51 has been shown to slow the translocation of GR into the nucleus, thus reducing GR activity (Wochnik et al. 2005). Thus, FKBP51 works in a short, negative feedback loop to regulate GR activity. This intimate relationship with GR activity, which has been previously linked to AD (Escribano et al. 2009), may be an indirect mechanism by which FKBP51 regulates tau and possibly also Aβ pathology.
FKBP51 has also been well characterized for its role in directly regulating tau biology (Blair et al. 2013, Jinwal et al. 2010). This work has shown that the over-expression of FKBP51 preserves tau, while knock-down of FKBP51 leads to tau degradation (Jinwal et al. 2010). Likewise, FKBP5−/− mice show reduced levels of endogenous tau (Blair et al. 2013). In this recent work from our group, FKBP51, in the presence of Hsp90, was found to preserve neurotoxic tau. This tau was confirmed to be both high in phospho-Thr231 and oligomer content (Blair et al. 2013). Thus, FKBP51 prevents tau degradation and stimulates its oligomerization.
Recent work using a high-throughput screen (HTS) has demonstrated that FKBP5 can bind Aβ oligomers (Olah et al. 2011); however, no functional outcome of this interaction has been described. Additional studies need to be done to determine the effects of modulating FKBP51 on Aβ. However, because there has not been a negative phenotype associated with FKBP5−/− mice (Hartmann et al. 2012, O'Leary et al. 2011), drugs which lower FKBP51 levels could help protect against tau-mediated neurotoxicity in AD.
FKBP38
Encoded by FKBP8, FKBP38 is a mitochondrial resident FKBP (Shirane & Nakayama 2003). FKBP38, unlike other immunophilins is calmodulin/calcium dependent and remains in a constitutively active state (Edlich et al. 2007). To date, data on tau regulation by FKBP38 are unavailable, even though it contains a TPR domain. FKBP38 has been shown to directly bind presenilins (Psns) and regulate apoptosis through B-cell lymphoma (Bcl-2) (Wang et al. 2005). Normally, FKBP38 forms a complex with Bcl-2 and may inhibit apoptosis via anchoring and stabilizing Bcl-2 to the membrane (Shirane & Nakayama 2003). However, it was found that Psns immunoprecipitated with both FKBP38 and Bcl-2, initiated their degradation, and subsequently activated apoptosis. Moreover, FKBP38, in association with Rheb, inhibits mTOR activity (Bai et al. 2007), a pathway which is involved in regulating AD pathology (Wang et al. 2014).
FKBP37
FKBP37, also known as X-associated protein 2 (XAP2) and Aryl hydrocarbon receptor interacting protein (AIP), is a TPR-containing cytosolic protein. Although XAP2 is able to bind Hsp90 through a TPR domain, it also has been shown to uniquely bind Hsp90, in part, through the PPIase domain (Linnert et al. 2013). Furthermore, XAP2 plays a role in estrogen and glucocorticoid receptor signaling (Laenger et al. 2009, Trivellin & Korbonits 2011). Although XAP2 has not been directly linked to AD pathology, it has been shown that histone deacetylase (HDAC) 6, which has been linked to both tau and Aβ regulation (Cook et al. 2012, Govindarajan et al. 2013, Selenica et al. 2014), regulates its activation (Kekatpure et al. 2009).
FKBP25
FKBP25, encoded by FKBP3, is a rare nuclear resident PPIase, which interacts with a diverse set of proteins. Based on interactions, FKBP25 plays an important role in DNA packaging, the remodeling of chromatin, splicing of mRNA, ribosomal assembly, as well as other aspects of neuronal signaling (Galat & Thai 2014, Jiang et al. 2001). FKBP25 is associated with a number of other proteins including HDACs, nucleolin, and protein kinase casein kinase II (CK2) (Gudavicius et al. 2013). FKBP25 has also been shown to be inhibited by p53 (Ahn et al. 1999) while also activating p53 activity (Ochocka et al. 2009). As a link to AD, p53 activity suppresses the levels of PS1 (Roperch et al. 1998). Furthermore, one study showed that there is a dramatic progressive increase in FKBP25 expression in the mouse hippocampus across the lifespan (Jiang et al. 2001). Taken together, these data suggest that FKBP25 could possibly regulate Aβ pathology, especially in the hippocampus, through the regulation of p53 and PS1. Further studies need to be done to elucidate if there is a connection between FKBP25 and tau.
FKBP22
FKBP22, encoded by FKBP14, is highly involved in development (van de Hoef et al. 2013), protein folding, and regulates and procollagens, proteins enriched in Pro residues (Baumann et al. 2012, Ishikawa & Bachinger 2014). FKBP22 is a predominantly endoplasmic reticulum (ER) resident protein (van de Hoef et al. 2013), which does not directly interact with tau or Aβ.However, FKBP22 has been shown to regulate Psn and Notch in a Drosophila screen for Psn and APP interactors (van de Hoef et al. 2009), suggesting this co-chaperone may modulate APP processing. FKBP22 stabilizes Psn, which increases the production of γ-secretase, promoting its association with notch. Therefore, higher levels of FKBP22 could enhance production of Aβ (1-42) pathogenic species.
FKBP12
The smallest FKBP associated with AD, FKBP12, is a ubiquitous, cytosolic PPIase with high central expression, particularly in neuronal cell bodies and neurites (Nigam et al. 1993, Sugata et al. 2009). FKBP12 has been shown to co-localize with tau in neurons and neurofibrillary tangles (NFT) and glial fibrillary acid protein (GFAP), an astrocytic marker (Sugata et al. 2009). Recently, FKBP12 was shown to bind to Aβ oligomers and regulate APP processing (Liu et al. 2014, Olah et al. 2011). In AD patients, FKBP12 expression was decreased and its localization altered. Specifically, FKBP12 expression in control brains was confined to neural cell bodies and neurites, while AD brains showed FKBP12 expression in reactive astrocytes, neuropil threads, and dystrophic neurites (Sugata et al. 2009).
Not only has FKBP12 been shown to co-localize with NFTs, it may also regulate tau aggregation. It was shown that FKBP12 prevented the aggregation of a peptide corresponding to the proposed aggregation region of tau (R3) in vitro. Interaction studies between FKBP12 and R3 suggested aggregation inhibition resulted from the PPIase activity of FKBP12 (Ikura & Ito 2013). These data suggest that FKBP12 could prevent tau oligomerization, since it prevents its aggregation. However, further studies would need to be performed to determine the role of FKBP12 on tau oligomerization.
Other FKBPs
There are many other FKBPs which have relatively high brain expression yet have not been associated with AD pathology to date, including the following: FKBP133, FKBP65, FKBP63, FKBP25, FKBP23, FKBP19, FKBP13, FKBP12.6 and FKBPL. Although these immunophilins have not been linked to AD, members such as FKBP133, FKBP65, and FKBP23 could likely play a role in tau or Aβ pathology.
FKBP133, encoded by FKBP15, is predominantly expressed in the developing hippocampus, cerebral cortex, and peripheral ganglia. This PPIase is localized to axonal shafts and colocalized with F-actin in growth cones (Nakajima et al. 2006). FKBP133 has yet to be connected with any disease.
There is little known about FKBP63, encoded by FKBP9. However, it was recently shown in an in vitro assay that FKBP9 was one of seven genes identified to play an important role in prion spreading and degradation (Brown et al. 2014). This new discovery, along with data suggesting Aβ and tau can spread in a prion fashion (Nussbaum et al. 2012, Sanders et al. 2014), may suggest that FKBP63 could potentially play a role in their propagation as well. Moreover, prions have been implicated as an initiating factor in AD pathology (Reiniger et al. 2011).
FKBP23, encoded by FKBP7, has been shown to interact with binding immunoglobulin protein (BiP), the resident Hsp70 in the ER (Zhang et al. 2004). FKBP23 regulates BiP ATPase activity through conformational changes of its ATPase domain (Feng et al. 2011). However, no work has been done to show if FKBP23 regulates tau or Aβ pathology.
Cyclophilins (Cyps)
Cyclophilins (Cyps) are a ubiquitously expressed family of conserved immunophilins, which, along with having PPIase activity, have a cyclophilin-like domain. Additionally, they bind the immunosuppressive drug Cyclosporin A (CsA) (Marks 1996). The cyps have a variety of members with ranging molecular weights and cellular locations; one member, Cyp-40, even has a TPR domain (Wang & Heitman 2005). Some of the cyclophilins have already been shown to be involved in regulating AD pathology, including Cyp-40. Other members such as CypC, CypE, CypG, CypH, and CypNK have not yet been shown to play a role in AD pathology.
Cyp40
One of the most unique and well-studied Cyps, Cyp40, also known as CypD, is encoded by PPID and located in the cytosol. CypF, encoded by PPIF, is also known as CypD. However, CypF, also called Cyp3, is located in the mitochondria where it associates with the mitochondrial permeability transport pore (mPTP) (Bergsma et al. 1991, Du et al. 2013). These proteins will be referred to as CypD in this review with differentiations made on location, when that is specified in the literature. This TPR-containing Cyp was shown to compete with FKBP52 for binding to Hsp90, each of which are components of estrogen and progesterone receptor signaling complexes (Pirkl & Buchner 2001, Ratajczak & Carrello 1996). Furthermore, CypD was shown to regulate the translocation of glucocorticoid receptors (GR) to the nucleus (Bourke et al. 2013).
Cytosolic CypD was also reported to be a strong binder of Aβ oligomers (Olah et al. 2011), while mitochondrial CypD was shown to interact with mitochondrial Aβ in the AD brain (Guo et al. 2013). Furthermore, CypD expression was associated with aging as well as Aβ-mediated oxidative stress. It was shown in primary cultures that the CypD-Aβ complex found in wild-type mice increases reactive oxygen species levels in comparison to to the same treatment in CypD-deficient neurons (Guo et al. 2013). It has further been shown that the CypD-Aβ complex disturbs the PKA/CREB signaling pathway, important in plasticity and memory, through Aβ-mediated oxidative stress, leading to synaptic damage (Du et al. 2013).
CypA
CypA, encoded by PPIA, is a ubiquitous, cytosolic protein (Wang & Heitman 2005). CypA has been implicated in many diseases including HIV, heart disease, rheumatoid arthritis, and various cancers (Kamada et al. 2009, Nigro et al. 2013). Moreover, CypA has been linked to diseases of the CNS, including AD and amyotrophic lateral sclerosis (Bell et al. 2012, Tanakaet al. 2011). In AD, CypA has been shown play a regulatory role in the maintenance of blood-brain barrier integrity via a complex with nuclear factor κB (NF-κB) and matrix metalloproteinase 9 (MMP9); however, this was dependent on the expression of apolipoproteins (Bell et al. 2012). This work demonstrated that CypA mediates BBB dysfunction in the presence of ApoE4, but was rescued upon treatment of CsA. However, a previous study showed that CypA was actually protective against Aβ-mediated toxicity by protecting against reactive oxidative species (ROS) (Ge et al. 2009), while other work showed that CypA promoted neuronal death (Cande et al. 2004). This work suggests that targeting CypA for the treatment of AD may produce differential effects depending on presence of the ApoE risk allele.
CypB
CypB is an ER-resident immunophilin, encoded by PPIB, which has been previously shown to protect against ER stress and play a role in synaptic transmission through synapsin signaling (Lane-Guermonprez et al. 2005, Kim et al. 2008). In response to ER stress, CypB is up-regulated along with BiP and Grp94 via an ER stress-specific response element indicating a role for CypB in the unfolded protein response. CypB is also found to reduce calcium ion leakage as well as reactive oxygen species during ER stress (Kim et al. 2008). CypB was shown to be protective against Aβ-induced neurotoxicity through regulating ROS levels and activation of kinases PI3K and MAPK (Oh et al. 2011). Although studies have yet to show a connection between CypB and tau directly, it would not be surprising if CypB could regulate tau hyperphosphorylation given the role of CypB in ER stress and kinase regulation.
PPIase-like proteins
There is also a class of proteins that are considered to be “PPIase-like”, which lack classical PPIase domains, but have PPIase-like activity. Many of the proteins that belong to this family are named to match this activity; for example, PPIase-like (PPIL)1 or Cyp-like 1 (CypL1), PPIL2 (Cyp60), PPIL3 (CypJ), PPIL4, PPIL5 (LRR1; leucine rich repeat protein1), and PPIL6 (Huang et al. 2005, Jang et al. 2001, Pushkarsky et al. 2005, Stegmann et al. 2010). Other PPIase-like proteins include Ran-binding protein 2 (RANBP2), peptidyl-prolyl isomerase containing WD40 repeat (PPWD1), Serologically Defined Colon Cancer Antigen 10 (SDCCAG-10 or CWC27), phosphatase two a phosphatase activator (PTPA), and protein phosphatase 5 (PP5) (Cho et al. 2014, Davis et al. 2008, Silverstein et al. 1997). To date, many of these PPIase-like proteins have not been well-studied. However, PPIL2 was identified in a HTS as a strong binder of Aβ oligomers (Olah et al. 2011) and a positive regulator of BACE1 in vitro (Espeseth et al. 2006), but no direct work on tau has been done. Very recently, RANBP2 was shown to regulate, in part, the degradation of proteins through the 26S proteasome (Cho et al. 2014). However, roles for RANBP2 and PPWD in tau biology have not been determined. PTPA and PP5, on the other hand, have been well studied in regards to AD.
PTPA is a unique PPIase which regulates the activity of protein phosphatase-2A (PP2A) (Jordens et al. 2006, Leulliot et al. 2006). PTPA has been shown to activate PP2A, which is the main phosphatases known to dephosphorylate tau (Martin et al. 2009). However, in AD, PTPA is found to be down-regulated about 50% in the brain, and thus, through subsequent decreased PP2A activity could be a factor leading to elevated phospho-tau species (Martin et al. 2013). Moreover, there has been a strong association between PP2A regulation and GSK-3β activity (Martin et al. 2013, Yao et al. 2011). Thus, PTPA is likely to play an important role in regulating both tau and Aβ pathology.
PP5 is a TPR-containing protein, which has been described to have both PPIase and immunophilin-like activities, and has been shown to play a major role in mature GR complexes with Hsp90 (Silverstein et al. 1997). PP5 has been shown to lead to tau dephosphorylation (Gong et al. 2004, Liu et al. 2005a), and its knock-down leads to tau clearance (Jinwal et al. 2013). Moreover, hyperphosphorylation of tau has been associated with lower activity of PP5 (Liu et al. 2005b). In addition, PP5 phosphatase activity was protective against neuronal toxicity caused by Aβ (Sanchez-Ortiz et al. 2009). To date, no studies have determined the effects of PP5 modulation on tau oligomerization, but taken together, these data suggest that increasing PP5 phosphatase activity could be beneficial for both pathologies found in AD.
Conclusions
PPIases are a unique family of protein chaperones. As described above, the levels of some of these PPIases are altered in the AD brain, such as the decreases of Pin1 and FKBP12 and the increase of FKBP51. Moreover, regulation of these chaperones have been shown to affect pathology found in other neurodegenerative disorders, like Parkinson's disease (Gerard et al. 2010), amyotrophic lateral sclerosis (Manabe et al. 2002), and multiple sclerosis (Lisi et al. 2013). This suggests that there is a balance of these PPIases that becomes altered in disease. This PPIase regulation of tau is particularly important since the bending of tau from cis to trans has been shown to change the associated toxicities, with trans tau being protective compared to cis. Previously, antibodies have been made to recognize the isomerization of phospho-Thr231 tau which could be used to determine the state of pathological tau in an AD or mild cognitive impairment (Nakamura et al. 2012a). This could be important for tracking disease progression, because this phospho-tau epitope is detectable in cerebrospinal fluid (Hampel et al. 2001). Likewise, APP isomerization to the trans state prevents production of Aβ (Pastorino et al. 2006). Thus, modulating these chaperones through drugs or vaccines targeted to restore tau and amyloid processing to non-pathogenic states or to prevent pathogenic processing could be effective in preventing AD progression.
In conclusion, PPIases regulate Aβ pathology, primarily through the isomerization of APP or through protection from the associated toxicities. This regulation can alter the accumulation of Aβ oligomers and plaques, which have been shown to regulate downstream tau oligomerization. PPIases can also directly regulate tau structure and function. Thus, PPIases could regulate tau in multiple ways; by modulating Aβ production, by regulating tau phosphorylation, and by altering tau structure. Each of these changes can culminate to protect against or accelerate the toxic multimerization of tau. Therefore, proteins with PPIase activity provide a unique set of potential drug targets that structurally modify and regulate phosphorylation of proteins like tau, which contain many Pro residues.
Acknowledgements
This work was supported by NIH/NINDS R01 NS073899.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer's disease
- AIP
Aryl hydrocarbon receptor interacting protein
- Akt
protein kinase B
- APP
amyloid precursor protein
- BACE
beta-secretase
- Bcl-2
B-cell lymphoma
- CK2
casein kinase II
- Cyps
cyclophilins
- CDK5
cyclin-dependent kinase 5
- DAPK1
death-associated protein kinase 1
- FKBP
FK506-binding protein
- FTLD-17
frontotemporal lobar dementia linked to chromosome 17
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- GSK3-β
glycogen synthase kinase 3 beta
- HDAC
histone deacetylase
- HTS
high-throughput screen
- IDP
intrinsically disordered protein
- IRS-1
insulin receptor substrate 1
- KO
knock-out
- MBD
microtubule binding domain
- MAPK
mitogen-activated protein kinase
- MMP9
matrix metalloproteinase 9
- mPTP
mitochondrial permeability transport pore
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor κB
- NIMA
never in mitosis
- PI3K
phosphoinositide 3-kinase
- Pin1
protein interacting with NIMA
- PP5
protein phosphatase 5
- PPIL
PPIase-like
- PPWD1
peptidyl-prolyl isomerase containing WD40 repeat
- Psn
presenilin
- PSP
progressive supranuclear palsy
- PTPA
phosphatase two a phosphatase activator
- RANBP2
Ran-binding protein 2
- SDCCAG-10
serologically defined colon cancer antigen 10
- TPR
tetratricopeptide repeat
- WISP39
WAF-1/CIP1 Stabilizing Protein 39
- Xap2
hepatitis B virus protein X-associated protein 2
Footnotes
conflict-of-interest disclosure
The authors have no conflicts of interest to declare.
References
- Ahn J, Murphy M, Kratowicz S, Wang A, Levine AJ, George DL. Down-regulation of the stathmin/Op18 and FKBP25 genes following p53 induction. Oncogene. 1999;18:5954–5958. doi: 10.1038/sj.onc.1202986. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta neuropathologica. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Balastik M, Lim J, Pastorino L, Lu KP. Pin1 in Alzheimer's disease: multiple substrates, one regulatory mechanism? Biochimica et biophysica acta. 2007;1772:422–429. doi: 10.1016/j.bbadis.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik S. Immunophilins: for the love of proteins. Cellular and molecular life sciences : CMLS. 2006;63:2889–2900. doi: 10.1007/s00018-006-6215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann M, Giunta C, Krabichler B, et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. American journal of human genetics. 2012;90:201–216. doi: 10.1016/j.ajhg.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsma DJ, Eder C, Gross M, et al. The cyclophilin multigene family of peptidylprolyl isomerases. Characterization of three separate human isoforms. J Biol Chem. 1991;266:23204–23214. [PubMed] [Google Scholar]
- Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Structural impact of proline-directed pseudophosphorylation at AT8, AT100, and PHF1 epitopes on 441-residue tau. Journal of the American Chemical Society. 2011;133:15842–15845. doi: 10.1021/ja205836j. [DOI] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34(Suppl 1):S186–195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochimica et biophysica acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Blair LJ, Nordhues BA, Hill SE, et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. The Journal of clinical investigation. 2013;123:4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA neurology. 2014;71:505–508. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke CH, Raees MQ, Malviya S, Bradburn CA, Binder EB, Neigh GN. Glucocorticoid sensitizers Bag1 and Ppid are regulated by adolescent stress in a sex-dependent manner. Psychoneuroendocrinology. 2013;38:84–93. doi: 10.1016/j.psyneuen.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CA, Schmidt C, Poulter M, Hummerich H, Klohn PC, Jat P, Mead S, Collinge J, Lloyd SE. In vitro screen of prion disease susceptibility genes using the scrapie cell assay. Human molecular genetics. 2014;23:5102–5108. doi: 10.1093/hmg/ddu233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulbarelli A, Lonati E, Cazzaniga E, Gregori M, Masserini M. Pin1 affects Tau phosphorylation in response to Abeta oligomers. Mol Cell Neurosci. 2009;42:75–80. doi: 10.1016/j.mcn.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. Pin1 in Alzheimer's disease. Journal of neurochemistry. 2006;98:1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- Cao W, Konsolaki M. FKBP immunophilins and Alzheimer's disease: a chaperoned affair. Journal of biosciences. 2011;36:493–498. doi: 10.1007/s12038-011-9080-7. [DOI] [PubMed] [Google Scholar]
- Chambraud B, Belabes H, Fontaine-Lenoir V, Fellous A, Baulieu EE. The immunophilin FKBP52 specifically binds to tubulin and prevents microtubule formation. Faseb J. 2007;21:2787–2797. doi: 10.1096/fj.06-7667com. [DOI] [PubMed] [Google Scholar]
- Chambraud B, Sardin E, Giustiniani J, Dounane O, Schumacher M, Goedert M, Baulieu EE. A role for FKBP52 in Tau protein function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2658–2663. doi: 10.1073/pnas.0914957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KI, Patil H, Senda E, et al. Differential loss of prolyl isomerase or chaperone activity of Ran-binding protein 2 (Ranbp2) unveils distinct physiological roles of its cyclophilin domain in proteostasis. J Biol Chem. 2014;289:4600–4625. doi: 10.1074/jbc.M113.538215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Gendron TF, Scheffel K, Carlomagno Y, Dunmore J, DeTure M, Petrucelli L. Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Human molecular genetics. 2012;21:2936–2945. doi: 10.1093/hmg/dds125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TL, Walker JR, Ouyang H, MacKenzie F, Butler-Cole C, Newman EM, Eisenmesser EZ, Dhe-Paganon S. The crystal structure of human WD40 repeat-containing peptidylprolyl isomerase (PPWD1). FEBS J. 2008;275:2283–2295. doi: 10.1111/j.1742-4658.2008.06381.x. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. Journal of neuroscience research. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinski K, Muir S, Cardenas M, Heitman J. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13093–13098. doi: 10.1073/pnas.94.24.13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Wu X, Sosunov AA, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochimica et biophysica acta. 2013 doi: 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich F, Fischer G. Pharmacological targeting of catalyzed protein folding: the example of peptide bond cis/trans isomerases. Handbook of experimental pharmacology. 2006:359–404. doi: 10.1007/3-540-29717-0_15. [DOI] [PubMed] [Google Scholar]
- Edlich F, Maestre-Martinez M, Jarczowski F, Weiwad M, Moutty MC, Malesevic M, Jahreis G, Fischer G, Lucke C. A novel calmodulin-Ca2+ target recognition activates the Bcl-2 regulator FKBP38. J Biol Chem. 2007;282:36496–36504. doi: 10.1074/jbc.M705061200. [DOI] [PubMed] [Google Scholar]
- Escribano L, Simon AM, Perez-Mediavilla A, Salazar-Colocho P, Del Rio J, Frechilla D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochemical and biophysical research communications. 2009;379:406–410. doi: 10.1016/j.bbrc.2008.12.071. [DOI] [PubMed] [Google Scholar]
- Espeseth AS, Huang Q, Gates A, et al. A genome wide analysis of ubiquitin ligases in APP processing identifies a novel regulator of BACE1 mRNA levels. Mol Cell Neurosci. 2006;33:227–235. doi: 10.1016/j.mcn.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Feng M, Gu C, Ma S, Wang Y, Liu H, Han R, Gao J, Long Y, Mi H. Mouse FKBP23 mediates conformer-specific functions of BiP by catalyzing Pro117 cis/trans isomerization. Biochemical and biophysical research communications. 2011;408:537–540. doi: 10.1016/j.bbrc.2011.04.050. [DOI] [PubMed] [Google Scholar]
- Forlenza OV, Torres CA, Talib LL, de Paula VJ, Joaquim HP, Diniz BS, Gattaz WF. Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer's disease. Journal of psychiatric research. 2011;45:220–224. doi: 10.1016/j.jpsychires.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Galat A, Thai R. Rapamycin-binding FKBP25 associates with diverse proteins that form large intracellular entities. Biochemical and biophysical research communications. 2014;450:1255–1260. doi: 10.1016/j.bbrc.2014.06.105. [DOI] [PubMed] [Google Scholar]
- Ge M, Pan XM. The contribution of proline residues to protein stability is associated with isomerization equilibrium in both unfolded and folded states. Extremophiles : life under extreme conditions. 2009;13:481–489. doi: 10.1007/s00792-009-0233-7. [DOI] [PubMed] [Google Scholar]
- Ge YS, Teng WY, Zhang CD. Protective effect of cyclophilin A against Alzheimer's amyloid beta-peptide (25-35)-induced oxidative stress in PC12 cells. Chinese medical journal. 2009;122:716–724. [PubMed] [Google Scholar]
- Gerard M, Deleersnijder A, Daniels V, et al. Inhibition of FK506 binding proteins reduces alpha-synuclein aggregation and Parkinson's disease-like pathology. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2454–2463. doi: 10.1523/JNEUROSCI.5983-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustiniani J, Chambraud B, Sardin E, et al. Immunophilin FKBP52 induces Tau-P301L filamentous assembly in vitro and modulates its activity in a model of tauopathy. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4584–4589. doi: 10.1073/pnas.1402645111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giustiniani J, Sineus M, Sardin E, Dounane O, Panchal M, Sazdovitch V, Duyckaerts C, Chambraud B, Baulieu EE. Decrease of the immunophilin FKBP52 accumulation in human brains of Alzheimer's disease and FTDP-17. Journal of Alzheimer's disease : JAD. 2012;29:471–483. doi: 10.3233/JAD-2011-111895. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Annals of neurology. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gong CX, Liu F, Wu G, Rossie S, Wegiel J, Li L, Grundke-Iqbal I, Iqbal K. Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5. Journal of neurochemistry. 2004;88:298–310. doi: 10.1111/j.1471-4159.2004.02147.x. [DOI] [PubMed] [Google Scholar]
- Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F, Lu J, Fischer A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer's disease. EMBO molecular medicine. 2013;5:52–63. doi: 10.1002/emmm.201201923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Molecular and cellular endocrinology. 2007;275:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:9047–9056. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K. Tau pathology generated by overexpression of tau. The American journal of pathology. 1999;155:1781–1785. doi: 10.1016/S0002-9440(10)65494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudavicius G, Soufari H, Upadhyay SK, Mackereth CD, Nelson CJ. Resolving the functions of peptidylprolyl isomerases: insights from the mutagenesis of the nuclear FKBP25 enzyme. Biochemical Society transactions. 2013;41:761–768. doi: 10.1042/BST20130013. [DOI] [PubMed] [Google Scholar]
- Guo L, Du H, Yan S, Wu X, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer's neurons. PloS one. 2013;8:e54914. doi: 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GS, Steiner JP. Immunophilins: beyond immunosuppression. Journal of medicinal chemistry. 1998;41:5119–5143. doi: 10.1021/jm980307x. [DOI] [PubMed] [Google Scholar]
- Hampel H, Buerger K, Kohnken R, Teipel SJ, Zinkowski R, Moeller HJ, Rapoport SI, Davies P. Tracking of Alzheimer's disease progression with cerebrospinal fluid tau protein phosphorylated at threonine 231. Annals of neurology. 2001;49:545–546. [PubMed] [Google Scholar]
- Hardy J. The relationship between amyloid and tau. Journal of molecular neuroscience : MN. 2003;20:203–206. doi: 10.1385/JMN:20:2:203. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science (New York, N.Y.) 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Wagner KV, Liebl C, et al. The involvement of FK506-binding protein 51 (FKBP5) in the behavioral and neuroendocrine effects of chronic social defeat stress. Neuropharmacology. 2012;62:332–339. doi: 10.1016/j.neuropharm.2011.07.041. [DOI] [PubMed] [Google Scholar]
- He Z, Li L, Luan S. Immunophilins and parvulins. Superfamily of peptidyl prolyl isomerases in Arabidopsis. Plant physiology. 2004;134:1248–1267. doi: 10.1104/pp.103.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LL, Zhao XM, Huang CQ, Yu L, Xia ZX. Structure of recombinant human cyclophilin J, a novel member of the cyclophilin family. Acta crystallographica. Section D, Biological crystallography. 2005;61:316–321. doi: 10.1107/S0907444904033189. [DOI] [PubMed] [Google Scholar]
- Ikura T, Ito N. Peptidyl-prolyl isomerase activity of FK506 binding protein 12 prevents tau peptide from aggregating. Protein engineering, design & selection : PEDS. 2013;26:539–546. doi: 10.1093/protein/gzt033. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Bachinger HP. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J Biol Chem. 2014;289:18189–18201. doi: 10.1074/jbc.M114.561944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang LK, Lee ZH, Kim HH, Hill JM, Kim JD, Kwon BS. A novel leucine-rich repeat protein (LRR-1): potential involvement in 4-1BB-mediated signal transduction. Molecules and cells. 2001;12:304–312. [PubMed] [Google Scholar]
- Jiang CH, Tsien JZ, Schultz PG, Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1930–1934. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Koren J, 3rd, Borysov SI, et al. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:591–599. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinwal UK, Koren J, 3rd, Dickey CA. Reconstructing the Hsp90/Tau Machine. Current enzyme inhibition. 2013;9:41–45. doi: 10.2174/1573408011309010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens J, Janssens V, Longin S, et al. The protein phosphatase 2A phosphatase activator is a novel peptidyl-prolyl cis/trans-isomerase. J Biol Chem. 2006;281:6349–6357. doi: 10.1074/jbc.M507760200. [DOI] [PubMed] [Google Scholar]
- Kamada K, Yamashita T, Hatcho K, Adachi A, Nomaguchi M. Evasion from CypA- and APOBEC-mediated restrictions is insufficient for HIV-1 to efficiently grow in simian cells. Microbes and infection / Institut Pasteur. 2009;11:164–171. doi: 10.1016/j.micinf.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science (New York, N.Y.) 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kekatpure VD, Dannenberg AJ, Subbaramaiah K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem. 2009;284:7436–7445. doi: 10.1074/jbc.M808999200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kim BM, You MH, Chen CH, Lee S, Hong Y, Hong Y, Kimchi A, Zhou XZ, Lee TH. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell death & disease. 2014;5:e1237. doi: 10.1038/cddis.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Choi TG, Ding Y, et al. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. Journal of cell science. 2008;121:3636–3648. doi: 10.1242/jcs.028654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Duffy LK, Dowling MM, Abraham C, McCluskey A, Selkoe DJ. Microtubule-associated protein 2: monoclonal antibodies demonstrate the selective incorporation of certain epitopes into Alzheimer neurofibrillary tangles. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:7941–7945. doi: 10.1073/pnas.81.24.7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laenger A, Lang-Rollin I, Kozany C, Zschocke J, Zimmermann N, Ruegg J, Holsboer F, Hausch F, Rein T. XAP2 inhibits glucocorticoid receptor activity in mammalian cells. FEBS letters. 2009;583:1493–1498. doi: 10.1016/j.febslet.2009.03.072. [DOI] [PubMed] [Google Scholar]
- Lane-Guermonprez L, Morot-Gaudry-Talarmain Y, Meunier FM, O'Regan S, Onofri F, Le Caer JP, Benfenati F. Synapsin associates with cyclophilin B in an ATP- and cyclosporin A-dependent manner. Journal of neurochemistry. 2005;93:1401–1411. doi: 10.1111/j.1471-4159.2005.03125.x. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. Faseb J. 2012;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Molecular biology of the cell. 2009;20:1533–1544. doi: 10.1091/mbc.E08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. The Journal of cell biology. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N, Vicentini G, Jordens J, Quevillon-Cheruel S, Schiltz M, Barford D, van Tilbeurgh H, Goris J. Crystal structure of the PP2A phosphatase activator: implications for its PP2A-specific PPIase activity. Molecular cell. 2006;23:413–424. doi: 10.1016/j.molcel.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Li TK, Baksh S, Cristillo AD, Bierer BE. Calcium- and FK506-independent interaction between the immunophilin FKBP51 and calcineurin. Journal of cellular biochemistry. 2002;84:460–471. doi: 10.1002/jcb.10026. [DOI] [PubMed] [Google Scholar]
- Li Y, Grupe A, Rowland C, et al. DAPK1 variants are associated with Alzheimer's disease and allele-specific expression. Human molecular genetics. 2006;15:2560–2568. doi: 10.1093/hmg/ddl178. [DOI] [PubMed] [Google Scholar]
- Lim J, Balastik M, Lee TH, et al. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. The Journal of clinical investigation. 2008;118:1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnert M, Lin YJ, Manns A, Haupt K, Paschke AK, Fischer G, Weiwad M, Lucke C. The FKBP-type domain of the human aryl hydrocarbon receptor-interacting protein reveals an unusual Hsp90 interaction. Biochemistry. 2013;52:2097–2107. doi: 10.1021/bi301649m. [DOI] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends in biochemical sciences. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi L, McGuire S, Sharp A, Chiosis G, Navarra P, Feinstein DL, Dello Russo C. The novel HSP90 inhibitor, PU-H71, suppresses glial cell activation but weakly affects clinical signs of EAE. Journal of neuroimmunology. 2013;255:1–7. doi: 10.1016/j.jneuroim.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. The European journal of neuroscience. 2005a;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, Gong CX. Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer's disease. J Biol Chem. 2005b;280:1790–1796. doi: 10.1074/jbc.M410775200. [DOI] [PubMed] [Google Scholar]
- Liu FL, Liu TY, Kung FL. FKBP12 regulates the localization and processing of amyloid precursor protein in human cell lines. Journal of biosciences. 2014;39:85–95. doi: 10.1007/s12038-013-9400-1. [DOI] [PubMed] [Google Scholar]
- Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- Luo J, Ma J, Yu DY, Bu F, Zhang W, Tu LH, Wei Q. Infusion of FK506, a specific inhibitor of calcineurin, induces potent tau hyperphosphorylation in mouse brain. Brain Res Bull. 2008;76:464–468. doi: 10.1016/j.brainresbull.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Ma SL, Pastorino L, Zhou XZ, Lu KP. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J Biol Chem. 2012;287:6969–6973. doi: 10.1074/jbc.C111.298596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur MW, Thornton JM. Influence of proline residues on protein conformation. Journal of molecular biology. 1991;218:397–412. doi: 10.1016/0022-2836(91)90721-h. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Annals of neurology. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neuroscience research. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Manabe Y, Warita H, Murakami T, Shiote M, Hayashi T, Omori N, Nagano I, Shoji M, Abe K. Early decrease of the immunophilin FKBP 52 in the spinal cord of a transgenic model for amyotrophic lateral sclerosis. Brain research. 2002;935:124–128. doi: 10.1016/s0006-8993(02)02466-6. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR. Cellular functions of immunophilins. Physiol Rev. 1996;76:631–649. doi: 10.1152/physrev.1996.76.3.631. [DOI] [PubMed] [Google Scholar]
- Martin L, Magnaudeix A, Esclaire F, Yardin C, Terro F. Inhibition of glycogen synthase kinase-3beta downregulates total tau proteins in cultured neurons and its reversal by the blockade of protein phosphatase-2A. Brain research. 2009;1252:66–75. doi: 10.1016/j.brainres.2008.11.057. [DOI] [PubMed] [Google Scholar]
- Martin M, Geudens I, Bruyr J, et al. PP2A regulatory subunit Balpha controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. The EMBO journal. 2013;32:2491–2503. doi: 10.1038/emboj.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. Journal of neuropathology and experimental neurology. 1999;58:188–197. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Mueller JW, Bayer P. Small family with key contacts: par14 and par17 parvulin proteins, relatives of pin1, now emerge in biomedical research. Perspectives in medicinal chemistry. 2008;2:11–20. doi: 10.4137/pmc.s496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JW, Kessler D, Neumann D, Stratmann T, Papatheodorou P, Hartmann-Fatu C, Bayer P. Characterization of novel elongated Parvulin isoforms that are ubiquitously expressed in human tissues and originate from alternative transcription initiation. BMC molecular biology. 2006;7:9. doi: 10.1186/1471-2199-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukrasch MD, Biernat J, von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J Biol Chem. 2005;280:24978–24986. doi: 10.1074/jbc.M501565200. [DOI] [PubMed] [Google Scholar]
- Nakajima O, Nakamura F, Yamashita N, et al. FKBP133: a novel mouse FK506-binding protein homolog alters growth cone morphology. Biochemical and biophysical research communications. 2006;346:140–149. doi: 10.1016/j.bbrc.2006.05.113. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012a;149:232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kosugi I, Lee DY, Hafner A, Sinclair DA, Ryo A, Lu KP. Prolyl isomerase Pin1 regulates neuronal differentiation via beta-catenin. Molecular and cellular biology. 2012b;32:2966–2978. doi: 10.1128/MCB.05688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Zhen Zhou X, Ping Lu K. Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion. 2013;7:117–120. doi: 10.4161/pri.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigam SK, Jin YJ, Jin MJ, Bush KT, Bierer BE, Burakoff SJ. Localization of the FK506-binding protein, FKBP 13, to the lumen of the endoplasmic reticulum. The Biochemical journal. 1993;294(Pt 2):511–515. doi: 10.1042/bj2940511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro P, Pompilio G, Capogrossi MC. Cyclophilin A: a key player for human disease. Cell death & disease. 2013;4:e888. doi: 10.1038/cddis.2013.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum JM, Schilling S, Cynis H, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary JC, 3rd, Dharia S, Blair LJ, et al. A New Anti-Depressive Strategy for the Elderly: Ablation of FKBP5/FKBP51. PloS one. 2011;6:e24840. doi: 10.1371/journal.pone.0024840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochocka AM, Kampanis P, Nicol S, Allende-Vega N, Cox M, Marcar L, Milne D, Fuller-Pace F, Meek D. FKBP25, a novel regulator of the p53 pathway, induces the degradation of MDM2 and activation of p53. FEBS letters. 2009;583:621–626. doi: 10.1016/j.febslet.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Oh Y, Kim EY, Kim Y, et al. Neuroprotective effects of overexpressed cyclophilin B against Abeta-induced neurotoxicity in PC12 cells. Free radical biology & medicine. 2011;51:905–920. doi: 10.1016/j.freeradbiomed.2011.05.036. [DOI] [PubMed] [Google Scholar]
- Olah J, Vincze O, Virok D, et al. Interactions of pathological hallmark proteins: tubulin polymerization promoting protein/p25, beta-amyloid, and alpha-synuclein. J Biol Chem. 2011;286:34088–34100. doi: 10.1074/jbc.M111.243907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino L, Ma SL, Balastik M, Huang P, Pandya D, Nicholson L, Lu KP. Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AbetaPP. Journal of Alzheimer's disease : JAD. 2012;30:277–297. doi: 10.3233/JAD-2012-111259. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Pirkl F, Buchner J. Functional analysis of the Hsp90-associated human peptidyl prolyl cis/trans isomerases FKBP51, FKBP52 and Cyp40. Journal of molecular biology. 2001;308:795–806. doi: 10.1006/jmbi.2001.4595. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Pushkarsky T, Yurchenko V, Vanpouille C, Brichacek B, Vaisman I, Hatakeyama S, Nakayama KI, Sherry B, Bukrinsky MI. Cell surface expression of CD147/EMMPRIN is regulated by cyclophilin 60. J Biol Chem. 2005;280:27866–27871. doi: 10.1074/jbc.M503770200. [DOI] [PubMed] [Google Scholar]
- Ratajczak T, Carrello A. Cyclophilin 40 (CyP-40), mapping of its hsp90 binding domain and evidence that FKBP52 competes with CyP-40 for hsp90 binding. J Biol Chem. 1996;271:2961–2965. doi: 10.1074/jbc.271.6.2961. [DOI] [PubMed] [Google Scholar]
- Reiniger L, Lukic A, Linehan J, Rudge P, Collinge J, Mead S, Brandner S. Tau, prions and Abeta: the triad of neurodegeneration. Acta neuropathologica. 2011;121:5–20. doi: 10.1007/s00401-010-0691-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JM, Wong CT, Clarke J. Coupled folding and binding of the disordered protein PUMA does not require particular residual structure. Journal of the American Chemical Society. 2014;136:5197–5200. doi: 10.1021/ja4125065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roperch JP, Alvaro V, Prieur S, et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat Med. 1998;4:835–838. doi: 10.1038/nm0798-835. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ortiz E, Hahm BK, Armstrong DL, Rossie S. Protein phosphatase 5 protects neurons against amyloid-beta toxicity. Journal of neurochemistry. 2009;111:391–402. doi: 10.1111/j.1471-4159.2009.06337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanokawa-Akakura R, Cao W, Allan K, et al. Control of Alzheimer's amyloid beta toxicity by the high molecular weight immunophilin FKBP52 and copper homeostasis in Drosophila. PloS one. 2010;5:e8626. doi: 10.1371/journal.pone.0008626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Huang R, Martin JA, Henley J, Mawal-Dewan M, Hurtig HI, Lee VM, Trojanowski JQ. Neurofibrillary tangles in progressive supranuclear palsy contain the same tau epitopes identified in Alzheimer's disease PHFtau. Journal of neuropathology and experimental neurology. 1996;55:534–539. doi: 10.1097/00005072-199605000-00006. [DOI] [PubMed] [Google Scholar]
- Schulke JP, Wochnik GM, Lang-Rollin I, Gassen NC, Knapp RT, Berning B, Yassouridis A, Rein T. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PloS one. 2010;5:e11717. doi: 10.1371/journal.pone.0011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- Schweiger R, Soll J, Jung K, Heermann R, Schwenkert S. Quantification of interaction strengths between chaperones and tetratricopeptide repeat domain-containing membrane proteins. J Biol Chem. 2013;288:30614–30625. doi: 10.1074/jbc.M113.493015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenica ML, Benner L, Housley SB, et al. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimer's research & therapy. 2014;6:12. doi: 10.1186/alzrt241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nature cell biology. 2003;5:28–37. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Galigniana MD, Chen MS, Owens-Grillo JK, Chinkers M, Pratt WB. Protein phosphatase 5 is a major component of glucocorticoid receptor.hsp90 complexes with properties of an FK506-binding immunophilin. J Biol Chem. 1997;272:16224–16230. doi: 10.1074/jbc.272.26.16224. [DOI] [PubMed] [Google Scholar]
- Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:868–873. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldin SJ, Murthy JN, Donnelly JG, Chen Y, Goodyear N. Immunophilin receptors for immunosuppressive drugs. Therapeutic drug monitoring. 1993;15:468–471. doi: 10.1097/00007691-199312000-00002. [DOI] [PubMed] [Google Scholar]
- Stegmann CM, Luhrmann R, Wahl MC. The crystal structure of PPIL1 bound to cyclosporine A suggests a binding mode for a linear epitope of the SKIP protein. PloS one. 2010;5:e10013. doi: 10.1371/journal.pone.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JP, Dawson TM, Fotuhi M, Glatt CE, Snowman AM, Cohen N, Snyder SH. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature. 1992;358:584–587. doi: 10.1038/358584a0. [DOI] [PubMed] [Google Scholar]
- Sugata H, Matsuo K, Nakagawa T, Takahashi M, Mukai H, Ono Y, Maeda K, Akiyama H, Kawamata T. A peptidyl-prolyl isomerase, FKBP12, accumulates in Alzheimer neurofibrillary tangles. Neuroscience letters. 2009;459:96–99. doi: 10.1016/j.neulet.2009.04.062. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu X, Peng Y, Goh JY, Liou YC, Lin D, Zhao Y. Solution structural analysis of the single-domain parvulin TbPin1. PloS one. 2012;7:e43017. doi: 10.1371/journal.pone.0043017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shimazaki H, Kimura M, Izuta H, Tsuruma K, Shimazawa M, Hara H. Apoptosis-inducing factor and cyclophilin A cotranslocate to the motor neuronal nuclei in amyotrophic lateral sclerosis model mice. CNS neuroscience & therapeutics. 2011;17:294–304. doi: 10.1111/j.1755-5949.2010.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatro ET, Everall IP, Kaul M, Achim CL. Modulation of glucocorticoid receptor nuclear translocation in neurons by immunophilins FKBP51 and FKBP52: implications for major depressive disorder. Brain research. 2009;1286:1–12. doi: 10.1016/j.brainres.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theillet F-X, Kalmar L, Tompa P, Han K-H, Selenko P, Dunker AK, Daughdrill GW, Uversky VN. The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disordered Proteins. 2013;1:e24360. doi: 10.4161/idp.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivellin G, Korbonits M. AIP and its interacting partners. The Journal of endocrinology. 2011;210:137–155. doi: 10.1530/JOE-11-0054. [DOI] [PubMed] [Google Scholar]
- Uchida T, Fujimori F, Tradler T, Fischer G, Rahfeld JU. Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl prolyl cis/trans isomerase. FEBS letters. 1999;446:278–282. doi: 10.1016/s0014-5793(99)00239-2. [DOI] [PubMed] [Google Scholar]
- van de Hoef DL, Bonner JM, Boulianne GL. FKBP14 is an essential gene that regulates Presenilin protein levels and Notch signaling in Drosophila. Development. 2013;140:810–819. doi: 10.1242/dev.081356. [DOI] [PubMed] [Google Scholar]
- van de Hoef DL, Hughes J, Livne-Bar I, Garza D, Konsolaki M, Boulianne GL. Identifying genes that interact with Drosophila presenilin and amyloid precursor protein. Genesis. 2009;47:246–260. doi: 10.1002/dvg.20485. [DOI] [PubMed] [Google Scholar]
- Wang C, Yu JT, Miao D, Wu ZC, Tan MS, Tan L. Targeting the mTOR signaling network for Alzheimer's disease therapy. Molecular neurobiology. 2014;49:120–135. doi: 10.1007/s12035-013-8505-8. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Nakaya Y, Du Z, et al. Interaction of presenilins with FKBP38 promotes apoptosis by reducing mitochondrial Bcl-2. Human molecular genetics. 2005;14:1889–1902. doi: 10.1093/hmg/ddi195. [DOI] [PubMed] [Google Scholar]
- Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederrecht G, Brizuela L, Elliston K, Sigal NH, Siekierka JJ. FKB1 encodes a nonessential FK 506-binding protein in Saccharomyces cerevisiae and contains regions suggesting homology to the cyclophilins. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:1029–1033. doi: 10.1073/pnas.88.3.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- Yao XQ, Zhang XX, Yin YY, et al. Glycogen synthase kinase-3beta regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. The Biochemical journal. 2011;437:335–344. doi: 10.1042/BJ20110347. [DOI] [PubMed] [Google Scholar]
- Yao Y, Chinnici C, Tang H, Trojanowski JQ, Lee VM, Pratico D. Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. Journal of neuroinflammation. 2004;1:21. doi: 10.1186/1742-2094-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nakatsu Y, Shinjo T, et al. Par14 protein associates with insulin receptor substrate 1 (IRS-1), thereby enhancing insulin-induced IRS-1 phosphorylation and metabolic actions. J Biol Chem. 2013;288:20692–20701. doi: 10.1074/jbc.M113.485730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang Y, Li H, Zhang W, Wu D, Mi H. The mouse FKBP23 binds to BiP in ER and the binding of C-terminal domain is interrelated with Ca2+ concentration. FEBS letters. 2004;559:57–60. doi: 10.1016/S0014-5793(04)00024-9. [DOI] [PubMed] [Google Scholar]
- Zhao L, Huang W, Liu H, Wang L, Zhong W, Xiao J, Hu Y, Li S. FK506-binding protein ligands: structure-based design, synthesis, and neurotrophic/neuroprotective properties of substituted 5,5-dimethyl-2-(4-thiazolidine)carboxylates. Journal of medicinal chemistry. 2006;49:4059–4071. doi: 10.1021/jm0502384. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Kops O, Werner A, et al. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Molecular cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]