

Abstract
The evolutionarily conserved DNA topoisomerase II beta-binding protein 1 (TopBP1) functions in DNA replication, DNA damage response, and cell survival. We analyzed the role of TopBP1 in human and bovine papillomavirus genome replication. Consistent with prior reports, TopBP1 co-localized in discrete nuclear foci and was in complex with papillomavirus E2 protein. Similar to E2, TopBP1 is recruited to the region of the viral origin of replication during G1/S and early S phase. TopBP1 knockdown increased, while over-expression decreased transient virus replication, without affecting cell cycle. Similarly, using cell lines harboring HPV-16 or HPV-31 genome, TopBP1 knockdown increased while over-expression reduced viral copy number relative to genomic DNA. We propose a model in which TopBP1 serves dual roles in viral replication: it is essential for initiation of replication yet it restricts viral copy number.
Keywords: papillomavirus, TopBP1, HPV, BPV, replication, E2, cell cycle, copy number
Introduction
Papillomaviruses are approximately 8 kb double stranded DNA viruses that infect basal cells of stratified epithelia and productively amplify in differentiated suprabasal cells (Bedell et al., 1991; Howley, 1996). Human papillomavirus (HPV) genomes replicate as low copy number autonomous episomes. Similarly, bovine papillomavirus 1 (BPV-1) transforms murine cells and is maintained as a stable multicopy nuclear plasmid (Law et al., 1981). Virally encoded E1 and E2 proteins are essential for its replication during S phase (Chiang et al., 1992; Mohr et al., 1990; Ustav and Stenlund, 1991), which depends on multiple host proteins such as DNA polymerase δ, Replication Protein A and Brd4 (Park et al., 1994; Wang et al., 2013). The E1 protein is a sequence specific DNA binding helicase (Hughes and Romanos, 1993; Yang et al., 1993). The E2 protein binds to specific DNA motifs in the viral regulatory region and is a transcriptional activator (Androphy et al., 1987; Phelps and Howley, 1987). E2 binds to E1; the transcriptional activity of E2 is not essential for viral replication (Grossel et al., 1996; Mohr et al., 1990). While metazoan origins typically span 50–250 kb, viral origins are defined by short sequences of less than 100 bp. The E2–E1 complex recognizes their respective adjacent motifs and this element acts as an origin of viral replication (ori). The subsequent ATP hydrolysis by E1 causes the E2 protein to dissociate and permits formation of hexameric E1 molecules on the ori (Abbate et al., 2004; Sanders and Stenlund, 1998).
Topoisomerase II- binding protein 1 (TopBP1), also known as Dpb11, Rad4, Cut5 (Yeast), Mus101 (Drosophila), is a functionally conserved protein with a BRCA1- C terminus (BRCT) domain [reviewed in (Garcia et al., 2005)]. Vertebrate TopBP1 participates in DNA replication and DNA damage repair processes (Balestrini et al., 2010; Kumagai et al., 2010; Makiniemi et al., 2001), is essential for cell survival (Yamane et al., 2002), and is required for embryonic development (Jeon et al., 2011). TopBP1 is also necessary for maintenance of genomic integrity (Kim et al., 2005) and has several roles in the G1/S transition such as loading of replication components and activation of CycE/Cdk2 (Jeon et al., 2007). Elevated expression of TopBP1 is believed to cause aggressive tumors by modulating the p53 pathway (Liu et al., 2009). TopBP1 is capable of activating the DNA damage response pathway (DDR) protein ATR (Kumagai et al., 2006) and is known to interact with Claspin to promote Chk1 activity (Liu et al., 2006).
The HPV-16 E2 protein is known to associate with TopBP1 and this interaction increases the transcriptional and replication activity of E2 protein (Boner et al., f). TopBP1 is believed to act as a chromatin receptor of E2 protein especially in late stages of mitosis (Donaldson et al., 2007). An HPV-31 genome with an E2 mutation crippled for TopBP1 binding exhibited reduced replicative ability (Donaldson et al., 2012). In U2OS cells transfected with HPV18 genome, TopBP1 co-localized with E1 containing foci suggesting association of TopBP1 with viral replication centers (Reinson et al., 2013). These results imply co-operation between HPV proteins and TopBP1 in facilitating viral replication. Although the DDR is activated by HPV E1 and E7 proteins (Banerjee et al., 2011; Fradet-Turcotte A, 2011; Sakakibara et al., 2011), chemical inhibitors of the DDR pathway did not alter HPV replication (King et al., 2010; Reinson et al., 2013). In this context we explored the role of TopBP1 in HPV and BPV replication by modulating the levels of TopBP1 in different cell types.
Materials and Methods
Cell culture, Plasmids, Chemicals
C127, BPV-A3 murine C127 cells with BPV-1 episomes (Voitenleitner and Botchan, 2002), C33A, and J2 3T3 cells were maintained in DMEM with 10% FBS and 1× Pen/Strep. H1299 cells were maintained in RPMI media with 10% FBS having 1× Pen/Strep. HPV-BP cells (Sprague et al., 2002) that maintain 10–50 copies of HPV-16 episomes were cultured in Keratinocyte Serum Free media (K-SFM) supplemented with bovine pituitary extract, EGF (Invitrogen) and 1× Pen/Strep. CIN612-9E cells (De Geest et al., 1993) that retain HPV-31 episomes were grown in E-Media on J2 3T3 feeders pre-treated with 5 μg/ml mitomycin C as described (Bedell et al., 1991). Cells were transfected with Lipofectamine 2000 (C33A, H1299, BPV-A3) or with FugeneHD (HPV-BP, CIN612) at 1:1 and at 4:1 ratio to DNA, respectively. We obtained plasmids HPV-31 E1, E2, pFLORI31 and pCI-Rluc (Fradet-Turcotte et al., 2010a), pSUPER-siScramble (knockdown control), pSUPER-siTopBP1 and full length human Flag-TopBP1 (Liu et al., 2004). The TopBP1 knockdown constructs used are human specific. pCG-BPV-E2 has been reported (Grossel et al., 1996). We used empty pcDNA3 as vector control. Mimosine and nocodazole were procured from Sigma, and thymidine was obtained from Alfa Aesar (Ward Hill, MA).
Antibodies
BPV anti-E2 (B201, II–I), anti-EE antibodies (Wang et al., 2009), and rabbit anti-TopBP1 (Donaldson et al., 2007) have been reported. Commercial antibodies used were anti-Actin, (Sigma Aldrich) and mouse anti-TopBP1 (BD Transduction Laboratories, 611875).
Luciferase assay for viral replication
Luciferase based replication protocols essentially followed as described (Fradet-Turcotte et al., 2010b). Briefly, 80 ng each of HPV-31 E1, E2 and pFLORI31 constructs were co-transfected along with 1 μg of TopBP1 shRNA (pSuper-siTopBP1) or over-expression (Flag-TopBP1) vectors or their respective controls (pSUPER-siSCramble, or pCDNA3) in 12 well plate. For normalization, pCI-RLuc construct was included at 16 ng per well. Cells were lysed in Dual-Glo luciferase reagent (Promega) and both firefly and renilla luciferase activities were determined using PHERAStar FS (BMG Labtech). Luciferase activities were measured 48 h and 72 h after transfection. Relative replication was expressed by the ratio of firefly to renilla luciferase activity normalized to vector control.
Viral genome copy number determination by real time PCR
Cells were transfected with 2 μg of TopBP1 over-expression or knockdown constructs or their respective controls. 72 h post-transfection, HPV-BP or CIN612 cells were lysed in 10 mM Tris – 1 mM EDTA (pH 8.0) with 0.1% SDS and 1× protease inhibitor cocktail, DNA was isolated using standard phenol:chloroform (PCA) method (Sambrook and Russell, 2006). Real time PCR was performed with a BioRad CFX using Sso Fast Evagreen mastermix (BioRad) according to manufacturer’s protocol. The PCR primer sets were HPV-16 LCR: forward 5′ GGGTGTGTGCAAACCGTTTTGGGTTA 3′ and reverse 5′ CCGATTTCGGTTACGCCCTTAGTTT 3′; HPV-31 LCR: forward 5′ CCTGCTCCTCCCAATAGTCAT 3′ and reverse 5′ AAACGGACCGGGTGTACA 3′. DNA primer set #23 (Mendoza-Maldonado et al., 2010) was used for genomic DNA copy number determination. Results were analyzed using CFX manager software (BioRad).
Chromatin Immunoprecipitation (ChIP) assay
BPV-A3 or HPV-BP cells were synchronized as follows. For double thymidine block, cells were kept in 2.0 mM thymidine overnight, released in the morning and again blocked overnight with 2.0 mM thymidine. For the mimosine block, cells were treated at 2 μM overnight. For G2 arrest, cells were treated at 100 nM of nocodazole overnight. After treatment, ChIP was performed as described in (Yu et al., 2007). Primer sets used for BPV-LCR were forward 5′ GCCTTCTCAGCCAAAATGAA 3′ and reverse 5′ TGAGGTTTTGATCCGCCTAC 3′
Flow cytometry
Synchronized cells were fixed in 90% ethanol and stored in 4°C. The following day, cells were washed in PBS, treated with 50 μg/ml of RNase A- 50 μg/ml Propidium iodide for 20 min, run on FACS Caliber and analyzed with FlowJo software. To test the effect of TopBP1 protein levels on cell cycle, C33A or H1299 cells were transfected with either over-expression or knockdown plasmids. After 48 h, cells were harvested and processed for flow cytometry.
Western blot of TopBP1
H1299 or C33A cells were transfected with pSUPER-siTopBP1 or pSUPER-siScramble at 3 μg per well of a six well plate. After 48 h, cells were lysed in urea buffer (6 M urea, 100 mM NaH2PO4, and 20 mM Tris pH-6.8). 20 μg of total protein were run on a 4–15% gradient gel (BioRad) and transferred onto PVDF membranes. Rabbit anti-TopBP1 (R1180) antibody was used at 1:2000, and anti-Actin antibody at 1:1000 dilutions. For over-expression studies, H1299 cells were transfected with vector control or Flag-TopBP1 plasmids at 3 μg per well of a six well plate.
In situ proximity ligation assay (PLA)
In situ PLA was performed using the PLA Red kit (Olink Biosciences). BPV-A3 cells were first blocked in S phase by double thymidine treatment, fixed in 4% para-formaldehyde for 10 min, permeabilized for 15 min in 0.5% Triton-X 100/PBS, washed in PBS, blocked with 5% goat serum in 0.2% Triton- X 100/PBS, then incubated overnight with primary antibody combinations (E2-TopBP1 or E1-E2) at 4°C . The PLA then followed the manufacturer’s protocol. Briefly, cover slips were washed in buffer A, incubated with PLA probe PLUS and MINUS for one hour at 37°C, washed twice and the probes were ligated for 30 min. Amplification was performed for 100 min at 37°C, washed twice in buffer A and B. Cover slips were mounted in Duolink in situ mounting media. Cells were analyzed with a Nikon microscope. In case of C127 cells, eGFP was co-transfected with or without pCG-E2. PLA signals were counted only from green fluorescent cells. Two independent transfections were carried out, the counts were averaged and calculated as percentage of transfected cells displaying PLA foci.
Statistical analysis
Unless specifically mentioned, all experiments were repeated thrice and results were averaged. Error bars denote the standard deviation. Statistical significance was determined using Student’s t-test.
Results
TopBP1 co-localizes with BPV E2 in nuclear foci
As it was previously reported that TopBP1 co-immunoprecipitates with E2, we sought to further characterize this interaction in vivo. To confirm the protein-protein interaction between TopBP1 and E2, in situ proximity ligation assays (PLA) were performed (Leuchowius et al., 2011). PLA is a modified immunofluorescence assay in which secondary antibody is conjugated to specific DNA sequences. When two secondary antibodies are in close proximity (~ 40 nm), DNA can be ligated, amplified and detected by hybridization with specific fluorescent probes. In BPV-A3 cells, we detected the in vivo interaction between the E2 protein and endogenous TopBP1 as demonstrated by formation of fluorescent spots inside the nucleus. We also observed similar interaction between E1 and E2 (Figure 1A). These cells harbor hundreds of copies of BPV genome due to high levels of an E2 protein with three serine to alanine mutations (McBride and Howley, 1991). We also repeated the assay in C127 cells transfected with (or without) BPV-E2. C127 cells transfected with BPV-E2 became positive in PLA reactions with TopBP1 (Figure 1B). Transfection efficiency was normalized to GFP expression and PLA signals were counted in transfected cells. As shown in the graph, not all C127 cells showed the PLA signal, possibly because of low level or a transient interaction between E2-TopBP1. More than 30% of the cells with PLA signal had more than one such fluorescent puncta.
Figure 1.

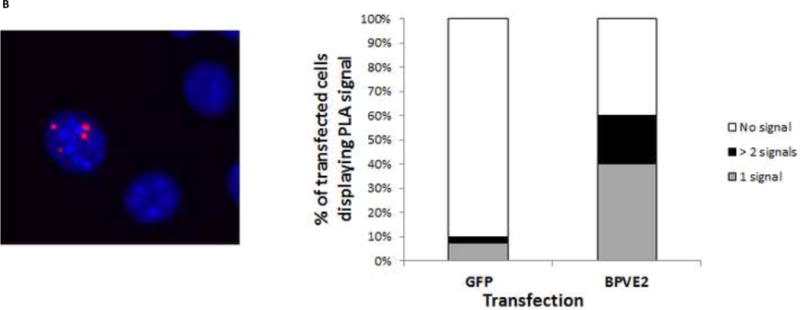
In vivo co-localization of BPV-E2 with TopBP1. (A) In situ proximity ligation assay (PLA) between E1 – E2 (left) and TopBP1 – E2 (right) in BPV-A3 cells synchronized by double-thymidine block. Fluorescent spots indicate interaction based PCR amplification occurring in the nucleus (stained blue). (B) PLA of transfected BPV E2 and endogenous TopBP1 in C127 cells synchronized by double thymidine. Fluorescent spot indicates interaction based PCR amplification. Graph shows the percentage distribution of PLA signals in GFP expressing transfected C127 cells.
TopBP1 is localized near the BPV and HPV ori in G1/S and early S phase
We next investigated whether TopBP1 is recruited to the viral genome. BPV-A3 cells harboring replicating BPV genomes were synchronized in different stages of cell cycle using mimosine, double thymidine or nocodazole. Cell cycle profiles were analyzed by flow cytometry. As shown in the inset of Figure 2A, cells accumulated in G1/S, early S and G2 after mimosine, double thymidine and nocodazole treatment respectively. Since papillomavirus oncoproteins are known to abrogate cell-cycle checkpoints (Heilman et al., 2009; Liu et al., 2007), we observed that BPV-A3 cells were not completely arrested and a small portion of cells progressed through cell cycle. ChIP was performed using BPV E2, TopBP1 or control EE antibodies. As expected (Melanson and Androphy, 2009), BPV E2 was present at the LCR region at very high levels in mimosine and double thymidine synchronized cells. Compared to the EE antibody control, TopBP1 ChIP was increased by 40 fold in mimosine treated G1/S cells and by 12 fold in double thymidine treated early S phase cells. Levels slightly above background (EE antibody) were detected in nocodazole treated mitotic arrested cells (Figure 2A). These results confirm enrichment of TopBP1 near the viral origin in G1/S or early S phases. TopBP1 is similarly localized to the LCR region in HPV-BP cells harboring episomal HPV-16 genome (Figure 2B) in a cell cycle dependent manner. The fold change in HPV-BP was not as high as that of BPV, likely due to antibody affinity and/or copy number difference. Our results also reaffirm that the virus replication is in synchrony with cell cycle and initiated in S phase during episomal maintenance. We could not detect the endogenous HPV-16 E2 protein in HPV-BP cells due to lack of a specific antibody.
Figure 2.
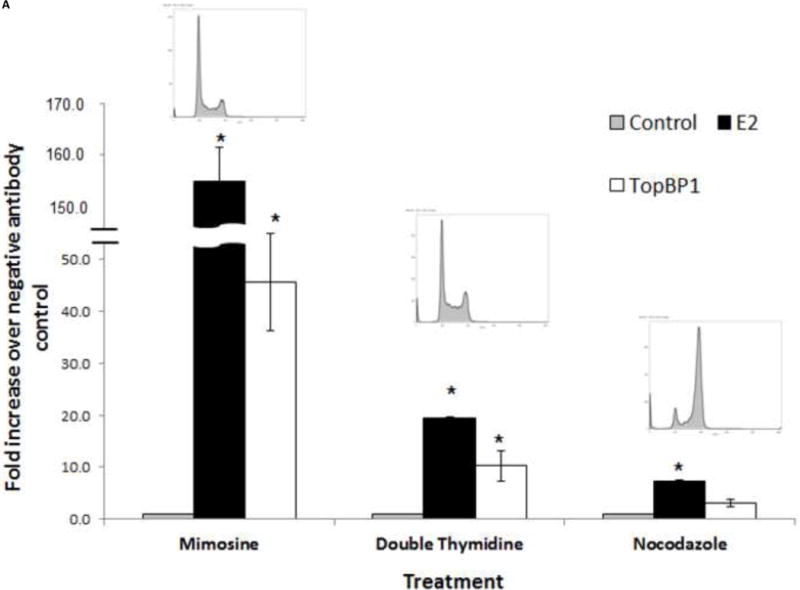
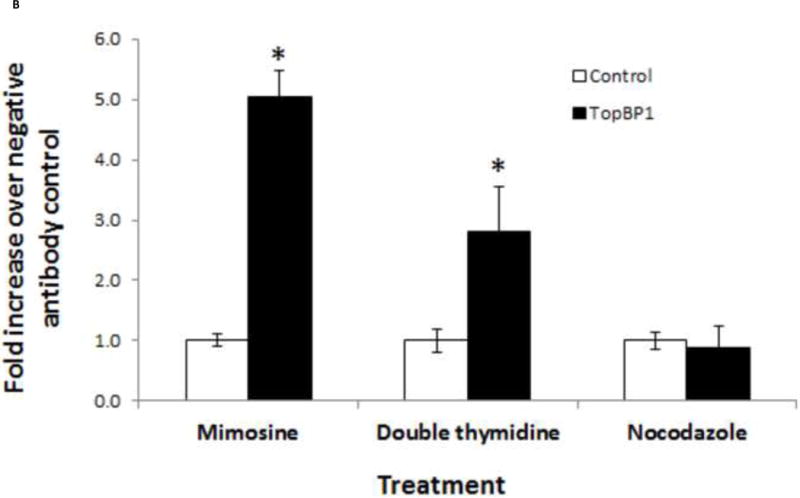
Both E2 and TopBP1 ChIP to the LCR region at G1/S and early S stages. (A) BPV-A3 cells were treated with mimosine, thymidine or nocodazole to arrest in different cell cycle stages. Inset diagram shows flow cytometry based cell cycle analysis. Bar indicates standard deviation, * p < 0.05 between TopBP1 antibody and control antibody. (B) TopBP1 is localized to HPV-16 LCR of HPV-BP cell line. Bar indicates standard deviation, * p < 0.05 between TopBP1 antibody and control antibody.
Over-expression of TopBP1 inhibits while its depletion promotes viral replication
We sought to understand the role TopBP1 might play in viral replication. We used two human cell lines to examine the effect of TopBP1 in transient replication assays: H1299, a non-small cell lung carcinoma cell line that has been successfully used for stable knockdown of TopBP1 (Liu et al., 2011) as well as for understanding HPV E6 and E7 functions (Mesplede et al., 2012; Todorovic et al., 2012) and C33A, a cervical carcinoma cell line often used in HPV replication assays. HPV-31 E1, E2 and the pFLORI31 reporter plasmids were co-transfected with either TopBP1 over-expression or knockdown constructs. There was significant reduction in protein level with the pSUPER-siTopBP1 construct compared to pSUPER-siScramble while over expression resulted in two to three fold increased TopBP1 (Figure 3A). At 48 h, knockdown of TopBP1 in both H1299 and C33A increased viral replication by 48% and 26% respectively. In contrast, its over-expression resulted in a nearly two fold decrease in HPV-31 replication in H1299 and 21% reduction in C33A cells. At 72 h post transfection, the effects were more pronounced with knockdown causing 60% and 40% increase in H1299 and C33A respectively, while over-expression decreased the viral replication by 60% and 46% in H1299 and C33A respectively (Figure 3B). As shown in Figures 4 A and B, modulation of TopBP1 protein levels did not significantly affect cell cycle compared to respective controls. We did observe that the pSUPER vector backbone slightly altered the “S” phase (as shown by pSUPER-siSCramble) in C33A cells, but the cell cycle patterns were similar for both pSUPER-siTopBP1 and pSUPERsi-Scrambled samples. Using trypan blue staining, we did not detect any significant cell death associated with TopBP1 manipulations in C33A or H1299, as treatments showed similar (94 ± 4 %) cell survival.
Figure 3.
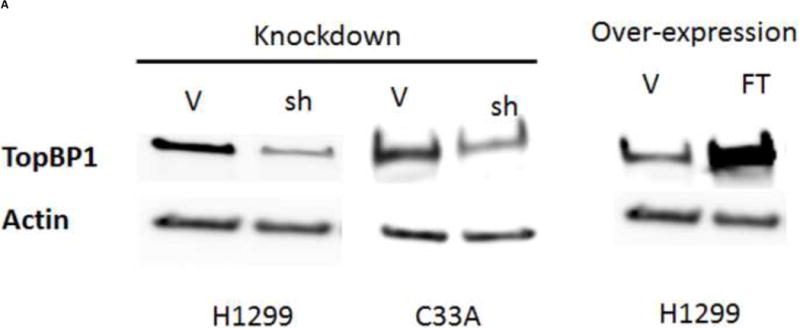
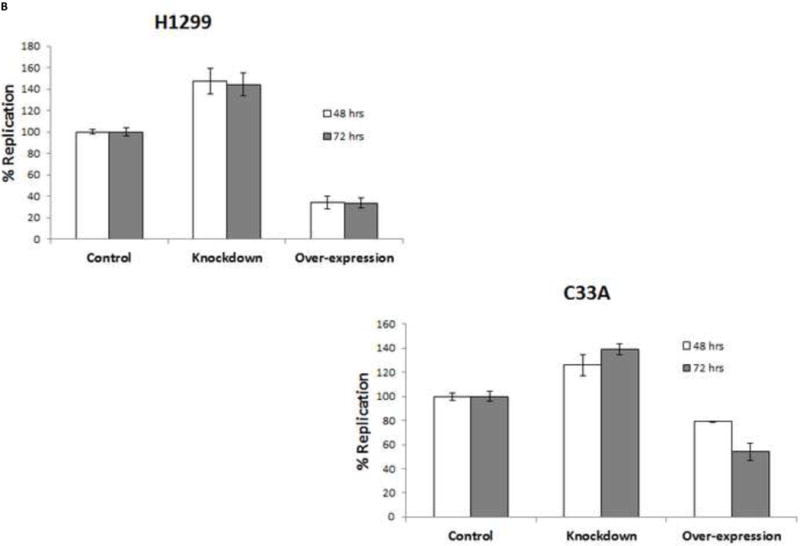
Transient HPV-31 replication was altered by modulating TopBP1 levels. (A) Western blot of TopBP1 in H1299 and C33A cells. Left panels show the western blot from H1299 and C33A cells transfected with pSUPER-siTopBP1 or its vector control. Right panel shows the western blot from H1299 cells transfected with vector or Flag-TopBP1 constructs. Actin was used as a loading control. Legend – V – vector controls, sh – pSUPER-siTopBP1, FT – Flag-TopBP1. (B) HPV-31 luciferase based assays in H1299 (upper) and C33A (lower) cells having either Knockdown or over-expression constructs of TopBP1. p < 0.05 between control and over-expression/knockdown at both time points for H1299, while it was significant at 72 hours for C33A.
Figure 4.
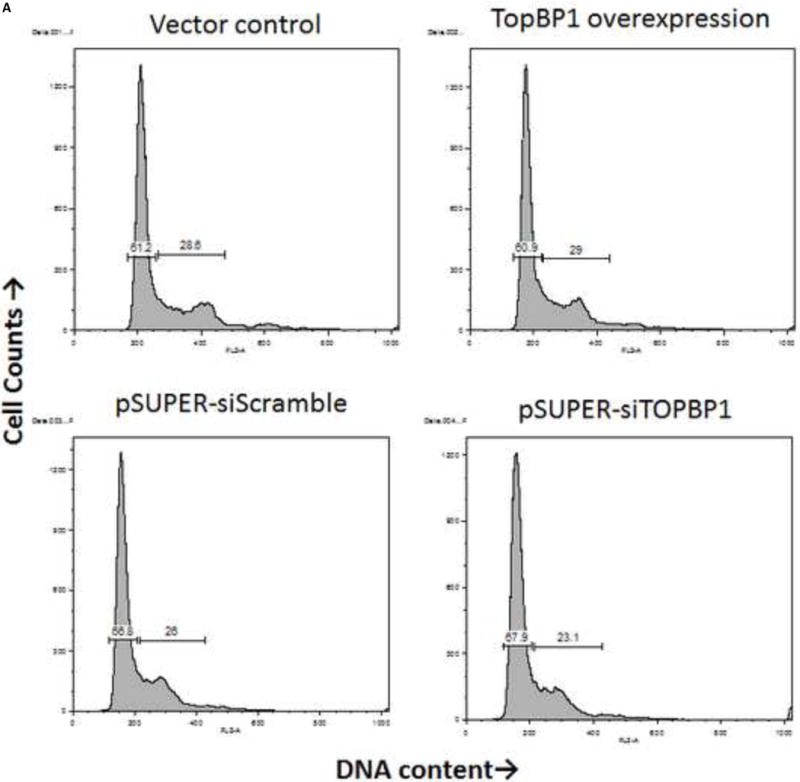
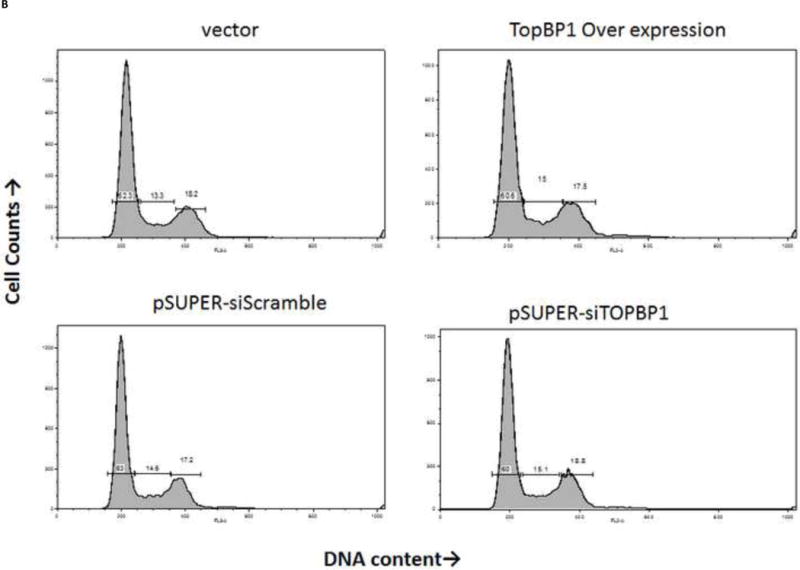
TopBP1 level does not alter cell cycle. C33A (A) and H1299 (B) cells were transfected with either over-expression or knockdown constructs (or respective controls). Flow analysis was carried out 48 h post-transfection.
Although we normalized the firefly luciferase (replication related) activity to renilla luciferase (transcription related) activity, we wished to confirm that the observed result was a result of genome replication. We repeated this assay by excluding HPV-31-E1 or E2 in this assay. As shown in Figure 5, the absence of E1 or E2 resulted in nearly 20 fold decrease in luciferase activity. In the absence of viral proteins, there was no significant difference in luciferase activity, suggesting an influential role of TopBP1 on E1-E2 dependent replication.
Figure 5.
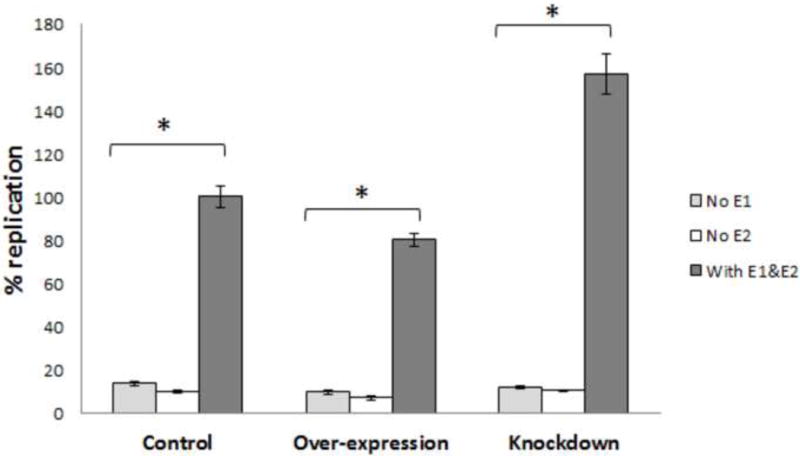
TopBP1 effect on HPV-31 replication depends on both E1 and E2. HPV-31 luciferase based assays in C33A cells with or without HPV-31 E1/E2 in TopBP1 knockdown or over-expressing cells. * p < 0.05.
To demonstrate the consequences of TopBP1 manipulation on a stable HPV replicon, HPV-BP cells were transfected with either over-expression or shRNA constructs of TopBP1. After 48 or 72 h post transfection, total DNA was isolated and viral copy number was determined by real time PCR using primers specific for the HPV-16 LCR region. A genomic DNA amplicon was included for normalization. Viral copy number increased to 150% at 48 h post-transfection and nearly threefold in TopBP1 knockdown cells at 72 h (Figure 6A and B). On the other-hand, over-expression of TopBP1 resulted in 20% and 50% reduction in copy number at 48 and 72 h respectively. We also tested whether analogous observations would hold in CIN612 cells. As shown in Figure 6B, heterologous expression of TopBP1 significantly decreased viral copy number relative to genomic DNA while knockdown increased viral content. Therefore, TopBP1 not only effects viral replication in transient assays, but also reduces viral copy number in cells maintaining HPV episomes.
Figure 6.
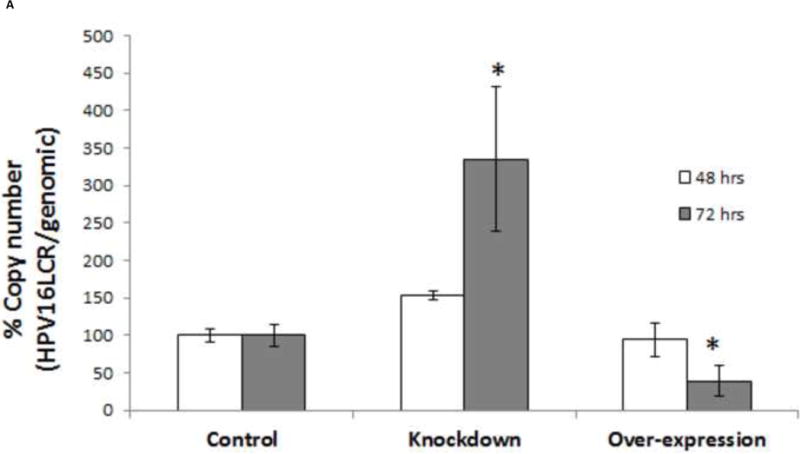
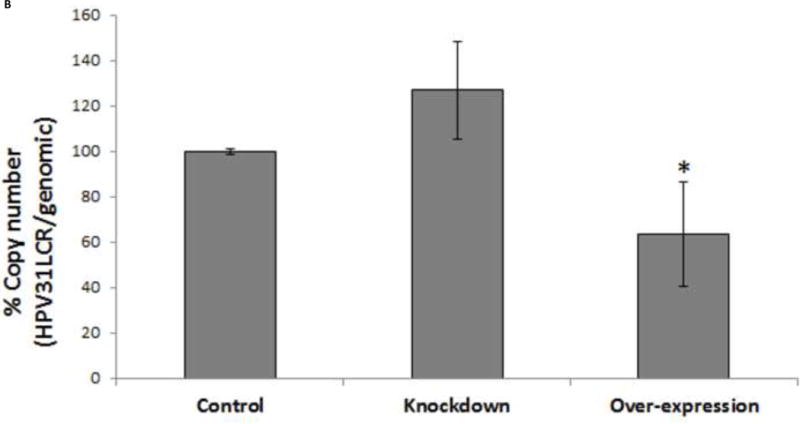
Episomal HPV replication is altered by changing TopBP1 levels. (A) Quantitative PCR of HPV-16 LCR in TopBP1 protein knockdown or over-expressing HPV-BP cells. HPV-16 LCR DNA values were normalized to PCR of genomic locus 23. * p < 0.05. (B) Quantitative PCR of HPV-31 LCR in TopBP1 protein knockdown or over-expressing CIN612 cells. HPV-31 LCR values were normalized to PCR of genomic locus 23. * p < 0.05.
Discussion
Papillomavirus genomes are maintained as low copy number episomes in basal epithelial cells and are amplified to thousands of copies in nuclei of upper level differentiating cells. While activation of the DDR pathway is known to stimulate the amplification stage, a role for DDR factors in the genome maintenance phase has not been observed. In the present study, we investigated the role of TopBP1 protein in HPV and BPV replication. TopBP1 plays an essential role during genomic DNA replication by facilitating loading of CDC45 at the origin (Kumagai et al., 2010). After replication initiates, TopBP1 is no longer necessary for continued DNA synthesis (Makiniemi et al., 2001). We demonstrate that the TopBP1 protein is present at the HPV and BPV origins of replication in G1/S phases of cell cycle (Figs. 2 A, B). This is presumably facilitated by its binding to the HPV E2 protein in vivo as shown here by in-situ PLA. We also observed interaction between BPV E1 and E2 by PLA in a murine cell line that stably maintains viral episomes (Figure 1A). In addition to strong signals, E1–E2 PLA showed a few faint signals, which were not observed in E2-TopBP1 interactions, possibly because of different affinities of proteins to each other and/or to the specific antibodies. The cell cycle specific localization of TopBP1 to the viral origin in G1/S implicates its role in the initiation of viral DNA replication. This is supported by a previous study wherein an HPV-16 genome encoding an E2 mutant crippled for TopBP1 failed to be maintained episomally in cultured keratinocytes (Donaldson et al., 2012). As the cells progressed through mitosis, the presence of TopBP1 near the viral ori decreased, indicating its displacement at this stage (Figure 2 A, B). It has been reported that the E2 binding protein Brd4 is also necessary for initiation of HPV replication and subsequently displaced from viral DNA foci (Sakakibara et al., 2013; Wang et al., 2013). Our results suggest a similar role for TopBP1.
Progression of a replication fork may stall upon collision with other DNA bound complexes, or when the template is damaged as occurs with nucleotide depletion or from chemical agents. Induction of DNA damage following HPV-18 E1 over-expression has been shown by comet assay (Reinson et al., 2013). This implies that E1 or an associated factor is capable of inducing genomic damage and the DDR. TopBP1 is recruited to the replisome by DDR mediators of the Mre11-Nbs1-Rad50 complex (Duursma et al., 2013; Makiniemi et al., 2001). Phosphorylated TopBP1 is necessary for a cascade of downstream protein phosphorylations that result in activation of Chk1, which restricts further origin firing (Feijoo et al., 2001). We observed that partial knockdown of TopBP1 consistently increased transient viral replication and viral episomes (Figures 3 and 6). The opposite outcome was observed following over-expression of TopBP1, which resulted in reduced viral replication in luciferase based reporter assays. We confirmed that the change in replication is a consequence of TopBP1 level on replication unrelated to cell cycle (Figure 4 A and B). Moreover, heterologous expression of TopBP1 consistently decreased viral copy number in keratinocytes stably carrying episomal HPV-16 or HPV-31 genomes (Figure 6). In the absence of E1 or E2, replication was not observed irrespective of TopBP1 protein level (Figure 5). We conclude that the sustained presence of TopBP1 hindered virus replication.
Targeting the TopBP1 pathway is not unprecedented in viral replication. Certain adenovirus serotypes inhibit ATR activation by targeting the TopBP1 protein for degradation, inferring that the DDR restricts adenoviral replication (Blackford et al., 2010). Another indirect link between TopBP1 and HPV replication may lie in the Fanconi Anemia (FA) pathway. FA is associated with increased susceptibility to HPV induced carcinogenesis (Kutler et al., 2003; Park et al., 2010). The viral E7 oncoprotein was reported to activate the FA pathway and accelerated chromosomal instability has been attributed to a defective FA pathway (Spardy et al., 2007). The FA pathway is believed to restrict the HPV life cycle as loss of FANCD2 component accelerates HPV amplification (Hoskins et al., 2012). Defective FANCM is also known to displace TopBP1 from chromatin (Schwab et al., 2010). Whether there is any link between HPV amplification and failure to retain TopBP1 on chromatin in FA is not very clear, but our results do indicate that after replication is initiated, removal of TopBP1 would facilitate HPV replication during episomal maintenance.
The experiments presented here suggest differential roles of TopBP1 in PV replication. Chromatin immunoprecipitation (ChIP) results support the essential role of TopBP1 for replication initiation, while over-expression of TopBP1 exerts a negative regulatory role. We believe that, as a key regulator of the DDR pathway, TopBP1 plays a critical role in fork stability during episomal replication. Since PV replication is presumed to occur by bidirectional DNA synthesis, the replication fork will converge on each episome. This poses the problem of colliding replisomes that require resolution. The DDR may be critical to appropriately resolve this possible fork collapse. A collapsed fork is likely to affect episomes more severely than the genomic DNA since the latter have multiple origins and thus could restart more efficiently. Alternatively, manipulation of the DDR by modulating the levels of TopBP1 may cause a replication block from which the episomal DNA cannot be rescued. A previous study using chemicals to alter HPV DNA structure showed that mounting of a DDR eliminated HPV episomes, possibly by activating Rad9-Hus1-Rad1 (9–1–1) (Edwards et al., 2013). It was suggested that the 9–1–1 complex may recruit enzymatic complexes that destroyed the HPV genomes. Our study also suggests that unscheduled or uncontrolled activation of TopBP1 is not conducive for PV replication, reconfirming the fine balance of host proteins for successful replication and episome maintenance. We also cannot rule out the possibility of TopBP1 playing a role during DNA segregation that affects episomal copy number. In differentiating cells where HPV undergoes rapid amplification, the DDR is a critical factor for promoting virus replication and the potential role of TopBP1 might be altogether different. Further studies are necessary to uncover the exact mechanism by which TopBP1 leads to episomal loss and/or replication block during HPV maintenance and how and when exactly TopBP1 is displaced during replication.
Research highlight.
Protein interaction study confirmed In situ interaction between TopBP1 and E2
TopBP1 present at papillomavirus ori in G1/S and early S phase of cell cycle
TopBP1 knockdown increased, over-expression reduced virus replication
TopBP1 protein level change did not influence cell survival or cell cycle
ToPBP1 displaced from papillomavirus ori after initiation of replication
Acknowledgments
We thank W.C. Lin for TopBP1 constructs, Al Klingenhultz for the HPV-BP cells, Michael Botchan for BPV-A3 cells and Jacques Archambault for HPV-31 luciferase assay plasmids. This study was supported by NIH R01 CA58376 to EJA.
Contributor Information
Sriramana Kanginakudru, Email: skangina@iu.edu.
Yanique Thomas, Email: ysthomas@umail.iu.edu.
Iain M. Morgan, Email: immorgan@vcu.edu.
Elliot J. Androphy, Email: eandro@iu.edu.
References
- Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes & development. 2004;18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androphy EJ, Lowy DR, Schiller JT. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature. 1987;325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- Balestrini A, Cosentino C, Errico A, Garner E, Costanzo V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nature cell biology. 2010;12:484–491. doi: 10.1038/ncb2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee NS, Wang HK, Broker TR, Chow LT. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. The Journal of biological chemistry. 2011;286:15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD, Laimins LA. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. Journal of virology. 1991;65:2254–2260. doi: 10.1128/jvi.65.5.2254-2260.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Patel RN, Forrester NA, Theil K, Groitl P, Stewart GS, Taylor AM, Morgan IM, Dobner T, Grand RJ, Turnell AS. Adenovirus 12 E4orf6 inhibits ATR activation by promoting TOPBP1 degradation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12251–12256. doi: 10.1073/pnas.0914605107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boner W, Taylor ER, Tsirimonaki E, Yamane K, Campo MS, Morgan IM. A Functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. The Journal of biological chemistry. 2002;277:22297–22303. doi: 10.1074/jbc.M202163200. [DOI] [PubMed] [Google Scholar]
- Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci U S A. 1992;89:5799–5803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Geest K, Turyk ME, Hosken MI, Hudson JB, Laimins LA, Wilbanks GD. Growth and differentiation of human papillomavirus type 31b positive human cervical cell lines. Gynecologic oncology. 1993;49:303–310. doi: 10.1006/gyno.1993.1131. [DOI] [PubMed] [Google Scholar]
- Donaldson MM, Boner W, Morgan IM. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. Journal of virology. 2007;81:4338–4342. doi: 10.1128/JVI.02353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson MM, Mackintosh LJ, Bodily JM, Dornan ES, Laimins LA, Morgan IM. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. Journal of virology. 2012;86:12806–12815. doi: 10.1128/JVI.01002-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duursma AM, Driscoll R, Elias JE, Cimprich KA. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TG, Vidmar TJ, Koeller K, Bashkin JK, Fisher C. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PloS one. 2013;8:e75406. doi: 10.1371/journal.pone.0075406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. The Journal of cell biology. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, B-L F, Moody CA, Lehoux M, Laimins LA, Archambault J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. Journal of virology. 2011:8996–9012. doi: 10.1128/JVI.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Moody C, Laimins LA, Archambault J. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. Journal of virology. 2010a;84:11747–11760. doi: 10.1128/JVI.01445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology. 2010b;399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Furuya K, Carr AM. Identification and functional analysis of TopBP1 and its homologs. DNA repair. 2005;4:1227–1239. doi: 10.1016/j.dnarep.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Grossel MJ, Sverdrup F, Breiding DE, Androphy EJ. Transcriptional activation function is not required for stimulation of DNA replication by bovine papillomavirus type 1 E2. Journal of virology. 1996;70:7264–7269. doi: 10.1128/jvi.70.10.7264-7269.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilman SA, Nordberg JJ, Liu Y, Sluder G, Chen JJ. Abrogation of the postmitotic checkpoint contributes to polyploidization in human papillomavirus E7-expressing cells. Journal of virology. 2009;83:2756–2764. doi: 10.1128/JVI.02149-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins EE, Morreale RJ, Werner SP, Higginbotham JM, Laimins LA, Lambert PF, Brown DR, Gillison ML, Nuovo GJ, Witte DP, Kim MO, Davies SM, Mehta PA, Butsch Kovacic M, Wikenheiser-Brokamp KA, Wells SI. The fanconi anemia pathway limits human papillomavirus replication. Journal of virology. 2012;86:8131–8138. doi: 10.1128/JVI.00408-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM. Papillomaviridae: the viruses and their replication 1996 [Google Scholar]
- Hughes FJ, Romanos MA. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic acids research. 1993;21:5817–5823. doi: 10.1093/nar/21.25.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Ko E, Lee KY, Ko MJ, Park SY, Kang J, Jeon CH, Lee H, Hwang DS. TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. The Journal of biological chemistry. 2011;286:5414–5422. doi: 10.1074/jbc.M110.189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Lee KY, Ko MJ, Lee YS, Kang S, Hwang DS. Human TopBP1 participates in cyclin E/CDK2 activation and preinitiation complex assembly during G1/S transition. The Journal of biological chemistry. 2007;282:14882–14890. doi: 10.1074/jbc.M609116200. [DOI] [PubMed] [Google Scholar]
- Kim JE, McAvoy SA, Smith DI, Chen J. Human TopBP1 ensures genome integrity during normal S phase. Molecular and cellular biology. 2005;25:10907–10915. doi: 10.1128/MCB.25.24.10907-10915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LE, Fisk JC, Dornan ES, Donaldson MM, Melendy T, Morgan IM. Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA damage signalling. Virology. 2010;406:95–102. doi: 10.1016/j.virol.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell. 2010;140:349–359. doi: 10.1016/j.cell.2009.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutler DI, Wreesmann VB, Goberdhan A, Ben-Porat L, Satagopan J, Ngai I, Huvos AG, Giampietro P, Levran O, Pujara K, Diotti R, Carlson D, Huryn LA, Auerbach AD, Singh B. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. Journal of the National Cancer Institute. 2003;95:1718–1721. doi: 10.1093/jnci/djg091. [DOI] [PubMed] [Google Scholar]
- Law MF, Lowy DR, Dvoretzky I, Howley PM. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:2727–2731. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchowius KJ, Weibrecht I, Soderberg O. In situ proximity ligation assay for microscopy and flow cytometry. In: Paul Robinson J, et al., editors. Current protocols in cytometry/editorial board. 2011. p. 36. Chapter 9, Unit 9. [DOI] [PubMed] [Google Scholar]
- Liu K, Bellam N, Lin HY, Wang B, Stockard CR, Grizzle WE, Lin WC. Regulation of p53 by TopBP1: a potential mechanism for p53 inactivation in cancer. Molecular and cellular biology. 2009;29:2673–2693. doi: 10.1128/MCB.01140-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ling S, Lin WC. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Molecular and cellular biology. 2011;31:4464–4481. doi: 10.1128/MCB.05574-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Luo Y, Lin FT, Lin WC. TopBP1 recruits Brg1/Brm to repress E2F1-induced apoptosis, a novel pRb-independent and E2F1-specific control for cell survival. Genes & development. 2004;18:673–686. doi: 10.1101/gad.1180204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Molecular and cellular biology. 2006;26:6056–6064. doi: 10.1128/MCB.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Heilman SA, Illanes D, Sluder G, Chen JJ. p53-independent abrogation of a postmitotic checkpoint contributes to human papillomavirus E6-induced polyploidy. Cancer research. 2007;67:2603–2610. doi: 10.1158/0008-5472.CAN-06-3436. [DOI] [PubMed] [Google Scholar]
- Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP, Syvaoja JE. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. The Journal of biological chemistry. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- McBride AA, Howley PM. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. Journal of virology. 1991;65:6528–6534. doi: 10.1128/jvi.65.12.6528-6534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson SM, Androphy EJ. Topography of bovine papillomavirus E2 protein on the viral genome during the cell cycle. Virology. 2009;393:258–264. doi: 10.1016/j.virol.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza-Maldonado R, Paolinelli R, Galbiati L, Giadrossi S, Giacca M. Interaction of the retinoblastoma protein with Orc1 and its recruitment to human origins of DNA replication. PloS one. 2010;5:e13720. doi: 10.1371/journal.pone.0013720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesplede T, Gagnon D, Bergeron-Labrecque F, Azar I, Senechal H, Coutlee F, Archambault J. p53 degradation activity, expression, and subcellular localization of E6 proteins from 29 human papillomavirus genotypes. Journal of virology. 2012;86:94–107. doi: 10.1128/JVI.00751-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science (New York, NY) 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- Park JW, Pitot HC, Strati K, Spardy N, Duensing S, Grompe M, Lambert PF. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer research. 2010;70:9959–9968. doi: 10.1158/0008-5472.CAN-10-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8700–8704. doi: 10.1073/pnas.91.18.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps WC, Howley PM. Transcriptional trans-activation by the human papillomavirus type 16 E2 gene product. Journal of virology. 1987;61:1630–1638. doi: 10.1128/jvi.61.5.1630-1638.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. Journal of virology. 2013;87:951–964. doi: 10.1128/JVI.01943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, Wu SY, Chiang CM, McBride AA. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS pathogens. 2013;9:e1003777. doi: 10.1371/journal.ppat.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara N, Mitra R, McBride AA. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. Journal of virology. 2011;85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol:chloroform. CSH protocols. 2006;2006 doi: 10.1101/pdb.prot4455. [DOI] [PubMed] [Google Scholar]
- Sanders CM, Stenlund A. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. The EMBO journal. 1998;17:7044–7055. doi: 10.1093/emboj/17.23.7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. The EMBO journal. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. Journal of virology. 2007;81:13265–13270. doi: 10.1128/JVI.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic B, Hung K, Massimi P, Avvakumov N, Dick FA, Shaw GS, Banks L, Mymryk JS. 2012 doi: 10.1128/JVI.01637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conserved region 3 of human papillomavirus 16 E7 contributes to deregulation of the retinoblastoma tumor suppressor. Journal of virology. 86:13313–13323. doi: 10.1128/JVI.01637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. The EMBO journal. 1991;10:449–457. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voitenleitner C, Botchan M. E1 protein of bovine papillomavirus type 1 interferes with E2 protein-mediated tethering of the viral DNA to mitotic chromosomes. Journal of virology. 2002;76:3440–3451. doi: 10.1128/JVI.76.7.3440-3451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Helfer CM, Pancholi N, Bradner JE, You J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. Journal of virology. 2013;87:3871–3884. doi: 10.1128/JVI.03068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Naidu SR, Sverdrup F, Androphy EJ. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. Journal of virology. 2009;83:2274–2284. doi: 10.1128/JVI.01791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Wu X, Chen J. A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Molecular and cellular biology. 2002;22:555–566. doi: 10.1128/MCB.22.2.555-566.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Peng YC, Androphy EJ. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus E2 during mitosis. Journal of virology. 2007;81:1736–1745. doi: 10.1128/JVI.01638-06. [DOI] [PMC free article] [PubMed] [Google Scholar]