

Abstract
Objective
The pathogenic events responsible for accelerated atherosclerosis in patients with chronic renal failure (CRF) are poorly understood. Here we investigate the hypothesis that concentrations of urea associated with CRF and increased ROS production in adipocytes might also increase ROS production directly in arterial endothelial cells, causing the same pathophysiologic changes seen with hyperglycemia.
Methods
Primary cultures of human aortic endothelial cells (HAEC) were exposed to 20 mM urea for 48 hrs. C57BL/6J wild-type mice underwent 5/6 nephrectomy or a sham operation. Randomized groups of 5/6 nephrectomized mice and their controls were also injected i.p. with a SOD/catalase mimetic (MnTBAP) for 15 days starting immediately after the final surgical procedure.
Results
Urea at concentrations seen in CRF induced mitochondrial ROS production in cultured HAEC. Urea-induced ROS caused the activation of endothelial pro-inflammatory pathways through the inhibition of GAPDH, including increased protein kinase C isoforms activity, increased hexosamine pathway activity, and accumulation of intracellular AGEs (advanced glycation end products). Urea-induced ROS directly inactivated the anti-atherosclerosis enzyme PGI2 synthase and also caused ER stress. Normalization of mitochondrial ROS production prevented each of these effects of urea. In uremic mice, treatment with MnTBAP prevented aortic oxidative stress, PGI2 synthase activity reduction and increased expression of the pro-inflammatory proteins TNFα, IL-6, VCAM1, Endoglin, and MCP-1.
Conclusions
Taken together, these data show that urea itself, at levels common in patients with CRF, causes endothelial dysfunction and activation of proatherogenic pathways.
Introduction
Cardiovascular disease risk is increased up to 30-fold in patients with chronic renal failure (CRF) compared with the general population (1,2), and remains 10 to 20 times higher after stratification for age, sex, and presence of diabetes (1). Overall, the 5-year mortality rate for patients on dialysis is 60% (2). Despite the well-established high incidence of atherosclerosis in CRF patients with uremia, the pathogenic events responsible for accelerated atherosclerosis are poorly understood. Although CRF patients have higher levels of general risk factors for cardiovascular disease (CVD) such as hypertension, diabetes, obesity, and lipid abnormalities (3), these traditional cardiovascular risk factors (CVRFs) do not fully account for the high risk of atherosclerosis, CVD, and total mortality in patients with CRF. Growing evidence supports a major role for nontraditional CVRFs in the pathogenesis of accelerated atherogenesis in this population (4). Several molecules whose levels rise as a consequence of decreased renal function have been associated with CVD in CRF, but mechanistic data demonstrating causality are lacking.
Recently, we showed that urea at concentrations seen in CRF can induce reactive oxygen species (ROS) production in cultured 3T3-L1 adipocytes, resulting in O-GlcNAc modification of several downstream insulin signaling effectors with reduction in insulin-stimulated IRS-1 (insulin receptor substrate-1) and Akt phosphorylation and glucose transport (5). Similarly, uremic mice also displayed increased ROS production, modification of insulin signaling molecules by O-GlcNAc, and increased insulin resistance and levels of insulin resistant adipokines. Moreover, urea infusion into normal rats induced insulin resistance with elevation of the insulin resistance–associated adipokines. As treatment with a SOD/catalase mimetic prevents these urea-induced abnormalities (5), we hypothesized that urea itself causes increased ROS in adipose tissue, which cause systemic insulin resistance.
Endothelial dysfunction has been shown to be the best predictor of subsequent cardiovascular events (6), and evidence of endothelial dysfunction has been detected in uremic patients from early stages of the disease (7). In an animal model of CRF-atherosclerosis, plasma urea itself was the only significant predictor of aortic plaque area fraction, and the antioxidant N-acetylcysteine prevented accelerated atherosclerosis in uremic apolipoprotein-E knockout mice (8). This observation suggested to us that the high levels of urea associated with chronic renal failure might also have direct pro-atherogenic effects on vascular cells, in addition to causing systemic insulin resistance. We therefore hypothesized that concentrations of urea associated with chronic renal failure and increased ROS production in adipocytes might also increase mitochondrial ROS production directly in arterial endothelial cells, damaging these cells by activating the same pro-inflammatory pathways and inactivating the same anti-atherosclerosis enzymes caused by diabetes (9,10). In this study, we demonstrate that urea induces endothelial cell ROS production, which causes the same pathophysiological changes reported to occur with hyperglycemia.
Materials And Methods
Cell culture conditions
Confluent primary human aortic endothelial cells (HAECs) from Cambrex, (East Rutherford, NJ) (passages 2–5) were maintained in EBM-2 medium (from Lonza, San Diego,CA) with 0.4% fetal bovine serum and all the supplements. Cells were incubated with either 20mM urea or with 20mM mannitol used as osmotic control, for 48 hours. The urea used in these experiments was certified to be free of LPS and heavy metals (Sigma Aldrich, St.Louis,MO). In subsequent experiments, cells were infected with UCP-1 adenovirus, MnSOD adenovirus, or control adenovirus at an MOI of 500, 4 hours before addition of 20mM urea containing medium.
Adenoviral vectors
UCP1 and SOD2, (obtained from Open Biosystems) were cloned into the shuttle vector pAd5CMVK-NpA, and adenoviral vectors and empty control virus were prepared by the Gene Transfer Vector Core at the University of Iowa.
Measurement of ROS generation
Treated cells seeded in a 96-well plate were incubated with 10 μmol/l CM-H2DCFDA (Molecular Probes-Life Technology, Brooklyn,NY) for 45 min at 37°C, and the intracellular formation of ROS was measured at excitation/emission wavelengths of 485/530 nm using a Wallac 1420 Fluorescent Plate Reader.
Measurement of NADPH oxidase activity
NADPH oxidase activity was determined using the lucigenin- enhanced chemiluminescence method as previously described (11). Superoxide production was measured as the rate of relative chemiluminescence (light) units per minute per microgram of protein (RLU/min mg) and expressed as fold increase compared to untreated cells.
Prostacyclin synthase activity
Activity was measured by determination of 6-keto-PGF-lα, a stable product produced by the nonenzymatic hydration of PGI2. A competitive immunoassay method (Correlate-EIA) was used for the quantitative determination of 6-keto-PGF-lα, according to the manufacturer's instructions (Assay Designs Enzo Life Sciences Farmingdale,NY). For 6-keto-PGF-lα determination in mouse aortae, the aortae were washed with PBS and incubated at 37°C for 3 hours in 400μl incubation buffer.
RT reaction and real-time quantitative PCR
Total RNA from treated cells was extracted using the RNeasy Mini Kit (QIAGEN Milan,IT), following the manufacturer's instructions. The mRNA was reverse transcribed by SuperScript III First Strand Synthesis System (Life Technology, Brooklyn,NY). Experiments were performed in quadruplicate in optical 96-well reaction plates on an iCycler iQ Multicolor Real-Time PCR Detector (Bio-Rad, Hercules,CA) with iQ SYBR green supermix (Bio-Rad). Expression levels of p65NFkB, MCP-1 VCAM1, BiP/GRP78 and XBP1 mRNA were normalized to β-actin levels in the same sample. Melting curves were analyzed to ensure that fluorescence signals solely reflected specific amplicons. PCR conditions were as follows: 7 minutes at 95°C and 45 cycles of 30 seconds at 95°C and 30 seconds at 60°C.
Determination of proinflammatory protein levels
Commercially available sandwich enzyme-linked immunosorbant assays were used to quantitate TNFα (Pierce, Rockford, IL), IL6 (Linco Research Inc.St.Charles,MO), MCP-1(R&D Systems,Minneapolis,MN) and VCAM1 (BioSource Camarillo, CA).
NFkB activity
Activity was measured by evaluating the expression of the NFkB-specific target genes (www.nf-kb.org) VCAM1, Endoglin, and VEGI by real time PCR as previously described (12).
PKC activity
PKC activity assay was performed in according to the manufacturer's instruction using the Protein Kinase C Assay System. (Enzo Life Sciences, Farmingdale,NY).
Hexosamine pathway activity
Dot Blot was perfomed using equal amount of cell lysates. Membranes were blotted with Anti-O-linked GlcNAc antibody (Affinity BioReagents Golden,CO). Immunoreactive protein-bound N-acetylglucosamine (GlcNAc) were visualized using an enhanced chemifluorescence kit according to the manufacturer's instructions (BioRad) and quantify using a VersaDoc Gel Imaging System (BioRad) and The Quantity One analytical software.
AGE formation
Equal amounts of cell extract protein were used for quantitative Dot blotting. Methylglyoxal-derived imidazole advanced glycation end products were detected using a monoclonal antibody to the major intracellular methylglyoxal-derived epitope, N-acetyl-N (5-hydro-5-methyl)-4-imidazolone. Immunocomplexes were visualized and quantified as described before.
GAPDH activity
GAPDH activity assay was performed in according to the manufacturer's instruction using GAPDH assay Kit from ScienCell Research Laboratories (Carlsbad,CA).
Quantitation of DNA strand breaks
DNA strand breaks were detected with a single-cell gel electrophoresis assay (CometAssay, Trevigen, Gaithersburg, MD) according to the manufacturer's instructions. DNA strand breaks were quantitated by examining the fixed and stained cells under a fluorescence microscope (Olympus IX70; Olympus, Melville,NY) with ×10 planoapo objectives. All analyses were performed with IP Lab Spectrum (Scanalytics, Fairfax,VA). The mean length of the DNA tail was determined by measuring 30 cells for each condition.
PARP activity
Activity was measured according to the manufacturer's Instructions using PARP assay Kit from TREVIGEN® (Gaithersburg, MD).
Mouse models
The experiments were approved by the Animal Experiments Inspectorate, Ministry of University, Rome, Italy. To induce moderate uremia 7 weeks old C57BL/6J wild-type mice (Harlan Italy S.r.1.) underwent 5/6 nephrectomy or a sham operation (5). A randomized group of 5/6 nephrectomized mice and their controls were injected i.p. with MnTBAP (Calbiochem-Merck KGaA, Darmstadt,Ge) at a dose of 10 mg/kg for 15 days starting immediately after the second surgical procedure. Three weeks after the first surgical procedure, animals were killed, and the aortae were dissected from the abdominal bifurcation to the aortic arch, to measure prostaciclyn synthase activity. Mouse blood glucose was determined using an ACCU_CHEK (Roche Diagnostics) active Blood Glucose monitoring system. Blood urea concentration was analyzed using the L-Type UN Kit (Wako Chemicals USA,Inc). Plasma fasting insulin levels were measured with monoclonal antibody-based sandwich ELISA (Linco). Enzymatic kits were used to determine plasma concentrations of triglycerides (GPO-TRINDER; Sigma) and total cholesterol (CHOD-PAP; Roche, Mannheim,Ge).
Aorta VCAM1 content
VCAM1 content in mouse aorta extracts was measured by quantitative WB as previously described (13).
Aorta oxidative stress
ROS levels were evaluated by quantifying 3-nitrotyrosine (3-NT) levels by immunoprecipitation–Western blot, using a highly selective and sensitive antibody against 3-NT (mAb Alexis 39B6 from Santa Cruz Biotechnology [Santa Cruz, Ca]), for 3-NT. The specificity of WB results using this antibody has been confirmed using an independent sensitive and quantitative method, high-performance liquid chromatography (HPLC) (14 ). Intensity of individual bands was measured using the ImajeJ program (http://imagej.nih.gov/ij/)
Statistical analysis
Data were analyzed using 1-factor ANOVA to compare the means of all the groups. The Tukey-Kramer multiple-comparisons procedure was used to determine which pairs of means were different. P value less than 0.01 was considered statistically significant.
Results
Urea induces mitochondrial reactive oxygen species production in endothelial cells
Since urea at a concentration similar to that seen in early and late CRF can induce ROS production in cultured 3T3-L1 adipocytes (5), we evaluated whether urea could also induce ROS generation in a primary culture of Human Aortic Endothelial cells (HAEC). Treatment of HAEC with 20 mM urea for 48 hrs increased ROS by 2.2 fold (Fig.1,bar3). Mannitol, used as an osmotic control, had no effect on the intracellular ROS production (data not showed). Since NADPH oxidases are major sources of superoxide in vascular cells and myocytes (15), and NADPH oxidase is upregulated in mouse aorta by experimental uremia (16), we measured NADPH oxidase activity in cells exposed to urea. Urea increased NADPH oxidase activity by 40% (Fig.1B, bar 3). ROS production was prevented in cells exposed to 20 mM urea by overexpression of either uncoupling protein 1 (UCP-1), a specific protein uncoupler of oxidative phosphorylation capable of collapsing the proton electrochemical gradient that drives superoxide production (17), or manganese superoxide dismutase (MnSOD) (9), the mitochondrial isoform of this enzyme (Fig. 1A,bars4 and 5). These data show that the initial ROS formed is superoxide, and that the mitochondrial electron transport chain is a source of urea-induced ROS production.
Figure 1. Urea induces mitochondrial reactive oxygen species in HAEC.
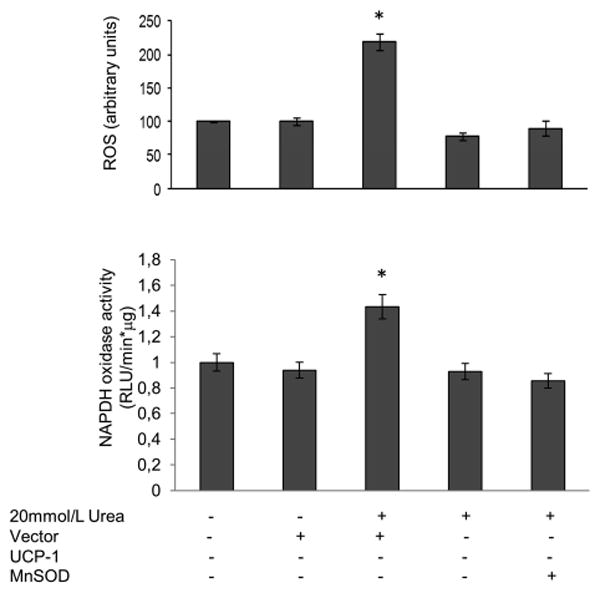
Cells were infected with adenoviral vectors expressing UCP-1 or MnSOD. After 48 hrs incubation with urea A) intracellular ROS generation and B) NADPH oxidase activity were measured. Each bar represents the mean±SEM of 5 for A and 3 for B separate experiments, each with n = 8. *p<0.01 compared with cells not treated with urea.
Overexpression of either UCP-1 or MnSOD also prevents the urea induced increase of NADPH oxidases activity, (Fig1B,bars3and4) indicating that the increased mitochondrial production of ROS is necessary for urea to increase NADPH oxidase activity
Urea-induced mitochondrial ROS cause pro-inflammatory changes in endothelial cells
We next determined whether urea-induced ROS cause pro-inflammatory and pro-atherogenic changes in HAEC. Remarkably, after incubation with 20 mM urea for 48 h the activity of the endothelial cell anti-atherosclerotic enzyme PGI2 synthase [18,19] was reduced by 88% (Fig. 2A, bar2). A direct concentration-response relationship between urea and decreased enzyme activity was observed between 10 mM and 40 mM urea (data not shown). Mannitol, used as an osmotic control, had no effect on the enzyme activity (data not shown). The reduction in the PGI2 synthase activity was associated with a 2.8-fold increase in mRNA expression of the major inflammatory mediator NFκB subunit p65 [20,21], while the mRNA expression of NFkB specific target genes VCAM1, Endoglin and VEGI increased by 2.9, 2.9 and 2.2-fold respectively, indicating that urea induced NFkB activation [12]. (Fig. 2B). Moreover the protein level of MCP-1 and VCAM1, two NFκB-dependent pro-atherogenic endothelial cell surface molecules expressed early in the development of atherosclerosis [22,23], increased by 1.6 and 3.2-fold respectively (Fig. 2C and D). Over-expression of either UCP-1 or MnSOD completely prevented the urea-induced reduction of PGI2 synthase activity (Fig. 2A) and the urea-induced increase of both NFkB activity, and protein levels of MCP-1 and VCAM1 in HAEC cells (Fig. 2B–D). These results indicate that urea-induced changes in endothelial cell proatherogenic gene expression are mediated by increased mitochondrial ROS production.
Figure 2. Urea-induced mitochondrial reactive oxygen species cause proinflammatory changes in endothelial cells.
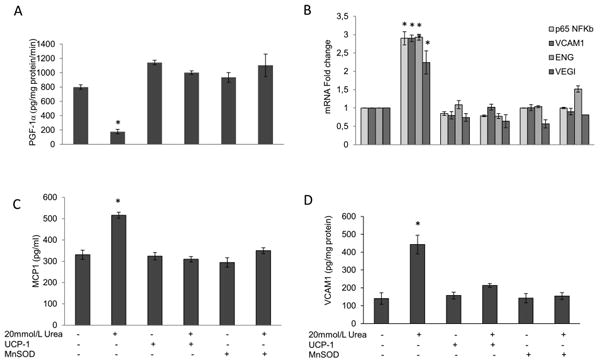
HAEC were incubated with 20mM urea alone or 20 mM urea after infection with adenoviral vectors expressing UCP-1 or MnSOD. After 48 hrs PGI2 Synthase activity (A) NFκB p65 and NFκB-specific target genes mRNA expression (B) MCP-1 (C) VCAM1 (D) protein levels were measured. Each bar represents the mean±SEM of 5 separate experiments, each with n = 3. *p<0.01 compared with cells not treated with urea.
Urea-induced ROS production increases PKC activity, hexosamine pathway activity, and AGE formation
We next investigated the effect of urea-induced ROS production on mechanisms which have been shown to cause endothelial cell damage in diabetes. Glucose-induced over production of ROS has been shown to cause a number of important pathologic alterations in vascular cells by increasing protein kinase C activity, hexosamine pathway flux, and advanced glycation end product (AGE) formation in HAECs (10,17). Incubation of HAEC cells with 20 mM urea increased PKC activity by 2.9-fold (Fig.3A). Similarly, urea increased intracellular protein modification by N-acetylglucosamine (O-GlcNAc), an indicator of hexosamine pathway activity, by 2.6-fold, and intracellular protein modification by the AGE precursor methylglyoxal by 1.8-fold (Fig.3B and C). Over expression of UCP-1 or MnSOD completely prevented urea-induced PKC activation (Fig. 3A), hexosamine pathway activity (Fig.3B) and intracellular AGE formation (Fig.3C).
Figure 3. Urea-induced ROS production increases PKC activity, hexosamine pathway activity and intracellular AGE formation in HAEC.
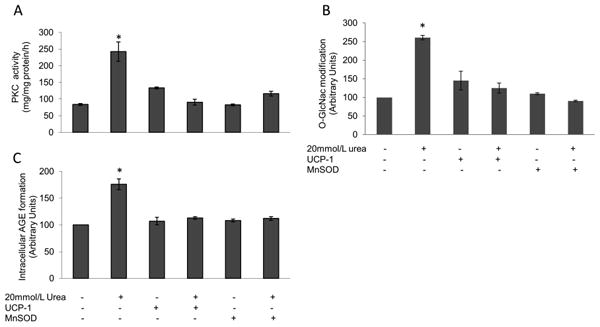
Cells were incubated in 20 mM Urea alone or in 20 mM Urea after infection with adenoviral vectors expressing UCP-1 or MnSOD. (A) PKC activity (B) hexosamine pathway activation and (C) Intracellular AGE formation. Each bar represents the mean±SEM of 5 separate experiments, each with n = 3. *p<0.01 compared with cells incubated in media alone.
Urea-induced ROS production inhibits GAPDH activity via activation of PARP
Over production of superoxide by the mitochondrial electron transport chain has been shown to activate the five major independent mechanisms of hyperglycemia-induced cellular damage by inhibiting activity of the glycolytic enzyme GAPDH [10]. As shown in Fig. 4A, exposure to 20 mM urea reduced HAEC GAPDH activity by 48%. This reduction was completely prevented by overexpression of UCP-1 or MnSOD. Since increased ROS have been reported to inhibit GAPDH activity by PARP-mediated polyADP-ribosylation of the enzyme as a result of PARP activation by ROS-induced DNA strand breaks [10], we next evaluated the effect of urea treatment on both endothelial cell DNA damage and PARP activity. In endothelial cells exposed to urea, DNA strand breaks increased by 2.21-fold (Fig. 4B) while PARP activity increased 2.1- fold (Fig. 4C). The urea induced increase in PARP activity was also prevented by over expression of UCP-1 or MnSOD, consistent with increased mitochondrial ROS production causing DNA strand breaks which activate the latent DNA repair enzyme poly(ADP-ribose)polymerase.
Figure 4. Urea-induced ROS decrease GAPDH activity, increase DNA strand breaks and increase PARP activity in HAEC.
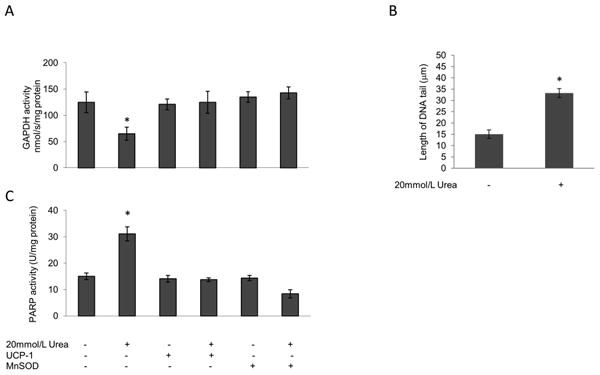
Cells were incubated in 20 mM Urea alone or in 20 mM Urea after infection with adenoviral vectors expressing UCP-1 or MnSOD. (A) GAPDH activity, (B) Quantitation of DNA strand breaks from single cell electrophoresis assay, (C) PARP activity. For A and C each bar represents the mean±SEM of 5 separate experiments, each with n = 3. *p <0.01 compared with cells incubated in media alone. For B bar represents the mean±SEM of 30 cells for each incubation condition. *p< 0.01 compared with cells incubated in media alone.
Urea induces endoplasmic reticulum (ER) stress in HAEC
Since glucose-induced over production of ROS has been shown to increase ER stress (24), we next investigated the capacity of urea- induced ROS to increase ER stress. The ER chaperone immunoglobulin heavy chain-binding protein/glucose-regulated protein 78-kDa (BiP/GRP78) increased by 3.6-fold, and the ER stress-induced spliced form of mRNA for the transcriptional activator X-box binding protein 1 (XBP-1) increased by 1.4-fold following urea treatment. Urea-induced increases in these ER stress markers were prevented by the over expression of UCP-1 or MnSOD, indicating that urea-induced ER stress were also mediated by the increased mitochondrial ROS production (Fig.5A and B).
Figure 5. Urea induces ER stress in HAEC.
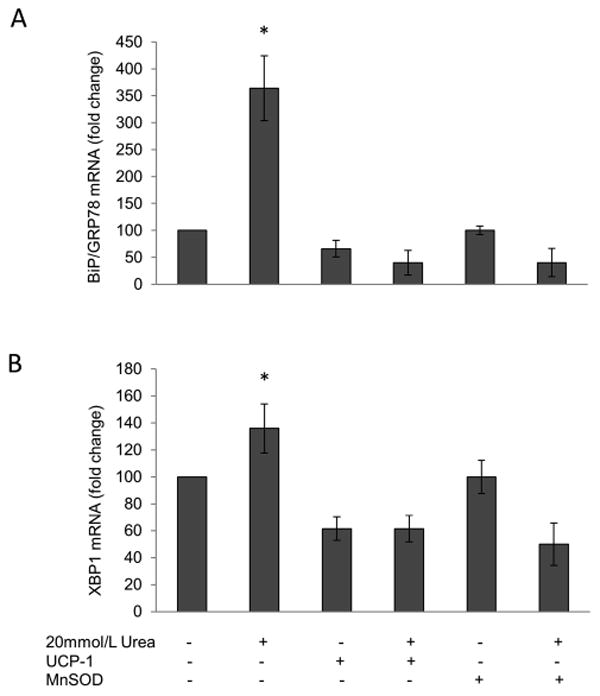
ER stress was evaluated in cells incubated in 20 mM Urea alone or in 20 mM Urea after infection with adenoviral vectors expressing UCP-1 or MnSOD by measuring both (A) BIP mRNA expression and(B) the splicing RNA of XBP-1. Each bar represents the mean±SEM of 5 separate experiments, each with n = 3. *p<0.01 compared with cells incubated in media alone.
Treatment of uremic mice with an SOD/catalase mimetic prevents uremia induced pro-atherosclerotic changes
Two wk after 5/6 nephrectomy, mice showed a 2.8-fold increase in plasma urea concentration, and they had already developed a systemic pro-atherosclerotic phenotype with significantly increased plasma insulin, triglyceride, total cholesterol levels (Table 1), and the serum pro-inflammatory cytokines TGFα and IL-6 (Fig.6A). In CFR mice the aortic PGI2 synthase activity was reduced by 94.4% (Fig.6B bar3), while the PGI2 protein levels did not change in the uremic mice compare with the control (data not shown). In these mice the aortic level of VCAM1 protein was 8 fold higher (Fig.6C bar 3) than the control, while the expression of relevant NFκB-specific target genes Vcam1, Eng and Vegi was augmented significantly(Fig.6D), demonstrating NFκB activation. This CRF mouse pro-atherosclerotic phenotype was associated with a 1.9-fold increase of oxidative stress markers in the aorta of uremic animals. (Fig.6E bar 3) To test in vivo the effect of uremia-generated ROS on endothelial pro-atherogenic changes, CRF mice and their controls were treated for two weeks with daily injections of the SOD/catalase mimetic tetrakis (4-benzoic acid) porphyrin (MnTBAP). MnTBAP has catalytic activities similar to the ROS-scavenging enzymes superoxide dismutase (SOD) and catalase. It protects mammalian cells from oxidative damage and complements loss-of-function SOD mutations in bacteria and mice (25). MnTBAP administration did not have any effect on plasma urea levels in either control or uremic mice (data not shown). However, MnTBAP treatment completely prevented oxidative stress in the aorta and all the pro-atherosclerotic changes caused by CRF (Fig.6 A,B,C,D,E).
Table 1.
Uremic mice have a systemic pro-atherosclerotic phenotype.
| C57BL/6J | Uremic C57BL/6J | p-value | |
|---|---|---|---|
| Plasma Urea nmol/L | 9.99 ± 11.01 | 27.83 ± 1.96 | <0.01 |
| Plasma Insulin (pg/ml) | 265.3 ± 29.20 | 1808.48 ± 31.83 | <0.01 |
| Plasma Glucose mg/dl | 88.92 ± 7.59 | 115.60 ± 13.91 | NS |
| Plasma Triglicerydes (mg/dl) | 58.23 ± 4,54 | 114.80 ± 18.85 | <0.01 |
| Plasma Cholesterol (mg/dl) | 66.59 ± 19,85 | 171.95 ± 19.82 | <0.01 |
Results are expressed as meam ±SEM n 15
Figure 6. Treatment of uremic mice with an SOD/catalase mimetic normalizes aorta pro-atherosclerotic changes.
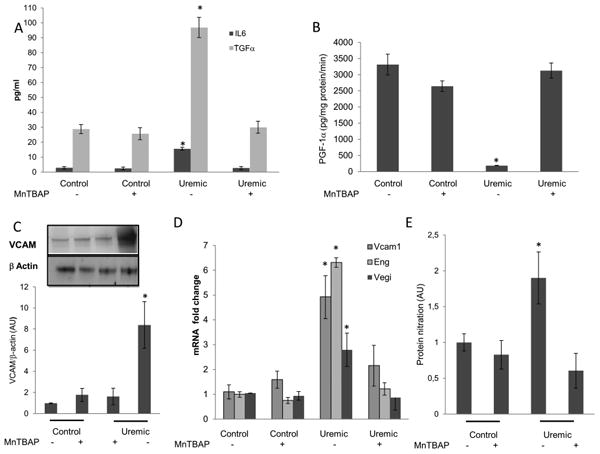
After 2 weeks of treatment with MnTBAP,A) serum TNFα and IL6, B) PGI2 synthase activity C) Aorta VCAM1 protein level D) NFκB-specific target gene expression and E) Aorta protein nitration content were measured in untreated and MnTBAP- treated control mice and in untreated and MnTBAP-treated uremic mice. Results represent the mean±SEM of 7 animals per group. *p<0.01 compared with controls.
Discussion
In the present study, we show that in cultured primary human aortic endothelial cells (HAEC), urea induces an increase of mitochondrial ROS production, which causes activation of pro-inflammatory pathways, increased expression of VCAM1, MCP-1, Endoglin and VGEI, inactivation of the anti-atherosclerosis enzyme PGI2 synthase, and ER stress. Normalization of mitochondrial ROS production prevented each of these effects of urea. In uremic mice, treatment with an SOD/catalase mimetic prevented aortic oxidative stress, PGI2 synthase activity reduction, NFkB activation and the increase of the pro-inflammatory protein VCAM1.
Whether excess urea is pathogenic has been a controversial issue. Due to the lack of acute organ injury after addition of urea to dialysate fluid (26) and the lack of impact on survival in patients whose urea reduction rate was increased from 66% to 75% by increased dialysis dose (27), it has been assumed that elevated levels of urea in CRF have negligible toxicity. However recent data have demonstrated that the survival of patients on daily hemodialysis is 2- to 3-fold greater than that of patients dialyzed less frequently (28), suggesting that one or more of the molecules cleared by dialysis plays an important role in accelerated CVD in patients with CRF. The observation that oral administration of urea accelerates atherogenesis in non-uremic ApoE/mice fed with a high-fat diet in the absence of other factors and toxins which accumulate in CRF (29) supports the hypothesis that urea itself plays an important role in the pathogenesis of accelerated cardiovascular disease in renal failure.
Recent proteomic studies of mitochondria by Mootha et al. showed that, of the ∼1,100 mitochondrial proteins, it appears that almost half are core components found in virtually all tissues, whereas the remaining half are distributed in a tissue-specific manner (30). Thus, it remains to be determined whether increased urea stimulates mitochondrial ROS production in all tissue types. Since in the vessel smooth muscle cells are also activated in the atherosclerotic process, we cannot exclude that urea affects them.
Like urea, hyperglycemia also increases ROS production, causing activation of pro-inflammatory pathways, inactivation of the anti-atherosclerosis enzyme PGI2 synthase, and ER stress. Hyperglycemia, like urea, also increases modification of insulin signaling molecules by O-GlcNAc, causing insulin resistance (9,17,24,31). In people without diabetes or impaired renal function, insulin resistance itself markedly increases cardiovascular disease risk, even after adjustment for known risk factors such as LDL, triglycerides, HDL, and systolic blood pressure (32,33). Among hemodialysis patients, the rate of death is higher in those with more severe insulin resistance (34).
Additive or synergistic effects of urea and hyperglycemia might explain the 2-fold increase in 5-year mortality of ESRD patients with diabetes compared with those without diabetes (35).
In summary, the present findings provide further insight into the underlying mechanisms that contribute to the enhanced cardiovascular risk associated with chronic renal failure. Urea, long considered to have negligible toxicity in patients with CRF, increases mitochondrial ROS production in arterial endothelial cells, thereby activating pro-atherosclerotic pathways and directly inactivating PGI2 synthase, a critical endothelial-specific anti-atherosclerotic enzyme in vitro and in vivo. Treatment with an SOD/catalase mimetic normalizes PGI2 synthase activity in mice with CRF. Since urea accelerates atherogenesis in non-uremic ApoE/mice, novel therapeutics that directly target urea-induced ROS may potentially help reduce the high morbidity and mortality caused by ESRD.
The urea, long considered to have negligible toxicity in patients with chronic renal failure (CRF), increases mitochondrial ROS production in arterial endothelial cells.
The Urea-induced ROS activate pro-atherosclerotic pathways through GAPDH inhibition.
The Urea-induced ROS directly inactivate PGI2 synthase, a critical endothelial-specific antiatherosclerotic enzyme in vitro and in vivo.
Treatment with an SOD/catalase mimetic normalizes PGI2 synthase activity in mice with CRF.
Acknowledgments
The authors thank Jeffrey Pessin for his assistance in editing the manuscript. This work was supported by grants from the Italian Ministry of Education PRIN PRT20082P8CCE_002 (I.Giardino) and by National Institute of Diabetes and Digestive and Kidney Diseases NIH grant R01DK74153-01 (M.Brownlee)
Nonstandard abbreviations used
- CRF
chronic renal failure
- ROS
reactive oxygen species
- GlcNAc
O-linked -N-acetylglucosamine
- UCP-1
uncoupling protein 1
- MnTBAP
manganese tetrakis (4-benzoic acid) porphyrin
Footnotes
The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32(5 Suppl 3):S112–19. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- 2.Schiffri EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116(1):85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 3.Rucker D, Tonelli M. Cardiovascular risk and management in chronic kidney disease. Nat Rev Nephrol. 2009;5(5):287–96. doi: 10.1038/nrneph.2009.42. [DOI] [PubMed] [Google Scholar]
- 4.Chade AR, Lerman A, Lerman LO. Kidney in early atherosclerosis. Hypertension. 2005;45(6):1042–49. doi: 10.1161/01.HYP.0000167121.14254.a0. [DOI] [PubMed] [Google Scholar]
- 5.D'Apolito M, Du X, Zong H, Catucci A, Maiuri L, Trivisano T, Pettoello-Mantovani M, Campanozzi A, Raia V, Pessin JE, Brownlee M, Giardino I. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J Clin Invest. 2010;120(1):203–13. doi: 10.1172/JCI37672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23 Suppl 1):III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 7.Annuk M, Zilmer M, Lind L, Linde T, Fellstro T. Oxidative Stress and Endothelial Function in Chronic Renal Failure. J Am Soc Nephrol. 2001;12(12):2747–52. doi: 10.1681/ASN.V12122747. [DOI] [PubMed] [Google Scholar]
- 8.Ivanovski O, Szumilak D, Nguyen-Khoa T, Ruellan N, Phan O, Lacour B, Descamps-Latscha B, Drüeke TB, Massy ZA. The antioxidant N-acetylcysteine prevents accelerated atherosclerosis in uremic apolipoprotein E knockout mice. Kidney Int. 2005;67(6):2288–94. doi: 10.1111/j.1523-1755.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 9.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 10.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabó C, Brownlee M. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage. J Clin Invest. 2003;112(7):1049–57. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Nox4 mediates angiotensinll induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol Renal Physiol. 2003;285:F219–F229. doi: 10.1152/ajprenal.00414.2002. [DOI] [PubMed] [Google Scholar]
- 12.Rothgiesser KM, Erener S, Waibel S, Lüscher B, Hottiger MO. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 2010;123(Pt 24):4251–8. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- 13.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Girault I, Kam AE, Schaper M, Barcellos-Hoff MH, Hagen T, Vogel DS, Ames BN, Christen S, Shigenaga MK. Immunodetection of 3-nitrotyrosine in the liver of zymosan-treated rats with a new monoclonal antibody: comparison to analysis by HPLC. Free Radic Biol Med. 2001;31(11):1375–87. doi: 10.1016/s0891-5849(01)00712-2. [DOI] [PubMed] [Google Scholar]
- 15.Griendling KK, Sorescu D, Ushio-Fukaiand M. NAD(P)H Oxidase Role in Cardiovascular Biology and Disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 16.Zanetti M, Barazzoni R, Bosutti A, Stocca A, Grassi G, Guarnieri G. Vascular sources of oxidative stress: implications for uremia-related cardiovascular disease. J Ren Nutr. 2007;17(1):53–6. doi: 10.1053/j.jrn.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 18.Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H (2) receptor mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51(1):198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 19.de Leval X, Hanson J, David JL, Masereel B, Pirotte B, Dogné JM. New developments on thromboxane and prostacyclin modulators part II: prostacyclin modulators. Curr Med Chem. 2004;11(10):1243–52. doi: 10.2174/0929867043365279. [DOI] [PubMed] [Google Scholar]
- 20.Thurberg BL, Collins T. The nuclear factor-kappa B/inhibitor of kappa B autoregulatory system and atherosclerosis. Curr Opin Lipidol. 1998;9(5):387–96. doi: 10.1097/00041433-199810000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Bu DX, Erl W, de Martin R, Hansson GK, Yan ZQ. IKKbeta-dependent NF-kappaB pathway controls vascular inflammation and intimal hyperplasia. FASEB J. 2005;19(10):1293–95. doi: 10.1096/fj.04-2645fje. [DOI] [PubMed] [Google Scholar]
- 22.El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, Cooper ME, Brownlee M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–17. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96(3):1395–403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Müller Cell–Derived Inflammatory Cytokine Production in Diabetes. Diabetes. 2012;61(2):492–504. doi: 10.2337/db11-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxidemediated injury. Arch Biochem Biophys. 1997;347(2):256–62. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- 26.Johnson WJ, Hagge WW, Wagoner RD, Dinapoli RP, Rosevear JW. Effects of urea loading in patients with far-advanced renal failure. Mayo Clin Proc. 1972;47(1):21–29. [PubMed] [Google Scholar]
- 27.Eknoyan G, Beck GJ, Cheung AK, Daugirdas JT, Greene T, Kusek JW, Allon M, Bailey J, Delmez JA, Depner TA, Dwyer JT, Levey AS, Levin NW, Milford Ornt DB, Rocco MV, Schulman G, Schwab SJ, Teehan BP, Toto R. Hemodialysis (HEMO) Study Group, Effect of dialysis dose and membrane flux in maintenance hemodialysis. N Engl J Med. 2002;347(25):2010–19. doi: 10.1056/NEJMoa021583. [DOI] [PubMed] [Google Scholar]
- 28.Vanholder R, Massy Z, Argiles A, Spasovski G, Verbeke F, Lamiere N. Chronic kidney disease as a cause of cardiovascular morbidity and mortality. Nephrol Dial Transpl. 2005;20(6):1048–56. doi: 10.1093/ndt/gfh813. [DOI] [PubMed] [Google Scholar]
- 29.Apostolov OE, Ray D, Savenka AV, Shah SV, Basnakian AG. Chronic Uremia Stimulates LDL Carbamylation and Atherosclerosis. J Am Soc Nephrol. 2010;21(11):1852–7. doi: 10.1681/ASN.2010040365. 10.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. doi: 10.1146/annurev-genom-082509-141720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116(4):1071–80. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yip J, Facchini FS, Reaven GM. Resistance to insulinmediated glucose disposal as a predictor of cardiovascular disease. J Clin Endocrinol Metab. 1998;83(8):2773–76. doi: 10.1210/jcem.83.8.5005. [DOI] [PubMed] [Google Scholar]
- 33.Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of IR in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care. 2002;25(7):1177–84. doi: 10.2337/diacare.25.7.1177. [DOI] [PubMed] [Google Scholar]
- 34.Bodlaj G, Ber J, Pichler R, Biesenbach G. Prevalence, severity and predictors of HOMA-estimated insulin resistance in diabetic and nondiabetic patients with end-stage renal disease. J Nephrol. 2006;19(5):607–12. [PubMed] [Google Scholar]
- 35.Koch M, Hollenbeck M, Trapp R, Kulas W, Grabensee B. Value of diabetes as an independent predictor of death in subj ects with end-stage renal disease. Med Klin (Munich) 2006;101(12):933–937. doi: 10.1007/s00063-006-1125-6. [DOI] [PubMed] [Google Scholar]