

Abstract
Group A rotaviruses (RVA) are among the main global causes of severe diarrhea in children under the age of 5 years. Strain diversity, mixed infections and untypeable RVA strains are frequently reported in Africa. We analysed rotavirus-positive human stool samples (n=13) obtained from hospitalised children under the age of 5 years who presented with acute gastroenteritis at sentinel hospital sites in six African countries, as well as bovine and porcine stool samples (n=1 each), to gain insights into rotavirus diversity and evolution. Polyacrylamide gel electrophoresis (PAGE) analysis and genotyping with G- (VP7) and P-specific (VP4) typing primers suggested that 13 of the 15 samples contained more than 11 segments and/or mixed G/P genotypes. Full-length amplicons for each segment were generated using RVA-specific primers and sequenced using the Ion Torrent and/or Illumina MiSeq next-generation sequencing platforms. Sequencing detected at least one segment in each sample for which duplicate sequences, often having distinct genotypes, existed. This supported and extended the PAGE and RT-PCR genotyping findings that suggested these samples were collected from individuals that had mixed rotavirus infections. The study reports the first porcine (MRC-DPRU1567) and bovine (MRC-DPRU3010) mixed infections. We also report a unique genome segment 9 (VP7), whose G9 genotype belongs to lineage VI and clusters with porcine reference strains. Previously, African G9 strains have all been in lineage III. Furthermore, additional RVA segments isolated from humans have a clear evolutionary relationship with porcine, bovine and ovine rotavirus sequences, indicating relatively recent interspecies transmission and reassortment. Thus, multiple RVA strains from sub-Saharan Africa are infecting mammalian hosts with unpredictable variations in their gene segment combinations. Whole-genome sequence analyses of mixed RVA strains underscore the considerable diversity of rotavirus sequences and genome segment combinations that result from a complex evolutionary history involving multiple host species.
Keywords: Rotavirus, mixed infections, reassortants, whole genome, Africa
1. Introduction
Group A rotaviruses (RVA) are the major cause of severe diarrhoea among infants and young children, as well as among the young of a variety of animal species globally (Estes and Kapikian, 2007). The burden of RVA diarrhoeal disease is highest in low income countries, mostly those in Africa and Asia. As of 2008, RVA-associated diarrhoea was estimated to cause 453,000 global deaths in children below 5 years of age, with over 50 % of these deaths occurring in Africa (Tate et al., 2012). In sub-Saharan Africa, RVA-associated gastroenteritis was responsible for an estimated 310,000 deaths annually (Sanchez-Padilla et al., 2009). According to World Health Organization (WHO) estimates, five African countries recorded highest RV mortality rates of > 300 per 100 000 children < 5 years of age (Tate et al., 2012). In South Africa alone, approximately 320 deaths were reported annually due to RV diarrhoea (Sanchez-Padilla et al., 2009; Seheri et al., 2010).
Vaccination is the most effective prevention measure against RV mortality. Two live-attenuated oral RVA vaccines, a pentavalent bovine-human reassortant vaccine (RotaTeq, Merck Co.) (Vesikari et al., 2006) and a monovalent human vaccine (Rotarix, GlaxoSmithKline) (Ruiz-Palacios et al., 2006) are in use. So far, 75 countries around the world have introduced either of the two RV vaccines through national immunization programmes, including 35 GAVI-eligible countries mostly in the African continent (PATH, January, 2015). With the effective implementation of these two RV vaccines in both developed and developing countries, the morbidity and mortality rates associated with RV disease have dramatically declined (Fernandes et al., 2014; Msimang et al., 2013; Seheri et al., 2012; Vesikari et al., 2013; Walker et al., 2013). In Africa, data from South Africa showed a significant delay in the RV season by up to 8 weeks when comparing pre- and post-vaccination parameters in some settings (Seheri et al., 2012).
Rotaviruses have dsRNA consisting of 11 genome segments which code for six structural proteins (VP1-VP4, VP6 and VP7) and six non-structural proteins (NSP1-NSP6), with a genome size of 18, 500 bp ranging from 3302 (genome segment 1) to 667 (genome segment 11) based on the molecular size (Estes and Kapikian, 2007). RVs are divided into groups or species based on the antigenic properties of the VP6 (Estes and Kapikian, 2007). Groups A to H has so far been identified (Matthijnssens et al., 2012). The RVA are classified based on the properties of the two outer capsid proteins VP7 (glycoprotein, G-types) and VP4 (protease sensitive, P-types). That way, 27 G and 37 P have been described (Matthijnssens et al., 2011; Trojnar et al., 2013). Worldwide, there is a great diversity of circulating RVA wild-type strains causing severe disease in different countries each year. In Africa, strain diversity is extensive, and several G and P genotype combinations have been reported from multiple countries. These include, but are not limited to, G1P[6], G1P[8], G2P[4], G2P[6], G3P[6], G8P[6], G9P[8] and G12P[8], as well as mixed/multiple G and/or P types and also stains regarded as untypeable with available primer sets (Abebe et al., 2014; Banga-Mingo et al., 2014; Boula et al., 2014; Hokororo et al., 2014; Kiulia et al., 2014; Mukaratirwa et al., 2014; Odiit et al., 2014; Page et al., 2010; Pukuta et al.,2014; Pursem et al., 2014; Seheri et al., 2014; Tsolenyanu et al., 2014). In addition, some studies have also reported the detection of animal or animal-human reassortant strains circulating among humans (Esona et al., 2009, Esona et al., 2010; Jere et al., 2012; 2014; Page et al., 2010; Nyaga et al., 2013; 2014).
Globally, from 1996 to 2007, RV infections caused by mixed G and P genotypes have increased from 7.9 to 11.7 % (Bányai et al., 2012). In Africa, mixed G and P genotype infections have been reported in numerous WHO-coordinated RV surveillance studies, and the overall detection rate ranges from 12 to 14 % (Bányai et al., 2012; Dóró et al., 2014; Mwenda et al., 2014; Seheri et al., 2014). The RVA genome has further been classified using a complete sequence-based system that allows each genome segment of the virus to be assigned a particular genotype. This way, genome segments VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5/6 are represented by Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx (x= ≥ 1) (Matthijnssens et al., 2008). With the increase in detection of mixed G and P genotypes in Africa, more investigations at the whole-genome level is necessary to determine the properties of mixed infections in all genome segments, understand their origin and backbone.
Based on whole-genome sequencing, only two studies have previously described the complete genome constellations of presumably mixed RVA co-infections globally. The first study reported the sequence of RVA/Human-wt/ZAF/2371WC/2008/G9P[8], which revealed co-infection by at least four different strains (Jere et al., 2011), and the second study sequenced the strain RVA/Simian-tc/ZAF/SA11-x/1958/G3P[2], where x represents two distinct sequences for genome segment 8 (NSP2), corresponding to genotypes N2 and N5 (Mlera et al., 2013). In addition, the number of RV whole genomes reported from Africa still remains relatively low compared to whole genomes from other parts of the world (Dennis et al., 2014; Heylen et al., 2014; Jere et al., 2011, 2012, 2014; Magagula et al., 2015; Nakagomi et al, 2013; Nyaga et al., 2013, 2014). Such data would aid our understanding of viral factors that could possibly assist in understanding the lower immunogenicity and efficacy of RV vaccines observed in developing countries (Armah et al., 2010; Madhi et al., 2010; Nakagomi et al, 2012; Sow et al., 2012; Steele et al., 2012).
Despite sub-Saharan Africa having high burden of RV disease due to high diversity of circulating strains, only limited country-specific data on RVA genotypes are available and these mostly target genome segments 4 (VP4) and 9 (VP7) (Mwenda et al., 2014; Seheri et al., 2014). This RVA diversity is generated by several mechanisms, which include (i) accumulation of point mutations (genetic drift) that can lead to antigenic changes; (ii) reassortment (genetic shift) which can occur as a result of exchange between two RVA strains (human-human, animal-animal or animal-human strains) leading to viruses with novel genetic and antigenic characteristics; (iii) direct transmission of animal strains into a human host (interspecies transmission); and (iv) gene rearrangement in the coding or noncoding regions, primarily in the non-structural genes (Estes and Kapikian, 2007; Kirkwood, 2010). Strain diversity is one of several suggested factors that could contribute to lower vaccine efficacy in Africa. Protection against rare, novel or mixed RVA strains by RV vaccines has not yet been conclusively determined (Kirkwood, 2010). However, mixed RV genome constellations (containing, for example, some segments of genotype 1 and some of genotype 2) may be less fit than parental strains and thus unable to compete with them (McDonald et al., 2009). Here, we analysed RV-positive samples from sub-Saharan Africa that appear to contain more than one infecting RV strain to determine the genetic diversity and possible origins of genome segments contained in mixed and/or co-infected RV samples.
2. Materials and methods
2.1 Ethics approval
Permission to conduct this study was sought from the Medunsa Research Ethics Committee (MREC) of the University of Limpopo, Medunsa Campus (MREC/P/108/2013: PG). To ensure confidentiality, patient names and records were dissociated from their unique laboratory identifiers.
2.2 Sample collection
Human stool samples (n=13) were obtained from children less than 5 years old who were hospitalised with acute gastroenteritis from six African countries: South Africa (n=7), Cameroon (n=1), Botswana (n=1), Senegal (n=1), Swaziland (n=1) and Zimbabwe (n=2). Bovine (n=1) and porcine (n=1) stool samples were from South Africa. Samples were stored at -20 °C at the Medical Research Council-Diarrhoeal Pathogens Research Unit (MRC-DPRU), WHO Rotavirus Regional Reference Laboratory in South Africa (WHO RRL-SA) until future use.
2.2 Sample inclusion criteria
All stool samples were previously screened for RVA antigen using a commercially available RV antigen kit (ProSpecT™ RV Microplate Kit, Oxoid Limited, United Kingdom). The mixed study samples were selected from a multitude of overall positive samples that exhibited single G and P genotypes and normal RNA profiles on polyacrylamide gel electrophoresis (PAGE) (Herring et al., 1982). They were selected either because they presented with more than 11 RV segments during PAGE, or demonstrated to harbour more than one RV strain upon G and P genotyping (Gentsch et al., 1992; Góuvea et al., 1990), or both. In summary, the study RV strains were assigned G and P genotype combinations before (conventional PCR) and after sequencing (Table 1). These genotypes were to some extent previously defined as common (e.g. G1P[8], G9P[8] and G9P[6]), rare (e.g. G1P[4] and G8P[4]), emerging (e.g. G12s), Mixed G and /or P-types (Bányai et al., 2012; Dóró et al., 2014; Santos and Hoshino, 2005; Seheri et al., 2014; Todd et al., 2010) and untypeable RV strains in human and animal populations.
Table 1.
Initial versus sequencing genotyping results for study strains. In addition, PAGE analyses of strains MRC-DPRU4680 and MRC-DPRU2605 displayed electropherotypes comparable to those of RVB and RVC, respectively.
| STRAIN NAME | INITIAL GT RESULTS | SEQUENCING GT RESULTS | ||
|---|---|---|---|---|
| VP7 (G) | VP4 (P) | VP7 (G) | VP4 (P) | |
| RVA/Human-wt/SEN/MRC-DPRU4680/2010/G1G6P[8] | G1, G9 | P[6] | G1, G6 | P[8] |
| RVA/Human-wt/ZAF/MRC-DPRU1893/2009/G8P[6] | untypeable | G8 | P[6] | |
| RVA/Human-wt/ZAF/MRC-DPRU228/2009/G2G1P[6] | untypeable | G2, G1 | P[6] | |
| RVA/Human-wt/CMR/MRC-DPRU3046/XXXX/G2G12P[4] | G2 | P[4], P[6] | G2, G12 | P[4] |
| RVA/Human-wt/BWA/MRC-DPRU2605/2007/G6G8P[6] | untypeable | G6, G8 | P[6] | |
| RVA/Human-wt/SWZ/MRC-DPRU4390/2010/G8G9P[6] | G8 | P[4] | G8, G9 | P[6] |
| RVA/Human-wt/ZAF/MRC-DPRU1255/2005/G9G12P[6] | G9 | P[6] | G9, G12 | P[6] |
| RVA/Human-wt/ZAF/MRC-DPRU2716/2008/G8P[4]P[8] | G1 | P[8] | G8 | P[4], P[8] |
| RVA/Human-wt/ZAF/MRC-DPRU1195/2009/G2P[6]P[8] | G1 | P[8] | G2 | P[6], P[8] |
| RVA/Human-wt/ZAF/MRC-DPRU309/2010/G12P[8] | untypeable | G12 | P[8] | |
| RVA/Human-wt/ZAF/MRC-DPRU1491/2010/G2P[4]P[8] | G1 | P[4] | G2 | P[4], P[8] |
| RVA/Human-wt/ZWE/MRC-DPRU1158/XXXX/G2G9P[6] | G12 | P[4], P[6] | G2, G9 | P[6] |
| RVA/Human-wt/ZWE/MRC-DPRU3348/2010/G9P[8] | G9 | P[8] | G9 | P[8] |
| RVA/Cow-wt/ZAF/MRC-DPRU3010/2009/G6P[5] | untypeable | G6 | P[5] | |
| RVA/Pig-wt/ZAF/MRC-DPRU1567/2008/G5P[6] | untypeable | G5 | P[6] | |
Italics, agreement between genotyping and sequencing data; Bold Italics, disagreement between genotyping and sequencing data; Underlined Italics, additional segment identified by sequencing but not by genotyping. RVA, Group A rotavirus; RVB, Group B rotavirus; RVC, Group C rotavirus; wt, wildtype; ZAF, South Africa; CMR, Cameroon; BWA, Botswana; SEN, Senegal; SWZ, Swaziland; ZWE, Zimbabwe; MRC, Medical Research Council; DPRU, Diarrhoeal Pathogens Research Unit; VP, viral structural protein; NSP, viral non-structural protein; GT, genotyping.
2.3 Whole-genome sequencing
The dsRNA genomes were extracted following previously described methods (Jere et al., 2011; 2012; Nyaga et al., 2013; Potgieter et al., 2009), and RNA sequencing performed as described previously (Magagula et al., 2015; Nyaga et al., 2014). Briefly, 11 one-step RT-PCR reactions (QIAGEN OneStep RT-PCR Kit, QIAGEN, Hilden, Germany) were performed to amplify each full-length genome segment using segment-specific primers as described elsewhere (Magagula et al., 2015; Martinez et al., 2014; Nyaga et al., 2014). The products were quantitated using a SYBR green dsDNA detection assay (SYBR Green I Nucleic Acid Gel Stain, Thermo Fisher Scientific, Waltham, MA, USA), and all 11 RT-PCR products for each genome were pooled in equimolar amounts. Pooled amplicons for each sample were barcoded and sequenced using the Ion Torrent (Thermo Fisher Scientific, Waltham, MA, USA) and/or MiSeq (Illumina, Inc., San Diego, CA, USA) next-generation sequencing (NGS) platforms, according to manufactures specifications. The Ion Torrent was the primary platform used on all samples and the MiSeq Illumina platform was used to fill gaps or sequence genome segments where the Ion Torrent did not yield enough sequence data.
The raw sequencing reads were de-multiplexed by barcode and, when available, reads from both NGS platforms were combined for each sample before assembly. Reads were trimmed and de novo assembled using CLC Bio's clc_novo_assemble program, and the resulting contigs were searched against custom full-length RVA segment databases to identify the closest reference sequence for each segment. Any contig that shared a percent nucleotide identity of 95 % or less was used to identify additional RV reference sequence(s) for use in the final mapping assembly. All sequence reads were then mapped to the selected reference segments using CLC Bio's clc_ref_assemble_long program. At loci where sequencing reads for the Illumina and/or Ion Torrent data agreed on a variation compared with the reference sequence, the reference sequence was updated to reflect the difference. A final mapping assembly of all reads to the updated reference sequences was performed with CLC Bio's clc_ref_assemble_long program.
2.4 Sequence analyses
For each genome segment, multiple sequence alignments were made using the MUSCLE algorithm implemented in MEGA 6 (Tamura et al., 2013). Maximum likelihood phylogenies were inferred for each genome segment based on their nucleotide sequences and included the study strains, as well as sequences of corresponding genome segments from selected RV strains available in GenBank, which were selected based on the genotype of the studied strains and most of which belonged to either of the three major genogroups; Wa, DS-1 and AU-1-like (Fig. 2 A-K). The best nucleotide substitution models were selected based on the corrected Akaike Information Criterion (AICc) value as implemented in MEGA 6. Models used in this study were GTR+I+G (NSP1, NSP2, NSP3, VP1, VP2, VP3, VP4, VP6 and VP7), TN93+G (NSP4) and GTR+G (NSP5). In addition, nucleotide and amino acid distance matrices for each genome segment were computed in MEGA 6 using the p-distance model.
Fig. 2.






A-K: Maximum likelihood phylogenetic trees inferred using MEGA 6 with bootstrapping show the evolutionary relationships among the complete coding nucleotide sequences for each rotavirus gene – VP7 (A), VP4 (B), VP6 (C), VP1 (D), VP2 (E), VP3 (F), NSP1 (G), NSP2 (H), NSP3 (I), NSP4 (J) and NSP5 (K) – from human and animal study strains and selected strains in GenBank. The scale for each tree indicates the average number of nucleotide substitutions per site, and bootstrap values ≥ 70% are shown for 500 replicates. The clades for each viral protein genotype are labelled in each tree. For the VP7 and VP4 trees, representative strains from different lineages were used to determine in which lineage the study strains clustered. The black squares represent the study strains in all trees.
2.5 Identification of genotype constellations
The online classification genotyping tool for RVA strains, RotaC v 2.0 (Maes et al., 2009), was used to assign genotypes to each genome segment of the study strains, which were subsequently employed to obtain the genome constellations.
2.6 Accession numbers
The nucleotide and deduced amino acid sequences of all genome segments were deposited in the National Centre of Biotechnology Information (NCBI) database, GenBank, under the accession numbers listed in Supplementary data 1. For simplicity, the common name for each study strain name has been used as follows: MRC-DPRU228, MRC-DPRU1893, MRC-DPRU3046, MRC-DPRU2605, MRC-DPRU3010, MRC-DPRU1567, MRC-DPRU4680, MRC-DPRU4390, MRC-DPRU1255, MRC-DPRU1255, MRC-DPRU2716, MRC-DPRU1195, MRC-DPRU309, MRC-DPRU1491, MRC-DPRU1158 and MRC-DPRU3348 (see full nomenclature in Table 1 and/or 2).
Table 2.
Complete genome constellations for the study strains displaying mixed/co-infected genotypes.
| STRAIN NAME | 1COMPLETE GENOTYPE CONSTELLATION | ASSIGNED GENOGROUP | 2GENOME SEGMENTS WITH > 2 SEQUENCES |
|---|---|---|---|
| RVA/Human-wt/SEN/MRC-DPRU4680/2010/G1G6P[8] | G1,G6-P[8]-I1,I2-R1,R2-C2-M1-A1,A2-N1,N1,N2-T1,T1,T2-E1,E2-H1 | Wa-like/DS-1-like | 1, 5, 6, 7, 8, 9 and 10 |
| RVA/Human-wt/ZAF/MRC-DPRU1893/2009/G8P[6] | G8-P[6]-I2,I2-R2,R1-C2-M2-A2-N2,N1-T2,T2-E2,E2-H2 | DS-1-like | 1, 6, 7, 8 and 10 |
| RVA/Human-wt/ZAF/MRC-DPRU228/2009/G2G1P[6] | G2,G1-P[6]-I2-R2-C2-M2-A2-N2-T2-E2-H2 | DS-1-like | 9 |
| RVA/Human-wt/CMR/MRC-DPRU3046/XXXX/G2G12P[4] | G2,G12-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2 | DS-1-like | 9 |
| RVA/Human-wt/BWA/MRC-DPRU2605/2007/G6G8P[6] | G6,G8-P[6]-I2-R2-C2-M2-A2-N2-T2-E2-H2 | DS-1-like | 9 |
| RVA/Human-wt/SWZ/MRC-DPRU4390/2010/G8G9P[6] | G8,G9P[6]-I2-R2-C2-M2-A2-N2,N1-T2-E2-H2 | DS-1-like | 8 and 9 |
| RVA/Human-wt/ZAF/MRC-DPRU1255/2005/G9G12P[6] | G9,G12-P[6]-I1-R1-C1-M1-A1,A1-N1-T1-E1-H1 | Wa-like | 5 and 9 |
| RVA/Human-wt/ZAF/MRC-DPRU2716/2008/G8P[4]P[8] | G8-P[4],P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2 | DS-1-like | 4 |
| RVA/Human-wt/ZAF/MRC-DPRU1195/2009/G2P[6]P[8] | G2-P[6],P[8]-I2-R2-C2-M2,M1-A2-N2-T2-E2-H2 | DS-1-like | 3 and 4 |
| RVA/Human-wt/ZAF/MRC-DPRU309/2010/G12P[8] | G12-P[8]-I1-R1-C1,C2-M1-A1,A2-N1,N2-T1-E1,E2-H1 | Wa-like | 2, 5, 8 and 10 |
| RVA/Human-wt/ZAF/MRC-DPRU1491/2010/G2P[4]P[8] | G2-P[4],P[8]-I2-R2-C2,C1-M2,M1-A2-N2-T2-E2-H2 | DS-1-like | 2, 4 and 3 |
| RVA/Human-wt/ZWE/MRC-DPRU1158/XXXX/G2G9P[6] | G2,G9-P[6]-I2-R2,R2-C2,C2-M2,M2-A2,A2-N2,N2-T2-E2,E2-H2 | DS-1-like | 1, 2, 3, 5, 8, 9 and 10 |
| RVA/Human-wt/ZWE/MRC-DPRU3348/2010/G9P[8] | G9-P[8]-I1-R1-C1-M1-A1-N1,N1,N2-T2-E1-H1 | Wa-like | 8 |
| RVA/Cow-wt/ZAF/MRC-DPRU3010/2009/G6P[5] | G6-P[5]-I2,I2-R2-C2-M2-A3-N2-T6-E2-H3,H3 | Bovine- like | 6 and 11 |
| RVA/Pig-wt/ZAF/MRC-DPRU1567/2008/G5P[6] | G5-P[6]-I5,I5-R1-C1,C1-M1,M1-A8,A8-N1,N1-T1,T7-E1-H1 | porcine-like | 2, 3, 5, 6, 7 and 8 |
VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5
The specific genome segments that had two or more sequences with the same or different genotypes are listed. The genome segments are numbered as follows: 1, VP1; 2, VP2; 3, VP3; 4, VP4; 5, NSP1; 6, VP6; 7, NSP3; 8, NSP2; 9, VP7; 10, NSP4; and 11, NSP5.
RVA, Group A rotavirus; wt, wildtype; ZAF, South Africa; CMR, Cameroon; BWA, Botswana; SEN, Senegal; SWZ, Swaziland; ZWE, Zimbabwe; MRC, Medical Research Council; DPRU, Diarrhoeal Pathogens Research Unit; VP, viral structural protein; NSP, viral non-structural protein.
3. Results
3.1 dsRNA electrophoretic patterns, nucleotide sequencing and assignment of genotypes
With the exception of strains MRC-DPRU228 and MRC-DPRU3046, study samples displayed more than 11 RVA segments with characteristic RVA electropherotype profiles on PAGE. The exhibited RVA RNA patterns were typical to those seen in children infected with more than one RVA strain (Fig. 1). Strains MRC-DPRU4680 and MRC-DPRU2605 displayed electropherotypes comparable to those of group B (RVB) and C rotaviruses (RVC), respectively (Data not shown). The two strains (MRC-DPRU4680 and MRC-DPRU2605) were regarded as possible inter-group co-infections with RVA because they were positive by EIA and for the former also by G and P priming, which are RVA specific (Table 1). The full-length nucleotide sequences for the coding region of all amplified RV genome segment present in each study sample were determined, and the genotypes for the genome segment encoding VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5 were assigned (Maes et al., 2009) (Table 2). In all 15 study samples, more than 11 segments were sequenced, and at least one sequence was obtained for each of the 11 RV genome segments. When two or more sequences existed for the same genome segment, they were either of the same genotype (e.g., the two N1 NSP2 gene segments of MRC-DPRU1567) or different genotypes (e.g., N1 and N2 for the NSP2 gene segment of MRC-DPRU1893) (Table 2). In all but one study sample, at least one segment was of the latter category and therefore was considered to have a mixed genotype. In the case of sample MRC-DPRU3010, no segment had a mixed genotype because the two duplicated segments (VP6 and NSP5) were of the same genotype. The detection of ≥ 2 sequences for the same genome segment in the study samples further supported the evidence of mixed infections suggested by the PAGE and/or RT-PCR genotyping findings. In order to differentiate multiple sequences for the same genome segment detected within a given study sample, the designation a, b or c was arbitrarily assigned and included after the common name in the strain nomenclature during sequence analyses (Fig. 2A-K and Supplementary data 2).
Fig. 1.
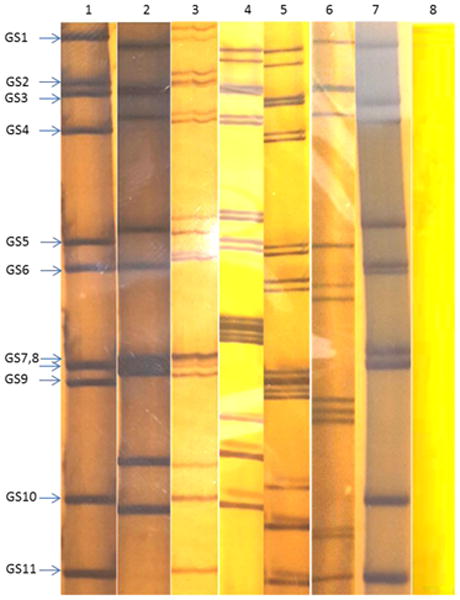
Rotavirus RNA migration patterns (electropherotypes) consisting of more than 11 rotavirus segments in some of the study samples using polyacrylamide gel electrophoresis (PAGE) compared with the typical RVA migration pattern of 4:2:3:2. The images are taken from different PAGE gels for normal 11-segmented control viruses and study strains from samples with presumed mixed rotavirus co-infections. Lane 1: control (normal long electropherotype pattern); lane 2: control (normal short electropherotype pattern); lane 3: strain MRC-DPRU1491 (short and long patterns); lane 4: strain MRC-DPRU1195 (mixed long and short pattern); lane 5: strain MRC-DPRU309 (mixed short and long patterns); lane 6: strain MRC-DPRU1158 (mixed short patterns); lane 7: strain MRC-DPRU3010 (mixed long pattern); and lane 8: negative control. The three controls were inserted in order to demonstrate how the normal long, normal short and negative appears but not for the purposes of comparing the migration sizes because the electrophoresis was performed distinctly. RVA, Group A rotavirus; GS, genome segment.
3.2 Nucleotide and amino acid sequence analysis of the study strains
3.2.1 Analysis of complete RV genome sequences derived from sample MRC-DPRU4680
Sample MRC-DPRU4680 was collected in 2010 from a 30-month-old female child in Senegal who was suspected to be infected by both RVA and RVB (Ina Peenze, unpublished data). When this sample was fully sequenced, both Wa-like (genotype 1) and DS-1-like (genotype 2) sequences were detected for genome segments 6 (VP6), 1 (VP1), 5 (NSP1), 8 (NSP2), 7 (NSP3) and 10 (NSP4). In addition, it contained two genome segments 9 (VP7) sequences belonging to genotypes G1 and G6. This suggests that the child was infected with two distinct strains, one of each belonging to the Wa-like and DS-1-like genogroups. The four genome segments having only a single sequence – genome segment 4 (VP4), 2 (VP2), 3 (VP3) and 11 (NSP5) – were either not amplified (potentially due to poor primer matching) or had sequences that were too similar to be distinguished. For two genome segments, 8 (NSP2) and 7 (NSP3), three sequences were generated of which two were Wa-like (genotype 1) but distinct from each other. The sequencing strategy employed would not rule out the possibility of a co-infecting RVB strain as well.
Phylogenetic analyses showed that most of the nucleotide sequences for RV MRC-DPRU4680 genome segments 1 to 11 clustered with those of the corresponding cognate human RVs, with the exception of one genome segment 6 (VP6) sequence. The DS-1-like VP6 and VP7sequences of MRC-DPRU4680a, shared ancestry with bovine RVs, as evidenced by clustering with the bovine study sample MRC-DPRU3010, the bovine WC3 strain and multiple RotaTeq strains that were all derived from bovine RVs (Matthijnssens et al., 2010). On the contrary, MRC-DPRU4680b, clustered with other human Wa-like strains, such as Matlab13 and 9J from Bangladesh and Nicaragua, respectively. This suggests that one of the strains infecting the child was potentially derived from reassortment events between bovine and human RVs (Fig. 2C).
Furthermore, strain MRC-DPRU4680a (DS-1-like genogroup) shared nucleotide (nt) amino acid (aa) similarities of nt (aa) 79.2-95.1 % (90.2-99.7 %) with other study strains under the DS-1-like backbone, while strain MRC-DPRU4680b (Wa-like genogroup) shared nt (aa) similarities of 83.4-96.6 % (91.4-99.7 %) with other study strains under the Wa-like backbone (Supplementary data 2A and B). Analogous similarity matrices were also created based on multiple sequence alignments for the genome segment 10 (NSP4) sequences MRC-DPRU4680a and MRC-DPRU4680b (Supplementary data 2C and D).
3.2.2 Analysis of complete RV genome sequences derived from sample MRC-DPRU1893
Sample MRC-DPRU1893 was collected in 2009 from a child in South Africa and was initially untypeable, although more than 11 segments were seen on PAGE, which displayed the short (DS-1-like) profile. Full genome sequencing identified two distinct sequences for genome segments 6 (VP6), 1 (VP1), 8 (NSP2), 7 (NSP3) and 10 (NSP4). For two genome segments, the sequences were different enough to be classified as two different genotypes, either DS-1-like (genotype R2 and N2) or Wa-like (genotype R1 and N1). All other duplicate genome segments were of the DS-1-like genotype (I2, T2 and E2). Therefore, overall, the viruses in this sample belong to the DS-1-like genogroup, although there appears to be some reassortment with Wa-like sequences for two segments from some viruses. Alternatively, albeit less likely, a fully Wa-like virus was present in this sample, but only two segments could be amplified and sequenced. Phylogenetic analyses of sequences from this sample reveal the relatedness of the entire genome with cognate regional human strains, mostly from the neighbouring countries of Botswana, Swaziland and Zimbabwe (Fig. 2A-K).
3.2.3 Analysis of complete RV genome sequences derived from sample MRC-DPRU228
This sample was collected in 2009 from a child in South Africa and was identified as RV positive, although it was initially untypeable and its PAGE displayed a pure short electropherotype (DS-1-like) profile with no evidence of mixed genome segments. Sequencing detected two different VP7 genotypes, G2 and G1, while only one sequence was detected for each of the remaining segments, which belong to the DS-1-like genogroup 2. Phylogenetically, genome segments from the MRC-DPRU228 sample generally cluster with strains from Senegal and Cameroon, which are located in western and central Africa, respectively, as well as with other strains from southern Africa (Fig. 2A-K).
3.2.4 Analysis of complete RV genome sequences derived from sample MRC-DPRU3046
Sample MRC-DPRU3046 was collected from a child in Cameroon who was suspected to be infected by more than one RV strain when the sample was initially genotyped as a G2, P[4] and P[6] (Table 1). Sequencing detected two VP7 sequences having genotypes G2 and G12. No P[6] VP4 genotype was detected by sequencing, and single DS-1-like sequences were found for all other segments. Phylogenetically, most MRC-DPRU3046 genome segments cluster with cognate human strains from South Africa, Zimbabwe and/or Senegal (Fig. 2 A-K).
3.2.5 Analysis of complete RV genome sequences derived from sample MRC-DPRU2605
Sample MRC-DPRU2605 was collected in 2007 from a child in Botswana and was initially hypothesized to be a RVC by PAGE. During sequencing, RVA-specific primers amplified 12 genome segments that were assigned to a DS-1-like genogroup. The duplicated VP7 genome segments were assigned to the G6 and G8 genotypes. Just like strain MRC-DPRU4680, the sequencing strategy employed does not rule out the possibility of a co-infecting RVC strain as well, although PAGE electropherotype does not provide evidence of an additional viral strain in this sample. Phylogenetically, the genome segments from this sample primarily clustered with the cognate human strain MRC-DPRU4390 Swaziland (Fig. 2A-K).
3.2.6 Analysis of complete RV genome sequences derived from sample MRC-DPRU4390
Sample MRC-DPRU4390 was collected in 2010 from a 2-month-old female child in Swaziland and initially genotyped as a G8P[4] strain, although PAGE electropherotype suggested more than 11 segments may be present. The complete nucleotide sequences that were recovered were for genome segments primarily belonging to the DS-1-like genogroups. These included duplicate sequences for genome segments 9 (VP7) and 8 (NSP2). The two genotypes for genome segment 9 were G8 and G9 – as originally identified by genotyping –, whereas the two genotypes for genome segment 8 were N2 (DS-1-like) and N1 (Wa-like). Although a P[4] genotype was originally assigned through RT-PCR genotyping, only a P[6] nucleotide was derived through NGS. Phylogenetically, this strain clusters with cognate human strains across the genome and was very closely related to strain MRC-DPRU2605 from Botswana in most genome segments (Fig. 2A-K).
3.2.7 Analysis of complete RV genome sequences derived from sample MRC-DPRU1255
Sample MRC-DPRU1255 was collected in 2005 from a child in South Africa and was originally genotyped as G9P[6]. Sequencing confirmed this initial genotyping and also yielded an additional genome segment 9 (VP7) G12 complete nucleotide sequence. All other segments belonged to the Wa-like genogroup, including duplicate NSP1 segments, which were distinct from each other but both had A1 genotypes. Phylogenetic analyses of this strain revealed that the entire genome is closely related to cognate global human strains (Fig. 2A-K).
3.2.8 Analysis of complete RV genome sequences derived from sample MRC-DPRU2716
Sample MRC-DPRU2716 was collected in 2008 from a 3-month-old female child in South Africa and was initially genotyped as G1P[8], although the PAGE electropherotype suggested more than 11 segments were present. Sequencing recovered segments primarily belonging to the DS-1-like genogroup and included duplicate sequences for genome segment 4 (VP4) that were assigned genotypes P[4] and P[8], with the latter matching the originally identified VP4 genotype. Sequencing presented the VP7 genotype as a G8 rather than G1, as originally identified by genotyping. Phylogenetic analyses of this strain revealed that the entire genome is primarily related to cognate regional human strains (Fig. 2A-K).
3.2.9 Analysis of complete RV genome sequences derived from sample MRC-DPRU1195
Sample MRC-DPRU1195 was collected in 2009 from a 9-month-old female child in South Africa and was initially genotyped as G1P[8], although PAGE suggested more than 11 segments may be present (Fig. 1). Sequencing recovered segments primarily belonging to the DS-1-like genogroup and included duplicate sequences for genome segments 4 (VP4) and 3 (VP3). The two genotypes assigned to the derived genome segment 4 were P[6] and P[8], with the latter matching the originally identified VP4 genotype; the two genotypes for genome segment 3 were M2 (DS1-like) and M1 (Wa-like). Sequencing indicated that the VP7 genotype were G2 rather than G1, as originally identified by genotyping. Phylogenetic analyses of this strain revealed clustering with cognate regional human strains, with the exception that one genome segment 3 (VP3) sequence also clusters closely with strain MRC-DPRU4680 from Senegal (West Africa) (Fig. 2A-K).
3.2.10 Analysis of complete RV genome sequences derived from sample MRC-DPRU309
Sample MRC-DPRU309 was collected in 2010 from a 7-month-old female child in South Africa. It was shown to have more than 11 RNA segments upon PAGE analysis (Fig. 1) and was initially untypeable. Sequencing identified duplicated nucleotide sequences for genome segments 2 (VP2), 5 (NSP1), 8 (NSP2) and 10 (NSP4). Wa-like (genogroup 1) segments were identified for all genome segments, while the second sequences for duplicated genome segments are DS-1-like (genogroup 2). Phylogenetic analyses of this strain revealed that the entire genome is related to cognate global human strains that were previously assigned a similar genotype (Fig. 2A-K).
3.2.11 Analysis of complete RV genome sequences derived from sample MRC-DPRU1491
Sample MRC-DPRU1491 was collected in 2010 from a 6-month-old female child in South Africa and was originally genotyped as G1P[4]. PAGE analysis showed more than 11 RNA segments were present (Fig. 1). Sequencing identified duplicated sequences for genome segments 4 (VP4), 2 (VP2) and 3 (VP3). The two genotypes assigned to nucleotide sequences derived for genome segment 4 were P[4] and P[8], with the former matching the originally identified VP4 genotype. Although a G1 genotype was originally assigned to sample MRC-DPRU1491 by genotyping, derived VP7 encoding nucleotide sequence was assigned a G2 genotype. DS-1-like (genogroup 2) segments were identified for all genome segments, while the second sequences for duplicated genome segments 2 and 3 were Wa-like (genogroup 1). Phylogenetic analyses of this strain revealed clustering with cognate regional human strains, with the exception that one sequence each of genome segments 2 (VP2) and 4 (VP4) clusters closely with cognate global strains from either Australia or Bangladesh that were previously assigned similar genotype (Fig. 2A-K).
3.2.12 Analysis of complete RV genome sequences derived from sample MRC-DPRU1158
Sample MRC-DPRU1158 was collected in Zimbabwe from a 7-month-old female child. It was originally genotyped as G12P[4]P[6] and had more than 11 RNA segments by PAGE analysis (Fig. 1). Sequencing identified duplicate sequences for genome segments 9 (VP7), 1 (VP1), 2 (VP2), 3 (VP3), 5 (NSP1), 8 (NSP2) and 10 (NSP4). The two genotypes assigned to genome segment 9 were G2 and G9, which did not match the originally identified VP7 genotype, G12. The VP4 genotype, P[6], identified by sequencing matches one of the VP4 genotypes originally identified by genotyping. All remaining genome segments, including duplicated sequences were DS-1-like (genogroup 2). Phylogenetically, this strain primarily clusters with cognate human strains with similar genotypes in all genome segments (Fig. 2A-K), with exceptions for one genome segment 3 (VP3) and one genome segment 9 (VP7) sequence. The G9 clade VI sequence for genome segment 9 (VP7) clustered most closely with strains JP29-6 and JP32-4 of porcine origin, while one M2 sequence for genome segment 3 (VP3) clustered most closely with strain OVR762 of ovine origin (Fig. 2A and F). The pairwise identity matrices for the ovine-like M2 genome segment 3 (VP3) nt and aa sequences showed they shared nt (aa) identities of 76.3-87.9 % (81.2-93.9 %) with other DS-1-like study strains (Supplementary data 2E and F). Similarly, the G9 nt and aa sequences of genome segment 9 (VP7) shared low nt (aa) identities of 73.0-92.2 % (74.1-96.7 %) with other study strains (Supplementary data 2G and F).
3.2.13 Analysis of complete RV genome sequences derived from sample MRC-DPRU3348
Sample MRC-DPRU3348 was collected in 2010 from a 9-month-old male child in Zimbabwe. It was originally genotyped as G9P[8] and presented with more than 11 RVA segments upon PAGE analysis. Sequencing confirmed the G9P[8] genotype and identified at least one Wa-like sequence (genogroup 1) for all other genome segments except genome segment 7 (NSP3), which has a DS-1-like T2 genotype. In addition, sequencing identified three distinct consensus sequences for genome segment 8 (NSP2) where two consensus sequences were typical Wa-like N1 genotypes, while the third consensus sequence had a DS-1-like N2 genotype. Phylogenetic analyses of this strain revealed that the entire genome is related to cognate global human strains that were previously assigned similar genotypes (Fig. 2A-K).
3.2.14 Analysis of complete RV genome sequences derived from sample MRC-DPRU3010
Sample MRC-DPRU3010 was collected in South Africa in 2009 from a calf and was initially untypeable, although duplicate RNA genome segment 6 (VP6) bands were visible with PAGE analysis (Fig. 1). Sequencing confirmed that two sequences were present for genome segment 6 (VP6), both with I2 genotypes. There were also two genome segments 11 (NSP5) sequences, both of genotype H3. Overall, the segments for this sample belong to the DS-1-like genogroup. Phylogenetic analyses of this strain revealed clustering with cognate bovine strains with several exceptions. The two genome segments 6 (VP6) sequences were in the same cluster with human strains from Indonesia and Senegal, although they are both part of a larger clade that includes bovine-derived VP6 RotaTeq vaccine sequences (Fig. 2C). The genome segment 2 (VP2) sequence for MRC-DPRU3010 forms a distinct branch that has roughly similar cophenetic distances with the human and bovine C2 clades (Fig. 2E). The genome segment 10 (NSP4) sequence groups most closely with human E2 strains from southern and western Africa (Fig. 2J).
3.2.15 Analysis of complete RV genome sequences derived from sample MRC-DPRU1567
Sample MRC-DPRU1567 was collected in South Africa in 2008 from a 2-month-old piglet and was initially reported as untypeable, although more than 11 faint RNA segments were visualised by PAGE analysis (gel not shown). Sequencing showed that the majority of genome segments from this sample belong to the Wa-like genogroup. Duplicate consensus sequences were obtained for genome segments 2 (VP2), 3 (VP3), 5 (NSP1), 6 (VP6), 7 (NSP3) and 8 (NSP2) and were assigned genotypes C1 and C1, M1 and M1, A8 and A8, I5 and I5, TI and T7 and N1 and N1, respectively. Phylogenetic analyses of this strain revealed clustering with cognate porcine strains that were assigned similar genotypes with two exceptions. The genome segment 4 (VP4) sequence was part of a monophyletic clade containing human sequences, although no other porcine VP4 sequences were included in this phylogeny (Fig. 2B). The T7 genome segment 7 (NSP3) sequence clusters with the only other available T7 sequence, which is of bovine origin (Fig. 2I). The genome segment 10 (NSP4) sequence clustered most closely with a human E9 strain, as well as a more distantly related porcine E9 sequence (Fig. 2J).
4. Discussion
Most RVA characterisation studies are focused mainly on G (VP7) and P (VP4) genotyping using RT-PCR and Sanger sequencing (Rahman et al., 2007). Detection of multiple segments for any of the other nine RV genome segments would thus go unnoticed. In addition, we have shown here that G and P genotyping is not always corroborated by full-genome sequencing in the case of mixed infections. However, this can be explained due to the possibility that some of the genotypes detected by conventional PCR could have been missed because sequence specific priming is likely to introduce selective bias of this sort. This is supported e.g. by strain MRC-DPRU1491, initially genotyped as G1P[4] but was sequenced as G2P[4], P[8]. The fact that sequencing picked an extra segment of P[8] (mostly associated with G1) and extra genotypes characteristic of C1 and M1 suggests that G1 probably existed in the stool sample but was missed by sequencing. Likewise, for some other study strains e.g. MRC-DPRU4680, there was a possibility that the initially typed G1,G9P[6] were also present in the sample although it was sequenced as G1,G6P[8]. This is so because both complete genotypes 1 and 2 were reported in the constellation. Furthermore, the primer set used for conventional genotyping only entailed common G types (G1-G4, G8, G9 and G12), thus did not contain a G6 specific primer, the reason why it was missed. In addition, cross reactivity among primers used for conventional PCR is common. Similarly, these could have been the case in other study samples. Therefore, we cannot conclusively say that the initial genotyping results were incorrect. However, the superiority of NGS over conventional RT-PCR was also attested by typing samples previously reported as untypeable (Table 1). Newer RV studies are taking advantage of NGS technologies, coupled with improved data analysis software, to obtain full-genome sequence data (Jere et al., 2011; Magagula et al., 2015; Mlera et al., 2013; Nyaga et al., 2013, 2014).
The specimens characterised in this study were part of strain surveillance studies conducted in Africa by the WHO RRL-SA. Over the years, this surveillance network has determined which strains are circulating in Africa using the RVA binary classification system by genotyping the two outer capsid proteins (G and P). This method has identified infections containing multiple G and/or P genotypes, as reported from different African countries (Abebe et al., 2014; Boula et al., 2014; Hokororo et al., 2014; Kiulia et al., 2014; Mukaratirwa et al., 2014; Odiit et al., 2014; Pursem et al., 2014; Seheri et al., 2014; Tsolenyanu et al., 2014). Most strains with G1P[8], G3P[8], G4P[8] and G9P[8] G and P genotype combinations usually belong to the Wa-like genogroup because they have a Wa-like genotype constellation (Gx-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1) composed primarily of genotype 1 genes, whereas G2P[4] strains are considered DS-1-like genogroup members because they have a DS-1-like constellation (G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2) composed of genotype 2 genes (Matthijnssens et al., 2008, 2011; Matthijnssens and Van Ranst, 2012). NGS of the 15 selected study samples, most of which had been previously reported as mixed G and P genotypes and/or samples with extra RV genome segments observed during PAGE analyses, provided full open reading frame (ORF) nucleotide sequences for all genome segments and confirmed that at least one genome segment in each sample had distinct duplicate consensus sequences.
Electropherotyping by PAGE is recognised as a useful and cost-effective method for RV epidemiological studies (Holmes, 1996). RNA genome segments of Wa- and DS-1-like genogroup strains exhibit either long or short electropherotypes. Long electropherotypes are most commonly associated with the Wa-like genogroup, while short electropherotypes are most often associated with the DS-1-like genogroup. There was concordance between the study samples shown to contain more than one RVA strain by PAGE and the sequencing results; furthermore, the identification of primarily Wa-like (long electropherotype) or DS-1-like (short electropherotype) genogroups by PAGE was upheld by the sequencing results. In some cases where there were both genotype 1 and 2 segments, PAGE results showed mixed long and short electropherotypes e.g. strain MRC-DPRU309.
Sequencing revealed distinct duplicated genome segment consensus sequences occurring across the RVA genomes. This study illustrates that infection with multiple RV strains of different genotypes may result in novel RV progeny following interspecies transmission and genome segment reassortment events. Furthermore, this study provides the first report from Africa of a G9 human strain whose genome segment 9 (VP7) is in lineage VI and is closely related to porcine G9 sequences; previously, all reported African human G9 strains have belonged to lineage III (Page et al., 2010; Nyaga et al., 2013; Esona et al; 2013). This clearly demonstrates that gene exchange mechanisms that generate such RVA strains could be a result of reassortment events and/or interspecies transmission occurring as a result of mixed infections. Interspecies transmission is also evident in one sequence for genome segment 6 (VP6) of strain MRC-DPRU4680, which clusters closely with cognate bovine strains. Some RV strains, such as the G8s and/or P[6]s, are believed to have originated from animals. The G8s are thought to be of bovine and Artiodactyla origin, while the P[6]s are believed to be of porcine origin (Martella et al., 2006; Papp et al., 2013). Gene segments of both strains are frequently detected in humans, especially in Africa (Seheri et al., 2014).
To our knowledge, this is the first study to analyse multiple RVA whole-genome sequences from individuals infected with more than one RVA strain using specific priming and not with the more sophisticated sequence independent amplification technique (Jere et al., 2011; Mlera et al., 2013; Nyaga et al., 2013). Interactions between host species could lead to reassortant strains as a result of interspecies transmission. Reassortant strains have been reported globally, including strains from Africa such as MRC-DPRU9317 (South Africa), GH018-08 and GH019-08 (Ghana), KisB332 (DRC) and MWI-860 (Malawi), which all had unusual genotype constellations (Dennis et al., 2014; Heylen et al., 2014; Nakagomi et al., 2013; Nyaga et al., 2013). Furthermore, the study also reports the first porcine (MRC-DPRU1567) and bovine (MRC-DPRU3010) mixed infections.
RVA vaccine effectiveness has been reported to be relatively low in developing countries compared with data from developed countries (Armah et al., 2010; Madhi et al., 2010; Sow et al., 2012; Steele et al., 2012). Mixed infections and reassortant strains may be a factor that contributes to low vaccine effectiveness that has not been extensively explored in Africa or other low income countries. Because most RV surveillance studies focused primarily on G and P genotyping, information on reassortment events occurring among other genome segments is missing but essential (Dóró et al., 2014). Evaluating the additional nine genome segments of the RV genome provided information on the genetic mechanisms contributing to RVA diversity, which could impact the pathogenicity, disease severity and transmission of RVs. By characterising the complete genome sequences of RV samples, this study has demonstrated that multiple RV strains from sub-Saharan Africa are infecting hosts with unpredictable variations in their genome segment combinations. This could certainly lead to genetic shifts resulting in new RV strains with unpredictable capacities for human-to-human spread, disease severity and abilities of current and future vaccines to protect against them.
The continued use of the current licenced RV vaccines is highly recommended as they have shown to provide good efficacy against both Wa- and DS-1-like genotypes, albeit lower for DS-1-like from some settings (Armah et al., 2010; Madhi et al., 2010). RotaTeq has DS-1-like backbone included in its formulation but not entirely (Vesikari et al., 2006), hence the question can a pure DS-1-like vaccine improve efficacy especially in Africa where the vaccine efficacy results are lower compared to the developed world? Combining the doses in the universal mass vaccination programs, e.g. Rotarix which is on a pure G1P[8] genetic backbone (Ruiz-Palacios et al., 2006), with another strain on a DS-1-like backbone would probably yield better efficacy. It could also be interesting to research on a vaccine candidate derived from a mixed genotype strain from regions with great diversity of RV genotypes to determine if it can provide better efficacy.
Supplementary Material
Highlights.
Full length amplicons for mixed infections from six African countries were sequenced.
Genomes had at least one segment in each sample for which duplicate sequences existed.
The first co-infections on porcine and bovine samples are reported.
The first report on African G9 human strain in lineage VI.
Acknowledgments
The financial assistance of the National Research Foundation (NRF), the South African Medical Research Council (MRC) and the Poliomyelitis Research Foundation (PRF), under grant numbers PRF14/05 (Major Impact) and PRF13/62 (PhD) are hereby acknowledged. The project was also funded in part by US federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C (Genome Sequencing Center for Infectious Diseases). Opinions expressed and conclusions arrived at, are those of the authors and are not necessarily to be attributed to the sponsors.
The data for this manuscript and its preparation was generated while DEW was employed at JCVI. The opinions expressed in this article are the authors’ own and do not reflect the views of the Centers for Disease Control, the Department of Health and Human Services, or the United States government.
We thank Kebareng Rakau, Nonkululeko Magagula and Dorah Nkolele for technical assistance at the MRC/Diarrhoeal Pathogens Research Unit. We also thank the Virology Group at JCVI, especially Ewen F. Kirkness, Nadia Fedorova and Susmita Shrivastava for their bioinformatics support. In addition, we thank all the sample collection teams from South Africa, Senegal, Swaziland, Botswana, Cameroon and Zimbabwe that provided the samples to the WHO RRL-SA.
Footnotes
Declaration of potential conflict of interest: No potential conflict of interest was declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Martin M. Nyaga, Email: wamodylan@gmail.com.
Khuzwayo C. Jere, Email: Khuzwayo.Jere@liverpool.ac.uk.
Mathew D. Esona, Email: mdi4@cdc.gov.
Mapaseka L. Seheri, Email: mpasi.seheri@gmail.com.
Karla M. Stucker, Email: kstucker@jcvi.org.
Rebecca A. Halpin, Email: RHalpin@jcvi.org.
Asmik Akopov, Email: aakopov@jcvi.org.
Timothy B. Stockwell, Email: tstockwell@jcvi.org.
Ina Peenze, Email: inapeenze@yahoo.com.
Amadou Diop, Email: amadoudioplaba@yahoo.fr.
Kader Ndiaye, Email: kadern@pasteur.sn.
Angeline Boula, Email: boulaayc@yahoo.fr.
Gugu Maphalala, Email: gpmaphalala@gmail.com.
Chipo Berejena, Email: cberejena@gmail.com.
Jason M. Mwenda, Email: mwendaj@afro.who.int.
A. Duncan Steele, Email: duncan.steele@gatesfoundation.org.
David E. Wentworth, Email: DWentworth@cdc.gov.
M. Jeffrey Mphahlele, Email: jeffrey.mphahlele@mrc.ac.za.
References
- Abebe A, Teka T, Kassa T, Seheri M, Beyene B, Teshome B, Kebede F, Habtamu A, Maake L, Kassahun A, Getahun M, Mitiku K, Mwenda JM. Hospital-based surveillance for rotavirus gastroenteritis in children younger than 5 years of age in Ethiopia: 2007-2012. Pediatr Infect Dis J. 2014;33(Suppl):S28–S33. doi: 10.1097/INF.0000000000000048. [DOI] [PubMed] [Google Scholar]
- Armah GE, Sow SO, Breiman RF, Dallas MJ, Tapia MD, Feikin DR, Binka FN, Steele AD, Laserson KF, Ansah NA, Levine MM, Lewis K, Coia ML, Attah-Poku M, Ojwando J, Rivers SB, Victor JC, Nyambane G, Hodgson A, Schödel F, Ciarlet M, Neuzil KM. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in sub-Saharan Africa: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;376:606–614. doi: 10.1016/S0140-6736(10)60889-6. [DOI] [PubMed] [Google Scholar]
- Banga-Mingo V, Waku-Kouomou D, Gody JC, Esona MD, Yetimbi JF, Mbary-Daba R, Dahl BA, Dimanche L, Koyazegbe TD, Tricou V, Cavallaro KF, Guifara G, Bowen MD, Gouandjika-Vasilache I. Molecular surveillance of rotavirus infection in Bangui, Central African Republic, October 2011-September 2013. Infect Genet Evol. 2014;14:S311–S316. doi: 10.1016/j.meegid.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bányai K, László B, Duque J, Steele AD, Nelson EA, Gentsch JR, Parashar UD. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: insights for understanding the impact of rotavirus vaccination programs. Vaccine. 2012;30(Suppl. 1):A122–A130. doi: 10.1016/j.vaccine.2011.09.111. [DOI] [PubMed] [Google Scholar]
- Boula A, Waku-Kouomou D, Njiki Kinkela M, Esona MD, Kemajou G, Mekontso D, Seheri M, Ngum Ndze V, Emah I, Ela S, Dahl BA, Kobela M, Cavallaro KF, Etoundi Mballa GA, Genstch JR, Bowen MD, Koki Ndombo P. Molecular surveillance of rotavirus strains circulating in Yaoundé, Cameroon, September 2007-December 2012. Infect Genet Evol. 2014;14:1567–1348. doi: 10.1016/j.meegid.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis FE, Fujii Y, Haga K, Damanka S, Lartey B, Agbemabiese CA, Ohta N, Armah GE, Katayama K. Identification of novel Ghanaian G8P[6] human-bovine reassortant rotavirus strain by next generation sequencing. PLoS One. 2014;9(6) doi: 10.1371/journal.pone.0100699. eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dóró R, László B, Martella V, Leshem E, Gentsch J, Parashar U, Bányai K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect Genet Evol. 2014;14:S1567–S1348. doi: 10.1016/j.meegid.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esona MD, Geyer A, Page N, Trabelsi A, Fodha I, Aminu M, Agbaya VA, Tsion B, Kerin TK, Armah GE, Steele AD, Glass RI, Gentsch JR. Genomic characterization of human rotavirus G8 strains from the African rotavirus network: relationship to animal rotaviruses. J Med Virol. 2009;81:937–951. doi: 10.1002/jmv.21468. [DOI] [PubMed] [Google Scholar]
- Esona MD, Mijatovic-Rustempasic S, Foytich K, Roy S, Banyai K, Armah GE, Steele AD, Volotão EM, Gomez MM, Silva MF, Gautam R, Quaye O, Tam KI, Forbi JC, Seheri M, Page N, Nyangao J, Ndze VN, Aminu M, Bowen MD. Gentsch JR. Human G9P[8] rotavirus strains circulating in Cameroon, 1999-2000: Genetic relationships with other G9 strains and detection of a new G9 subtype. Infect Genet Evol. 2013;18:315–324. doi: 10.1016/j.meegid.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esona MD, Steele D, Kerin T, Armah G, Peenze I, Geyer A, Page N, Nyangao J, Agbaya VA, Trabelsi A, Tsion B, Aminu M, Sebunya T, Dewar J, Glass R, Gentsch J. Determination of the G and P types of previously nontypeable rotavirus strains from the African Rotavirus Network, 1996-2004: Identification of unusual G types. J Infect Dis. 2010;202:S49–54. doi: 10.1086/653552. [DOI] [PubMed] [Google Scholar]
- Estes MK, Kapikian AZ. Rotaviruses. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. 5th. Vol. 2. Lippincott Williams and Wilkins/Wolter Kluwer; Philadelphia: 2007. pp. 1917–1974. [Google Scholar]
- Fernandes EG, Sato HK, Leshem E, Flannery B, Konstantyner TC, Veras MA, Patel MM. Impact of rotavirus vaccination on diarrhea-related hospitalizations in São Paulo State, Brazil. Vaccine. 2014;14:S0264–S410. doi: 10.1016/j.vaccine.2014.04.015. [DOI] [PubMed] [Google Scholar]
- Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J, Das BK, Bhan MK. Identification of group A rotavirus gene 4 types by polymerase chain reaction. J Clin Microbiol. 1992;30:1365–1373. doi: 10.1128/jcm.30.6.1365-1373.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Góuvea V, Glass RI, Woods P, Taniguchi K, Clark HF, Forrester B, Fang ZY. Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J Clin Microbiol. 1990;28:276–282. doi: 10.1128/jcm.28.2.276-282.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring AJ, Inglis NF, Ojeh CK, Snodgrass DR, Menzies JD. Rapid diagnosis of rotavirus infection by direct detection of viral nucleic acid in silver-stained polyacrylamide gels. J Clin Microbiol. 1982;16:473–477. doi: 10.1128/jcm.16.3.473-477.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heylen E, Batoko Likele B, Zeller M, Stevens S, De Coster S, Conceição-Neto N, Van Geet C, Jacobs J, Ngbonda D, Van Ranst M, Matthijnssens J. Rotavirus surveillance in kisangani, the democratic republic of the congo, reveals a high number of unusual genotypes and gene segments of animal origin in non-vaccinated symptomatic children. PLoS One. 2014 doi: 10.1371/journal.pone.0100953. eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokororo A, Kidenya BR, Seni J, Mapaseka S, Mphahlele J, Mshana SE. Predominance of rotavirus G1[P8] genotype among under-five children with gastroenteritis in Mwanza, Tanzania. J Trop Pediatr. 2014 doi: 10.1093/tropej/fmu028. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes IH. Development of rotavirus molecular epidemiology: electropherotyping. Arch Virol Suppl. 1996;12:87–91. doi: 10.1007/978-3-7091-6553-9_10. [DOI] [PubMed] [Google Scholar]
- Jere KC, Mlera L, Page NA, van Dijk AA, O'Neill HG. Whole genome analysis of multiple rotavirus strains from a single stool specimen using sequence-independent amplification and 454® pyrosequencing reveals evidence of intergenotype genome segment recombination. Infect Genet Evol. 2011;8:2072–2082. doi: 10.1016/j.meegid.2011.09.023. [DOI] [PubMed] [Google Scholar]
- Jere KC, Mlera L, O'Neill HG, Peenze I, van Dijk AA. Whole genome sequence analyses of three African bovine rotaviruses reveal that they emerged through multiple reassortment events between rotaviruses from different mammalian species. Vet Microbiol. 2012;159:245–250. doi: 10.1016/j.vetmic.2012.03.040. [DOI] [PubMed] [Google Scholar]
- Jere KC, Esona MD, Ali YH, Peenze I, Roy S, Bowen MD, Saeed IK, Khalafalla AI, Nyaga MM, Mphahlele J, Steele D, Seheri ML. Novel NSP1 genotype characterised in an African camel G8P[11] rotavirus strain. Infect Genet Evol. 2014;21:58–66. doi: 10.1016/j.meegid.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Kirkwood CD. Genetic and antigenic diversity of human rotaviruses: potential impact on vaccination programs. J Infect Dis. 2010;202:S43–S48. doi: 10.1086/653548. [DOI] [PubMed] [Google Scholar]
- Kiulia NM, Nyaga MM, Seheri ML, Wolfaardt M, van Zyl WB, Esona MD, Irimu G, Inoti M, Gatinu BW, Njenga PK, Taylor MB, Nyachieo A. Rotavirus G and P types circulating in the eastern region of Kenya: predominance of G9 and emergence of G12 genotypes. Pediatr Infect Dis J. 2014;33(Suppl. 1):S85–88. doi: 10.1097/INF.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes P, Matthijnssens J, Rahman M, Van Ranst M. RotaC: a web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009;9:238. doi: 10.1186/1471-2180-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhi SA, Cunliffe NA, Steele D, Witte D, Kirsten M, Louw C, Ngwira B, Victor JC, Gillard PH, Cheuvart BB, Han HH, Neuzil KM. Effect of human rotavirus vaccine on severe diarrhea in African infants. N Engl J Med. 2010;362:289–298. doi: 10.1056/NEJMoa0904797. [DOI] [PubMed] [Google Scholar]
- Magagula NB, Esona MD, Nyaga MM, Stucker KM, Halpin RA, Stockwell TB, Seheri ML, Steele AD, Wentworth DE, Mphahlele MJ. Whole genome analyses of G1P[8] rotavirus strains from vaccinated and non-vaccinated South African children presenting with diarrhea. J Med Virol. 2015;87:79–101. doi: 10.1002/jmv.23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V, Bányai K, Ciarlet M, Iturriza-Gómara M, Lorusso E, De Grazia S, Arista S, Decaro N, Elia G, Cavalli A, Corrente M, Lavazza A, Baselga R, Buonavoglia C. Relationships among porcine and human P[6] rotaviruses: evidence that the different human P[6] lineages have originated from multiple interspecies transmission events. Virology. 2006;344(20056):509–519. doi: 10.1016/j.virol.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Martinez M, Galeano ME, Akopov A, Palacios R, Russomando G, Kirkness EF, Parra GI. Whole-genome analyses reveals the animal origin of a rotavirus G4P[6] detected in a child with severe diarrhea. Infect Genet Evol. 2014;27:156–162. doi: 10.1016/j.meegid.2014.07.020. [DOI] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Bányai K, Estes MK, Gentsch JR, Iturriza-Gómara M, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Patton JT, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Desselberger U, Van Ranst M. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol. 2008;153:1621–1629. doi: 10.1007/s00705-008-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Joelsson DB, Warakomski DJ, Zhou T, Mathis PK, van Maanen MH, Ranheim TS, Ciarlet M. Molecular and biological characterization of the 5 human-bovine rotavirus (WC3)-based reassortant strains of the pentavalent rotavirus vaccine, RotaTeq. Virology. 2010;403:111–127. doi: 10.1016/j.virol.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gómara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreño V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG) Arch Virol. 2011;156:1397–1413. doi: 10.1007/s00705-011-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Van Ranst M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr Opin Virol. 2012;4:426–433. doi: 10.1016/j.coviro.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R. VP6-sequence-based cut off values as a criterion for rotavirus species demarcation. Arch Virol. 2012;157:1177–1182. doi: 10.1007/s00705-012-1273-3. [DOI] [PubMed] [Google Scholar]
- McDonald SM, Matthijnssens J, McAllen JK, Hine E, Overton L, Wang S, Lemey P, Zeller M, Van Ranst M, Spiro DJ, Patton JT. Evolutionary dynamics of human rotaviruses: balancing reassortment with preferred genome constellations. PLoS Pathog. 2009;10:e1000634. doi: 10.1371/journal.ppat.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlera L, O'Neill HG, Jere KC, van Dijk AA. Whole-genome consensus sequence analysis of a South African rotavirus SA11 sample reveals a mixed infection with two close derivatives of the SA11-H96 strain. Arch Virol. 2013;158:1021–1030. doi: 10.1007/s00705-012-1559-5. [DOI] [PubMed] [Google Scholar]
- Msimang VM, Page N, Groome MJ, Moyes J, Cortese MM, Seheri M, Kahn K, Chagan M, Madhi SA, Cohen C. Impact of rotavirus vaccine on childhood diarrheal hospitalization after introduction into the South African public immunization program. Pediatr Infect Dis J. 2013;32:1359–1364. doi: 10.1097/INF.0b013e3182a72fc0. [DOI] [PubMed] [Google Scholar]
- Mukaratirwa A, Berejena C, Nziramasanga P, Shonhai A, Mamvura TS, Chibukira P, Mucheuki I, Mangwanya D, Kamupota M, Manangazira P, Tapfumaneyi C, Gerede R, Munyoro M, Mwenda JM, Mphahlele JM, Seheri ML, Peenze I, Gonah AN, Maruta A, Tengende MB. Epidemiologic and genotypic characteristics of rotavirus strains detected in children less than 5 years of age with gastroenteritis treated at 3 pediatric hospitals in Zimbabwe during 2008-2011. Pediatr Infect Dis J. 2014;(Suppl. 33):S45–S48. doi: 10.1097/INF.0000000000000050. [DOI] [PubMed] [Google Scholar]
- Mwenda JM, Tate JE, Parashar UD, Mihigo R, Agócs M, Serhan F, Nshimirimana D. African rotavirus surveillance network: a brief overview. Pediatr Infect Dis J. 2014;33:S6–S8. doi: 10.1097/INF.0000000000000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi T, Nakagomi O, Dove W, Doan YH, Witte D, Ngwira B, Todd S, Duncan Steele A, Neuzil KM, Cunliffe NA. Molecular characterization of rotavirus strains detected during a clinical trial of a human rotavirus vaccine in Blantyre, Malawi. Vaccine. 2012;30(Suppl 1):A140–51. doi: 10.1016/j.vaccine.2011.09.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi T, Doan YH, Dove W, Ngwira B, Iturriza-Gómara M, Nakagomi O, Cunliffe NA. G8 rotaviruses with conserved genotype constellations detected in Malawi over 10 years (1997-2007) display frequent gene reassortment among strains co-circulating in humans. J Gen Virol. 2013;6:1273–1295. doi: 10.1099/vir.0.050625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyaga MM, Jere KC, Peenze I, Mlera L, van Dijk AA, Seheri ML, Mphahlele MJ. Sequence analysis of the whole genomes of five African human G9 rotavirus strains. Infect Genet Evol. 16:62–77. doi: 10.1016/j.meegid.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Nyaga MM, Stucker KM, Esona MD, Jere KC, Mwinyi B, Shonhai A, Tsolenyanu E, Mulindwa A, Chibumbya JN, Adolfine H, Halpin RA, Roy S, Stockwell TB, Berejena C, Seheri ML, Mwenda JM, Steele AD, Wentworth DE, Mphahlele MJ. Whole-genome analyses of DS-1-like human G2P[4] and G8P[4] rotavirus strains from Eastern, Western and Southern Africa. Virus Genes. 2014 doi: 10.1007/s11262-014-1091-7. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odiit A, Mulindwa A, Nalumansi E, Mphahlele MJ, Seheri LM, Mwenda JM, Kisakye A. Rotavirus prevalence and genotypes among children younger than 5 years with acute diarrhea at Mulago National Referral Hospital, Kampala, Uganda. Pediatr Infect Dis J. (Suppl. 33):S41–S44. doi: 10.1097/INF.0000000000000070. [DOI] [PubMed] [Google Scholar]
- Page N, Esona M, Armah G, Nyangao J, Mwenda J, Sebunya T, Basu G, Pyndiah N, Potgieter N, Geyer A, Steele AD. Emergence and characterization of serotype G9 rotavirus strains from Africa. J Infect Dis. 2010;(Suppl. 202):S55–S63. doi: 10.1086/653551. [DOI] [PubMed] [Google Scholar]
- Papp H, László B, Jakab F, Ganesh B, De Grazia S, Matthijnssens J, Ciarlet M, Martella V, Bányai K. Review of group A rotavirus strains reported in swine and cattle. Vet Microbiol. 2013;165:190–199. doi: 10.1016/j.vetmic.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATH. 2014 http://sites.path.org/rotavirusvaccine/rotavirus-vaccines/
- Potgieter AC, Page NA, Liebenberg J, Wright IM, Landt O, van Dijk AA. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J Gen Virol. 2009;90:1423–1432. doi: 10.1099/vir.0.009381-0. [DOI] [PubMed] [Google Scholar]
- Pukuta ES, Esona MD, Nkongolo A, Seheri M, Makasi M, Nyembwe M, Mondonge V, Dahl BA, Mphahlele MJ, Cavallaro K, Gentsch J, Bowen MD, Waku-Kouomou D, Muyembe JJ, SURVAC Working Group Molecular surveillance of rotavirus infection in the Democratic Republic of the Congo August 2009 to June 2012. Pediatr Infect Dis J. 2014;33:355–359. doi: 10.1097/INF.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursem VN, Peeroo BM, Mangar TI, Sohawon FM, Seheri LM, Mphahlele MJ, Mwenda JM, Manraj SS. Epidemiology of rotavirus diarrhea and diversity of rotavirus strains among children less than 5 years of age with acute gastroenteritis in Mauritius: June 2008 to December 2010. Pediatr Infect Dis J. 2014;(Suppl. 33):S49–S53. doi: 10.1097/INF.0000000000000051. [DOI] [PubMed] [Google Scholar]
- Rahman M, Matthijnssens J, Yang X, Delbeke T, Arijs I, Taniguchi K, Iturriza-Gómara M, Iftekharuddin N, Azim T, Van Ranst M. Evolutionary history and global spread of the emerging G12 human rotaviruses. J Virol. 81:2382–2390. doi: 10.1128/JVI.01622-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Palacios GM, Pérez-Schael I, Velázquez FR, Abate H, Breuer T, Clemens SC, Cheuvart B, Espinoza F, Gillard P, Innis BL, Cervantes Y, Linhares AC, López P, Macías-Parra M, Ortega-Barría E, Richardson V, Rivera-Medina DM, Rivera L, Salinas B, Pavía-Ruz N, Salmerón J, Rüttimann R, Tinoco JC, Rubio P, Nuñez E, Guerrero ML, Yarzábal JP, Damaso S, Tornieporth N, Sáez-Llorens X, Vergara RF, Vesikari T, Bouckenooghe A, Clemens R, De Vos B, O'Ryan M. Human Rotavirus Vaccine Study Group. Safety and efficacy of an attenuated vaccine against severe rotavirus gastroenteritis. N Engl J Med. 2006;354:11–22. doi: 10.1056/NEJMoa052434. [DOI] [PubMed] [Google Scholar]
- Santos N, Hoshino Y. Global distribution of rotavirus serotypes/genotypes and its implication for the development and implementation of an effective rotavirus vaccine. Rev Med Virol. 2005;15:29–56. doi: 10.1002/rmv.448. [DOI] [PubMed] [Google Scholar]
- Sanchez-Padilla E, Grais RF, Guerin PJ, Steele AD, Burny ME, Luquero FJ. Burden of disease and circulating serotypes of rotavirus infection in sub-Saharan Africa: systematic review and meta-analysis. Lancet Infect Dis. 2009;9:567–576. doi: 10.1016/S1473-3099(09)70179-3. [DOI] [PubMed] [Google Scholar]
- Seheri LM, Page N, Dewar JB, Geyer A, Nemarude AL, Bos P, Esona M, Steele AD. Characterization and molecular epidemiology of rotavirus strains recovered in Northern Pretoria, South Africa during 2003-2006. J Infect Dis. 2010;202:S139–S147. doi: 10.1086/653559. [DOI] [PubMed] [Google Scholar]
- Seheri LM, Page NA, Mawela MP, Mphahlele MJ, Steele AD. Rotavirus vaccination within the South African Expanded Programme on Immunisation. Vaccine. 2012;30:S14–S20. doi: 10.1016/j.vaccine.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Seheri M, Nemarude L, Peenze I, Netshifhefhe L, Nyaga MM, Ngobeni HG, Maphalala G, Maake LL, Steele AD, Mwenda JM, Mphahlele JM. Update of rotavirus strains circulating in Africa from 2007 through 2011. Pediatr Infect Dis J. 2014;(Suppl. 331):S76–S84. doi: 10.1097/INF.0000000000000053. [DOI] [PubMed] [Google Scholar]
- Sow SO, Tapia M, Haidara FC, Ciarlet M, Diallo F, Kodio M, Doumbia M, Dembélé RD, Traoré O, Onwuchekwa UU, Lewis KD, Victor JC, Steele AD, Neuzil KM, Kotloff KL, Levine MM. Efficacy of the oral pentavalent rotavirus vaccine in Mali. Vaccine. 2012;(Suppl. 30):A71–78. doi: 10.1016/j.vaccine.2011.11.094. [DOI] [PubMed] [Google Scholar]
- Steele AD, Neuzil KM, Cunliffe NA, Madhi SA, Bos P, Ngwira B, Witte D, Todd S, Louw C, Kirsten M, Aspinall S, Van Doorn LJ, Bouckenooghe A, Suryakiran PV, Han HH. Human rotavirus vaccine Rotarix™ provides protection against diverse circulating rotavirus strains in African infants: a randomized controlled trial. BMC Infect Dis. 2012;12:213. doi: 10.1186/1471-2334-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD. WHO-coordinated Global Rotavirus Surveillance Network. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12:136–141. doi: 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- Todd S, Page NA, Duncan Steele A, Peenze I, Cunliffe NA. Rotavirus strain types circulating in Africa: Review of studies published during 1997-2006. J Infect Dis. 2010;(Suppl. 202):S34–S42. doi: 10.1086/653555. [DOI] [PubMed] [Google Scholar]
- Trojnar E, Sachsenröder J, Twardziok S, Reetz J, Otto PH, Johne R. Identification of an avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J Gen Virol. 2013;94:136–142. doi: 10.1099/vir.0.047381-0. [DOI] [PubMed] [Google Scholar]
- Tsolenyanu E, Seheri M, Dagnra A, Djadou E, Tigossou S, Nyaga M, Adjeoda E, Armah G, Mwenda JM, Atakouma Y. Surveillance for rotavirus gastroenteritis in children less than 5 years of age in Togo. Pediatr Infect Dis J. 2014;(Suppl. 33):S14–S18. doi: 10.1097/INF.0000000000000046. [DOI] [PubMed] [Google Scholar]
- Vesikari T, Matson DO, Dennehy P, Van Damme P, Santosham M, Rodriguez Z, Dallas MJ, Heyse JF, Goveia MG, Black SB, Shinefield HR, Christie CD, Ylitalo S, Itzler RF, Coia ML, Onorato MT, Adeyi BA, Marshall GS, Gothefors L, Campens D, Karvonen A, Watt JP, O'Brien KL, DiNubile MJ, Clark HF, Boslego JW, Offit PA, Heaton PM. Rotavirus Efficacy and Safety Trial (REST) Study Team. Safety and efficacy of a pentavalent human-bovine (WC3) reassortant rotavirus vaccine. N Engl J Med. 2006;5354:23–33. doi: 10.1056/NEJMoa052664. [DOI] [PubMed] [Google Scholar]
- Vesikari T, Uhari M, Renko M, Hemming M, Salminen M, Torcel-Pagnon L, Bricout H, Simondon F. Impact and effectiveness of RotaTeq® vaccine based on 3 years of surveillance following introduction of a rotavirus immunization program in Finland. Pediatr Infect Dis J. 2013;32:1365–1373. doi: 10.1097/INF.0000000000000086. [DOI] [PubMed] [Google Scholar]
- Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE. Global burden of childhood pneumonia and diarrhoea. Lancet. 2013;381:1405–1416. doi: 10.1016/S0140-6736(13)60222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.