

Abstract
Purpose
To determine whether the degree of tumor anaplasia has prognostic value by evaluating its correlation with high-risk histopathologic features and clinical outcomes in a series of retinoblastoma patients.
Design
Retrospective clinicopathologic study.
Methods
The clinical and pathologic findings in 266 patients who underwent primary enucleation for retinoblastoma were reviewed. The histologic degree of anaplasia was graded as retinocytoma, mild, moderate, or severe as defined by increasing cellular pleomorphism, number of mitoses, nuclear size, and nuclear hyperchromatism. Nuclear morphometric characteristics were measured. The clinical and pathologic data of 125 patients were compared using Kaplan-Meier estimates of survival. Fisher's exact test and multivariate regression were used to analyze the association between anaplasia grade and high-risk histologic features.
Results
Increasing grade of anaplasia was associated with decreased overall survival (p=0.003) and increased risk of metastasis (p=0.0007). Histopathologic features that were associated with anaplasia included optic nerve invasion (p<0.0001), choroidal invasion (p=<0.0001), and anterior segment invasion (p=0.04). Multivariate analysis considering high-risk histopathology and anaplasia grading as predictors of distant metastasis and death showed that high-risk histopathology was statistically significant as an independent predictor (p=0.01 for metastasis, p=0.03 for death) but anaplasia was not (p=0.63 for metastasis, p=0.30 for death). In the absence of high-risk features, however, severe anaplasia identified an additional risk for metastasis (p=0.0004) and death (p=0.01).
Conclusion
Grading of anaplasia may be a useful adjunct to standard histopathologic criteria in identifying retinoblastoma patients who do not have high-risk histologic features but still have an increased risk of metastasis and may need adjuvant therapy.
Keywords: anaplasia, retinoblastoma, high-risk histopathology, metastasis, survival
Introduction
Retinoblastoma is the most common primary intraocular malignancy of childhood.1 Risks of metastasis and mortality in retinoblastoma have been shown to be associated with certain histopathologic features including tumor invasion of the optic nerve2, choroid3, and anterior chamber.4 The presence of any of these high-risk histologic features warrants administration of adjuvant chemotherapy after enucleation.5 There are occasional cases that are negative for high-risk histologic findings which develop metastases, although these cases are probably underreported. In such cases, we believe that there may be cytologic characteristics that are not routinely assessed which may yield prognostic information.
Anaplasia refers to the cytologic features of cells considered to be characteristic of malignant neoplasms. Anaplastic cells display marked pleomorphism, high nuclear to cytoplasmic ratios, hyperchromatic nuclei, abnormal nuclear contours, prominent nucleoli, and loss of normal polarity. The term anaplasia also implies an increased capacity for cellular multiplication. Mitoses are often numerous in highly anaplastic tumors.6 Neoplastic cell populations may show considerable genetic instability and manifest progressive increases in mutation rates. Consequently, clonogenic subpopulations emerge which become preferentially selected so as to form the dominant proliferating cell pool, resulting in an increased tumor growth fraction. This malignant transformation is phenotypically expressed as cellular anaplastic change.7, 8 Increasing degrees of anaplasia are thought to translate to more aggressive tumor invasion and progression.
Retinoblastoma is considered to be one of the types of embryonal central nervous system tumors, a group which includes medulloblastoma, neuroblastoma, pineoblastoma, and medulloepithelioma, among others. The grading of anaplasia is a well-established criteria used in the classification of medulloblastomas and has been shown to be associated with aggressive clinical behavior.9 In this study, we describe a grading system for anaplasia in retinoblastoma and determine whether the grading of anaplasia correlates with histologic features known to have prognostic significance regarding clinical outcomes (metastasis and survival). We hypothesize that increasing severity of cellular anaplasia is associated with high-risk histologic features and progressively worse clinical outcomes in retinoblastoma.
Methods
We obtained Emory University Institutional Review Board approval for this study (IRB #00069328). Cases were identified by searching through the records of the L.F. Montgomery Laboratory at the Emory Eye Center from January 1940 through August 2013 for enucleation specimens with a diagnosis of retinoblastoma. Exclusion criteria were as follows: cases that were treated with any modality (chemotherapy, cryotherapy, laser, and/or radiotherapy) prior to enucleation; specimens with less than two microscopic low power fields (20×) of tumor; and specimens with diffuse growth patterns.10, 11 Patient medical records were reviewed for demographic information (age at presentation, age at enucleation, gender, and race) and clinical findings including family history, laterality, symptoms, ophthalmologic findings, Reese-Ellsworth (RE) Classification12, International Classification of Retinoblastoma (ICRB)13, length of follow up, treatments, and clinical outcomes. Clinical outcomes were documented including local recurrence, distant metastasis, secondary tumors, and final disposition (alive with no active disease, alive with metastasis, death from disease, and death from other causes).
Histopathologic Review
Microscopic examination of the enucleation specimens was conducted independently by two ophthalmic pathologists (H.E.G and C.S.S). The reviewers were masked to individual patient data at the time of histopathologic assessment. For all cases, standard pupil-optic nerve sections and transverse sections of the optic nerve at the surgical margin were reviewed. Additional sections from calottes were also reviewed when available.14, 15 The histopathologic review was performed in accordance with the American Joint Committee on Cancer (AJCC) pathologic classification,15 and included only the T (tumor) tumor size (greatest basal dimension and thickness of tumor in millimeters), growth pattern (exophytic, endophytic, or combined), level of differentiation (undifferentiated, poorly differentiated, moderately differentiated, or well differentiated), tumor seeding (vitreous, subretinal, or both), extent of tissue invasion (anterior segment, optic nerve, choroid, and/or extrascleral), degree of apoptosis (percentage of apoptotic cells within tumor), degree of anaplasia (mild, moderate or severe), and presence/absence of retinocytoma. A mitotic index was performed by counting the number of mitotic figures in ten randomly selected high-power fields using a 40× objective lens (Olympus, Tokyo, Japan).
Optic nerve invasion was characterized as: pre-laminar (superficial invasion anterior to the lamina cribrosa), laminar (invasion to the level of the lamina cribrosa), post-laminar (invasion posterior to the lamina cribrosa and anterior to the surgical margin of resection), or invasion to the surgical resection margin of resection. Choroidal invasion was classified as: non-massive (<3mm in maximum diameter adjacent to the sclera), or massive (≥3mm in diameter adjacent to the sclera). Anterior segment invasion was considered positive if tumor invasion was seen in the anterior chamber, iris, and/or ciliary body. High-risk histologic features were defined based on consensus guidelines set by the International Retinoblastoma Staging Working Group as the presence of one or more of the following: post-laminar optic nerve invasion, massive choroidal invasion, a combination pre-laminar or laminar optic nerve invasion and non-massive choroidal invasion, and tumor invasion into the anterior segment.14,16
Anaplasia grading (see Figure 1) was described based on the following cytologic features: cell shape, cell wrapping, nuclear size, nuclear contour, nuclear chromaticity, and frequency of mitoses. The grading of anaplasia was as follows: retinocytoma – nuclei not enlarged, evenly dispersed chromatin, no pleomorphism, no mitotic figures, abundant eosinophilic cytoplasm, and prominent photoreceptor differentiation (fleurettes); mild anaplasia – nuclei not enlarged, mild pleomorphism, rare mitotic figures, photoreceptor differentiation (Flexner-Wintersteiner and Homer Wright rosettes); moderate anaplasia – unambiguously enlarged nuclei, definite pleomorphism, frequent mitotic figures, moderate to poor differentation; severe anaplasia – very large hyperchromatic nuclei, extreme pleomorphism (angular, rhomboid, or fusiform), cell wrapping, numerous mitotic figures, poor differentiation. In all tumors, the highest grade of anaplasia encountered that occupied at least 10% of the tumor was used to classify the tumor. For example, a tumor with diffuse moderate anaplasia but with a focal region of severe anaplasia that occupied 10% of the tumor was designated as severely anaplastic.
Figure 1.
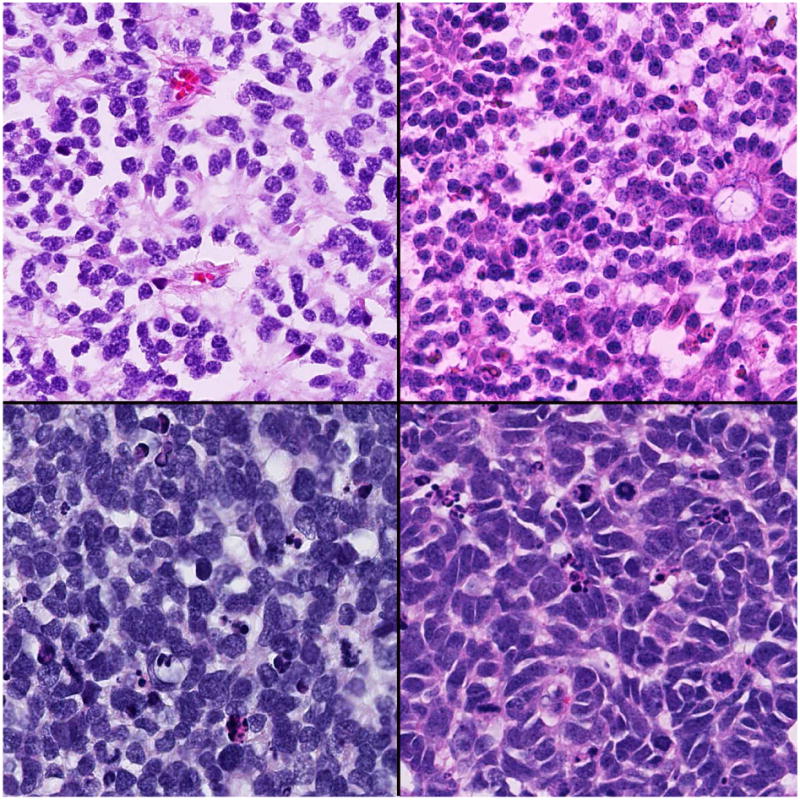
Grading of anaplasia in retinoblastoma. Top left: Retinocytoma-the nuclei are not enlarged, there is no pleomorphism nor mitotic activity, cells have abundant eosinophilic cytoplasm, and prominent photoreceptor differentiation (fleurettes); Top right: Mild anaplasia-the nuclei are not enlarged, but there can be mild pleomorphism, occasional rare mitotic figures, and photoreceptor differentiation (Flexner-Wintersteiner and Homer Wright rosettes); Bottom left: Moderate anaplasia-there is unambiguous enlargement of nuclei compared to mild anaplasia, definite pleomorphism, frequent mitotic figures, and poor differentation; Bottom right: Severe anaplasia-there is enlargement of nuclei similar in size to moderate anaplasia, extreme pleomorphism (angular, rhomboid, or fusiform), cell wrapping, numerous mitotic figures, and poor differentiation. (hematoxylin and eosin: top left, 250×; top right 250×; bottom left, 250×; bottom right, 250×)
There is no current standard definition for the levels of differentiation in retinoblastoma. For this study, we did separate analyses for 2 different ways of defining differentiation based on: the types of rosettes/fleurettes (Scheme 1) and the presence of rosettes/fleurettes considering the extent of these within the tumor (Scheme 2). For Scheme 1, level of differentiation was defined as: undifferentiated (no rosettes or fleurettes), poorly differentiated (presence of Homer Wright rosettes only), moderately differentiated (presence of Homer Wright and/or Flexner-Wintersteiner rosettes), or well differentiated (presence of Homer Wright, Flexner-Wintersteiner and/or fleurettes). For Scheme 2, the level of differentiation was defined as: undifferentiated (no rosettes or fleurettes), poorly differentiated (Homer Wright and/or Flexner-Wintersteiner rosettes seen in <1/3 of the tumor), moderately differentiated (Homer Wright and/or Flexner-Wintersteiner rosettes seen in 1/3-2/3 of the tumor), or well differentiated (Homer Wright, Flexner-Wintersteiner and/or fleurettes seen in >2/3 of the tumor).
Statistical Analysis
Fisher's exact test was used to analyze the association between anaplasia grade and ordinal categorical variables of optic nerve invasion (absent, pre-laminar, laminar, post-laminar, or surgical margin), choroidal invasion (absent, non-massive, or massive), and anterior segment invasion (absent or present). The association of anaplasia grade and clinical features such as age at enucleation and tumor size were also analyzed. Logistic regression analysis was performed to identify factors predictive of outcome (metastasis and death from disease). The factors found to be significant on univariate analysis were considered for multivariate analysis. Those significant at the 5% level on multivariate analysis (proportional hazards regression) were reported. Pathologic and clinical data were compared using Kaplan-Meier estimates of survival. A P-value of <0.05 was considered to be statistically significant. All analyses were performed using SAS version 9.2 (SAS Institute Inc., Cary, North Carolina).
Nuclear Morphometric Analysis
Glass slides of hematoxylin and eosin-stained tumor tissue were digitally scanned using the Hamamatsu Nanozoomer 2.0HT (Hamamatsu Photonics, Hamamatsu, Japan). The resulting images had a resolution of 0.2265 micron per pixel. Regions of interest equivalent to 2 high power fields at 40× magnification were outlined for each slide. Nuclei within the regions of interest were identified through color deconvolution17, cell similarity values18, and hysteresis thresholds.19 Analysis was done on a total of 5260 nuclei (911 from retinocytoma, 1389 from mild, 1432 from moderate, and 1528 from severe). For each segmented nucleus, computations were carried out for features pertaining to nuclear size (area – total number of pixels within a nucleus, and perimeter – arc length of a nuclear boundary), shape (circularity – degree of nuclear circularity/round shape, extent – degree of nuclear shape regularity, and eccentricity – degree of nuclear elongation), and hyperchromaticity (mean intensity – mean brightness of pixels within a nucleus) using Matlab (Mathworks Inc, Natick, Massachusetts). The nuclear diameter in microns was determined based on the largest distance between two nuclear edge points. For statistical analysis, the mean was computed for each nuclear feature and One-way Analysis of Variance (ANOVA) and Multiple Comparison Test (MCT) were used to analyze differences between group means.
Results
Clinical Features and Histopathologic Findings
Three hundred fourteen cases were reviewed and 48 cases were excluded due to the previously mentioned exclusion criteria. Histopathology slides were available for all remaining 266 cases. The clinical and pathologic findings in our cases are listed in Table 1. The mean age at time of enucleation was 21.9 months (range 1 – 84). Patients with bilateral tumors presented at a younger age (mean 11.6 months) compared to those with unilateral tumors (mean 24.3 months). Of the 266 patients, 147 (55.3%) were male and 119 (44.7%) were female. The majority, 231 (86.8%), of patients had unilateral retinoblastoma and 249 (93.6%) had a negative family history of retinoblastoma. The most common sign at presentation was leukocoria in 165 (62%) followed by strabismus in 37 (13.9%). Tumors in the enucleated eyes classified using the Reese-Ellsworth scheme were either Group Va or Vb, while those classified using the International Classification of Retinoblastoma (ICRB) scheme were all either Group D or Group E. The mean percentage of apoptotic cells in tumors was 36% (range 0-90%). Combined exophytic and endophytic growth pattern was commonly seen in 119 (44.7%). Our results showed a variable association between anaplasia grade and level of differentiation. For Scheme 1, there was no significant association between degree of anaplasia and differentiation (p=0.06). In contrast, Scheme 2 showed a statistically significant correlation between anaplasia and differentiation (p=0.001). One or more high-risk histopathologic features were present in 76 (28.6%) of cases, the most common of which was post-laminar optic nerve invasion in 48 (18.0%), followed by massive choroidal invasion in 40 (15.0%), anterior segment invasion in 18 (6.7%), and combined non-postlaminar optic nerve and non-massive choroidal invasion in 15 (5.6%).
Table 1. Demographic, Clinical, and Histopathologic Features of 266 Cases of Retinoblastoma.
| Gender | n (%) |
|---|---|
| Male | 147 (55.3) |
| Female | 119 (44.7) |
| Laterality | |
| Unilateral | 231 (86.8) |
| Bilateral | 35 (13.2) |
| Age at Enucleation (months) | |
| Overall | 21.9 (range 1-84) |
| Unilateral | 24.3 |
| Bilateral | 11.6 |
| Less than 1 year | 62 (23.3) |
| 1-2 years | 92 (34.6) |
| Greater than 2 years | 112 (42.1) |
| Race | |
| Caucasian/White | 158 (59.4) |
| African-American | 87 (32.7) |
| Hispanic | 12 (4.5) |
| Asian | 5 (1.9) |
| African | 3 (1.1) |
| Middle Eastern | 1 (0.4) |
| Family History | |
| Negative | 249 (93.6) |
| Positive | 17 (6) |
| Presenting Signs | |
| Leukocoria | 165 (62.0) |
| Strabismus | 37 (13.9) |
| Decreased vision | 24 (9.0) |
| Eye redness | 15 (5.6) |
| Periorbital swelling | 14 (5.3) |
| Proptosis | 5 (1.9) |
| Buphthalmos | 3 (1.1) |
| Cloudy cornea | 2 (0.8) |
| Asymptomatic, screened for family history | 1 (0.4) |
| Degree of Anaplasia | |
| Mild | 29 (10.9) |
| Moderate | 171 (64.3) |
| Severe | 66 (24.8) |
| Level of Differentiation (mean age in months) | |
| Undifferentiated (30.4) | 106 (39.8) |
| Poorly differentiated (20.9) | 84 (31.6) |
| Moderately differentiated (19.5) | 48 (18.0) |
| Well differentiated (12.0) | 28 (10.5) |
| High-Risk Histopathology | 76 (28.6) |
| Post-laminar optic nerve invasion | 48 (18.0) |
| Massive choroidal invasion | 40 (15.0) |
| Anterior segment invasion | 18 (6.7) |
| Non-postlaminar optic nerve + nonmassive choroidal invasion | 15 (5.6) |
| Growth Pattern | |
| Exophytic | 108 (40.6) |
| Endophytic | 39 (14.6) |
| Combined | 119 (44.7) |
| AJCC Pathologic Staging | |
| pT1 | 110 (41.4) |
| pT2a | 82 (30.8) |
| pT2b | 15 (5.6) |
| pT3a | 30 (11.3) |
| pT3b | 16 (6.0) |
| pT4 | 5 (1.9) |
| pT4a | 4 (1.5) |
| pT4b | 6 (2.3) |
AJCC – American Joint Committee on Cancer
Complete clinical data and follow-up reports were available for 125 cases and these were analyzed for survival and metastasis. The mean length of follow-up was 84.7 months, with a range of 6 - 581 months. Distant metastasis occurred in 11 of 125 (8.8%) patients, and 7 of 125 (5.6%) died from metastatic retinoblastoma. The findings for the cases with metastasis are summarized in Table 2. Secondary tumors developed in 5 (4.0%) patients and included 1 pinealoma, 2 osteosarcomas, 1 soft tissue sarcoma, and 1 renal cell carcinoma. One patient died from osteosarcoma which metastasized to the lungs.
Table 2. Summary of Findings in 11 Cases of Retinoblastoma with Metastasis.
| Case Number | Year of Diagnosis | Laterality | High-Risk Features (level of tumor invasion) | Age at Enucleation (months) | Anaplasia Grade | Adjuvant Chemotherapy | Site of Metastasis | Death from Disease |
|---|---|---|---|---|---|---|---|---|
| 1 | 1947 | Unilateral | Margin optic nerve, massive choroidal, anterior segment | 19 | Severe | No | CNS | Yes |
| 2 | 1965 | Bilateral | Post-laminar optic nerve, massive choroidal | 2 5 | Severe | No | CNS, bone | No |
| 3 | 1975 | Bilateral | Post-laminar optic nerve, massive choroidal | 24 | Severe | No | CNS | Yes |
| 4 | 1993 | Bilateral | Laminar optic nerve, non-massive choroidal | 16 | Severe | No; received external beam radiation to the opposite eye | Bone | Yes |
| 5 | 2009 | Unilateral | Post-laminar optic nerve, massive choroidal | 68 | Severe | Yes | CNS | Yes |
| 6* | 2012 | Unilateral | None | 33 | Severe | No | Bone, liver | No |
| 7* | 2012 | Unilateral | None | 27 | Severe | No | Bone, liver, CNS | Yes |
| 8 | 2013 | Unilateral | Post-laminar optic nerve, massive choroidal, anterior segment | 31 | Severe | Yes | CNS | No |
| 9 | 1966 | Unilateral | Margin optic nerve, massive choroidal, anterior segment | 20 | Moderate | No | CNS | Yes |
| 10 | 1999 | Unilateral | Massive choroidal | 23 | Moderate | No | Bone | No |
| 11 | 2001 | Unilateral | Post-laminar optic nerve, | 28 | Moderate | Yes | CNS | Yes |
Patients with no high-risk histologic features but developed metastasis
CNS – central nervous system
Anaplasia Grading
Based on evaluation of 266 enucleation specimens, 171 (64.3%) retinoblastomas were moderately anaplastic, followed 66 (24.8%) severe, and 29 (10.9%) mild. Eight tumors contained retinoblastoma with an associated retinocytoma component. Higher ranges and mean numbers of mitoses corresponded to increasing severity of anaplasia. The mitotic counts were as follows: mild anaplasia – mean: 2.4 and range: 1.5 – 3.2; moderate anaplasia – mean: 5.7 and range: 3.1 – 8.1; severe anaplasia – mean: 7.4; range: 4.2 – 21.2. Using the Fisher's exact test, histopathologic features that were statistically associated with anaplasia were the degree of optic nerve invasion (p=<0.0001), degree of choroidal invasion (p=<0.0001), and anterior segment invasion (p=0.04) (see Figures 2 and 3, Supplemental Tables 1-3). There were cases in which the anaplasia grade did not coincide with the presence or absence of high-risk features: 4 cases of mild anaplasia that had post-laminar optic nerve, one of which had concurrent massive choroidal invasion; and 23 cases of severe anaplasia that did not have any histologic high-risk features (see Figure 4). The mean age at enucleation was progressively older as tumors became more anaplastic (p = 0.05). Factors that were not significant were race (p=0.07), laterality (p=0.26), growth pattern (p=0.17), and tumor size (p=0.05).
Figure 2.
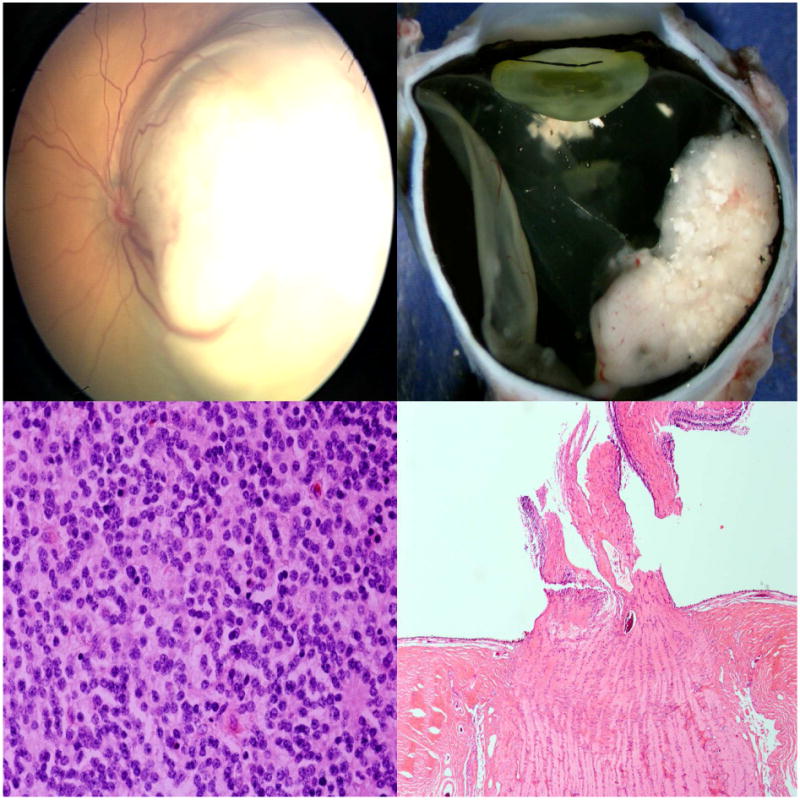
Mild anaplasia without high-risk features in retinoblastoma. Top left: A whitish, elevated lesion in the left eye involves the macula and temporal retina; Top right: Gross sectioning of the enucleated eye shows a white retinal mass with calcifications; Bottom left: Histologic evaluation reveals mild anaplasia; Bottom right: There is no optic nerve nor choroidal invasion. (hematoxylin and eosin: bottom left, 100×; bottom right, 10×)
Figure 3.
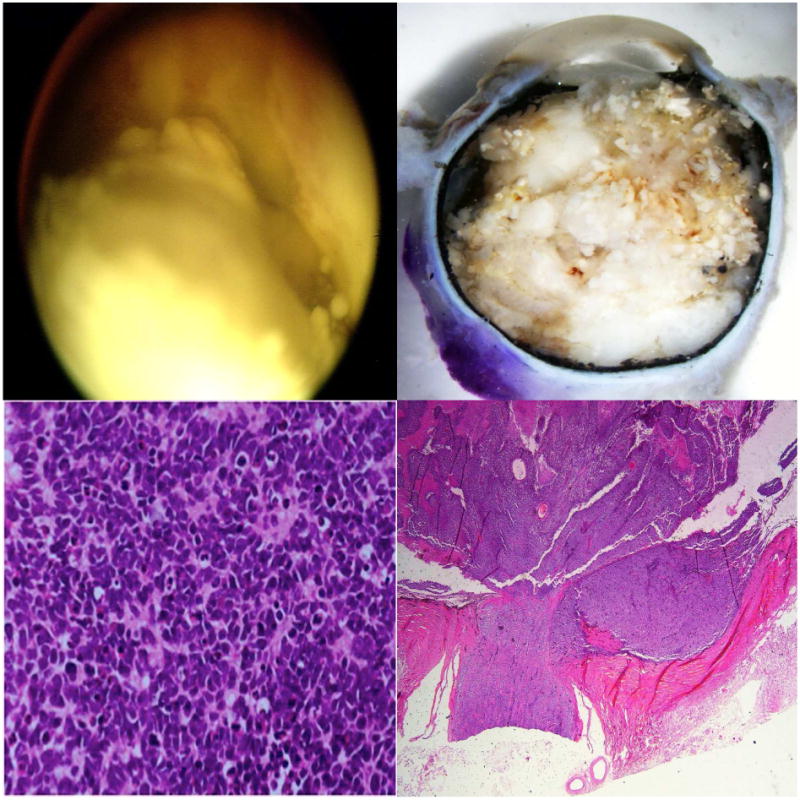
Severe anaplasia with high-risk features in retinoblastoma. Top left: Clinical findings show a Group E retinoblastoma; Top right: Gross findings of the enucleated eye show a whitish mass filling the entire posterior compartment; Bottom left: Histologic evaluation shows severe anaplasia; Bottom right: There is post-laminar optic nerve and massive choroidal invasion. (hematoxylin and eosin: bottom left, 100×; bottom right, 10×)
Figure 4.
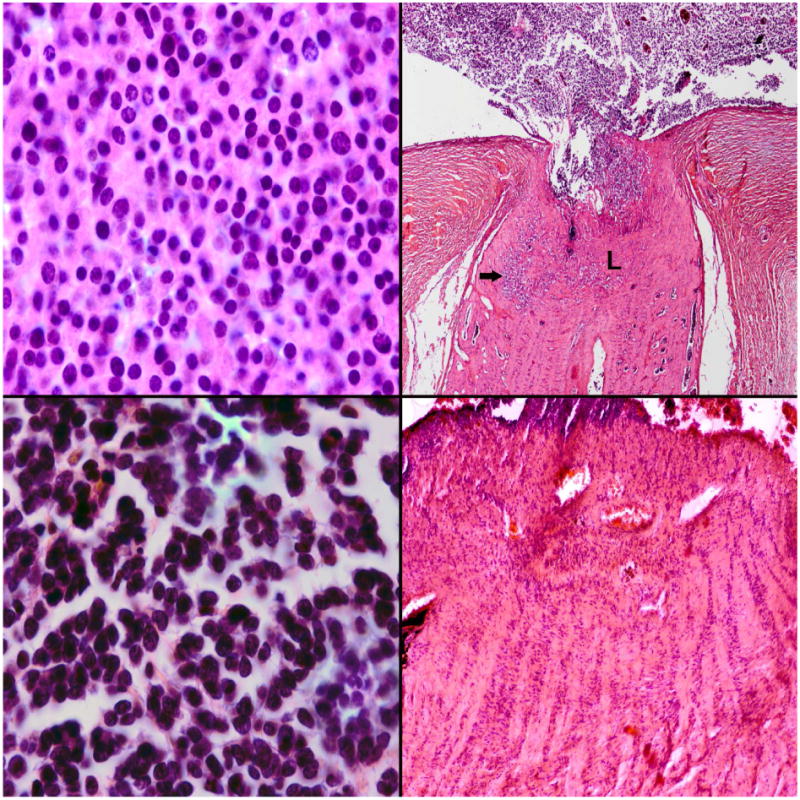
Mild anaplasia with high-risk features in retinoblastoma. Top left: This tumor was graded as mild anaplasia; Top right: Despite being mildly anaplastic, there was post-laminar optic nerve invasion noted (arrow). L-lamina cribrosa. Severe anaplasia without high-risk features. Bottom left: This tumor was graded as severe anaplasia; Bottom right: Although highly anaplastic, there was no optic nerve invasion and no choroidal invasion. (hematoxylin and eosin; top and bottom left, 100×, top right 10×, bottom right 25×)
The extent of anaplasia was variable within the specimens. We observed that tumors could have extensive areas of a certain anaplasia grade with a focal area of a different grade. The classification was further subdivided into 5 categories for the 125 cases with complete clinical data: 15 out of 125 (12%) were mild diffuse, 39 out of 125 (31.2%) were mild with focal moderate, 38 out of 125 (30.4%) were moderate diffuse, 27 out of 125 (21.6%) were moderate with focal severe, and 6 out of 125 (4.8%) were severe diffuse. In general surgical pathology, neoplasms are graded histologically using the highest grade region identified. We utilized this evaluation scheme in our study; for example a tumor classified as “moderate with focal severe” was simply designated as “severe”. Our findings showed a greater incidence of metastasis among those with severe diffuse anaplasia at 4 out of 6 (66.7%) compared to moderate with focal severe anaplasia at 4 out of 27 (14.8%). We also noted a difference in the moderate anaplasia group. None of the mild with focal moderate cases developed metastasis, while 3 out of 38 (7.89%) in the moderate diffuse group developed metastasis.
Among the 62 patients who were less than 1 year old, 43 (69.4%) had moderate anaplasia, followed by 17 (27.4%) mild, and only 2 (3.2%) had severe anaplasia. However, among the moderately anaplastic tumors, 15 out of 43 (34.9%) cases had focal areas of severe anaplasia. Based on differentiation Scheme 2, 8 (12.9%) were undifferentiated, 23 (37.1%) poorly, 14 (22.6%) moderately, and 17 (27.4%) well-differentiated. We had one documented case of a unilateral non-familial retinoblastoma diagnosed at 7 months of age with molecular testing revealing no mutations in the RB1 gene but with MYCN amplification as described by Rushlow et al.20 The enucleated eye from that patient showed an undifferentiated tumor having moderate with focal severe anaplasia and no optic nerve nor choroidal invasion.
Based on follow-up data in 125 retinoblastoma patients, Kaplan-Meier evaluations showed that increasing grade of anaplasia was associated with decreased survival (p=0.003) (see Figure 5) and increased risk of metastasis (p=0.0007) (see Figure 6). The presence of high-risk features was associated with decreased survival (p<0.0001) and increased risk of metastasis (p<0.0001). Multivariate analysis considering high-risk histopathology and anaplasia grading as predictors of distant metastasis and death showed that high-risk histopathology is statistically significant as an independent predictor (p=0.01 for metastasis, p=0.03 for death) but anaplasia is not (p=0.63 for metastasis, p=0.30 for death). In cases that have no high-risk features, severe anaplasia identifies an additional risk for metastasis (p=0.0004) and death (p=0.01). A limitation of our study was the insufficient statistical power to make any definite conclusions due to the small number of cases with metastasis and death. The distribution of cases with/without metastasis who were/were not treated with adjuvant therapy with regard to severe anaplasia severe and high-risk histopathology are shown in Table 3.
Figure 5.
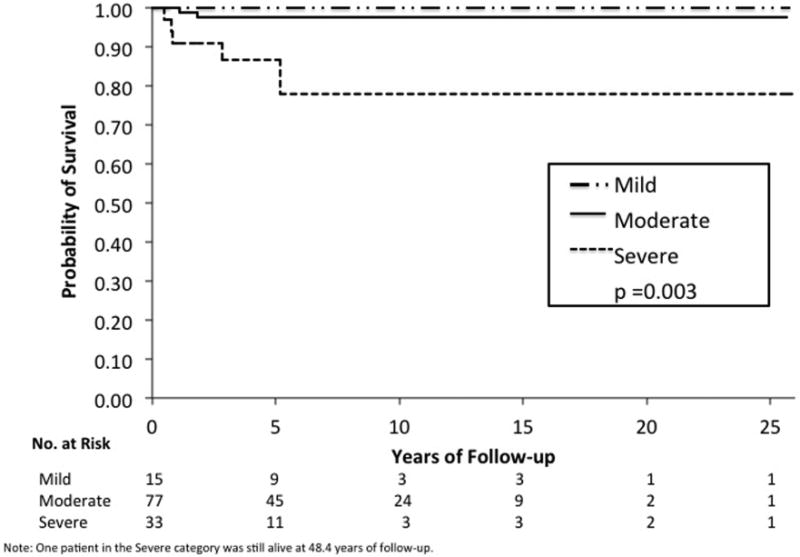
Kaplan-Meier estimate shows that increasing severity of anaplasia grade is associated with decreased survival in retinoblastoma (p = 0.003).
Figure 6.
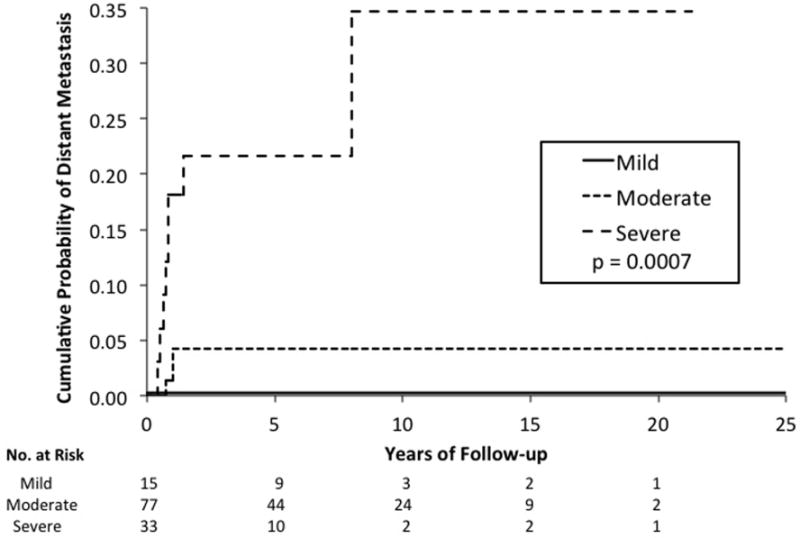
Kaplan-Meier estimate shows that increasing severity of anaplasia grade is associated with increased risk of metastasis in retinoblastoma (p = 0.0007).
Table 3. Adjuvant Treatment and Metastasis in Tumors with Severe Anaplasia and High-Risk Histopathology Among 125 Cases of Retinoblastoma*.
| Severe Anaplasia N=33 of 125 n (%) | High-Risk Histopathology N=42 of 125 n (%) | |
|---|---|---|
| With Adjuvant Treatment | 16 (48.5) | 27 (64.3) |
| Without Adjuvant Treatment | 17 (51.5) | 15 (35.7) |
| Without metastasis | 25 (75.8) | 33 (78.6) |
| With metastasis | 8 (24.2) | 9 (21.4) |
based on 125 cases with complete clinical data; adjuvant treatment was either systemic chemotherapy or systemic chemotherapy plus external beam radiation
Nuclear Morphometric Analysis
A total of 47 retinoblastoma cases were chosen for nuclear morphometric analysis: 8 with retinocytoma components, and 13 selected randomly from each anaplastic grade (mild, moderate, and severe) previously categorized by microscopic examination. Mean nuclear diameter in microns for the different groups were as follows: 6.3 for retinocytoma, 5.8 for mild, 8.0 for moderate, and 9.3 for severe anaplasia. For parameters relating to size (area and perimeter), there was no significant difference between retinocytoma and mild anaplasia, as well as moderate and severe anaplasia groups; however both retinocytoma and mild anaplasia groups had significantly smaller nuclear sizes compared to moderate and severe anaplasia groups based on perimeter. There was no significant difference in area between retinocytoma and moderate anaplasia, but there was a trend toward larger area in the moderate group. In terms of shape, the nuclei became progressively more elongated, less circular, and less regular with increasing severity of anaplasia. With regard to hyperchromaticity, retinocytoma nuclei were significantly lighter or less hyperchromatic than anaplastic nuclei based on mean intensity. There were no significant differences in hyperchromaticity of nuclei between the mild, moderate, and severe anaplasia groups (see Figure 7 and Supplemental Table 4).
Figure 7.
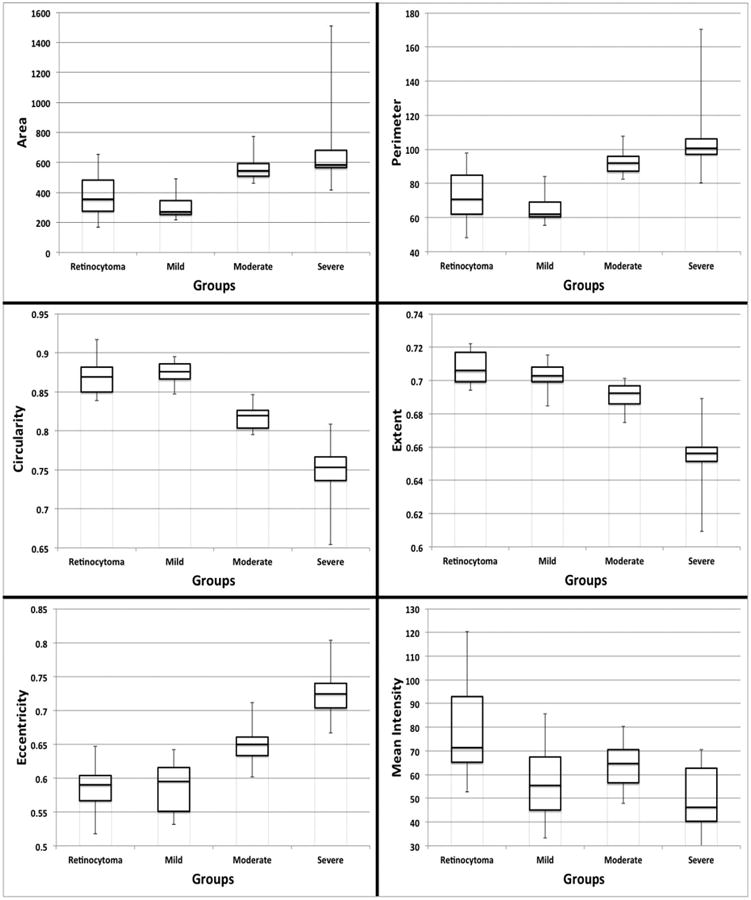
Box plots showing differences in nuclear morphometric features of retinocytoma, mild, moderate and severe anaplasia grades of retinoblastoma. Top left and top right: Analysis of nuclear size in terms of area and perimeter shows no significant difference between retinocytoma and mild anaplasia, as well as between moderate and severe anaplasia groups; however both retinocytoma and mild anaplasia groups had significantly smaller nuclear sizes compared to moderate and severe anaplasia groups. Middle left, middle right, and bottom left: Tumor nuclei became progressively less round based on measurements of circularity, less regular based on extent, and more elongated based on eccentricity with increasing severity of anaplasia. Bottom right: Retinocytoma nuclei were significantly lighter or less hyperchromatic than retinoblastoma nuclei based on mean intensity. There were no significant differences in mean intensity of nuclei between the mild, moderate, and severe anaplasia groups. (horizontal lines represent median values, bars represent standard error)
Discussion
Retinoblastoma is a success story in ophthalmology and pediatric oncology.21 Advances in diagnosis and management have increased survival approaching 100% in developed countries.22 Early recognition of risk factors for metastasis followed by prompt treatment has significantly contributed to improved survival.23 Clinicopathologic24, 25 and genetic26, 27 risk factors for metastasis have been identified. Well-established histologic features with prognostic significance for survival include the following: invasion of optic nerve, particularly if tumor is present at the surgical margin; invasion of choroid especially if with extension into or beyond the sclera or orbit; and involvement of the anterior segment.2-4, 28 Although clinical evaluation can suggest optic nerve and extraocular extension, evaluation of the pathologic features in enucleated eyes, especially for optic nerve and choroidal invasion, plays an essential role in identifying risk features for metastasis.29-31 Histologic factors that are potentially associated with metastasis but not as well-established are tumor size, tumor vascularity, seeding of vitreous, degree of differentiation, and growth pattern29, 32,33
Despite the fact that cellular anaplasia is currently a widely adopted criteria for the stratification of other embryonal central nervous system tumors that are closely related to retinoblastoma8, until now grading of anaplasia for retinoblastoma has not been evaluated. Unlike choroidal melanoma, cell type, degree of differentiation, and necrosis have not been defined (in themselves) as risk factors for metastasis in retinoblastoma.31 We now report the association of anaplasia grading with high-risk histologic features and metastasis in retinoblastoma. Assessment of anaplasia grade is based on histomorphology of routinely stained hematoxylin and eosin sections with no special studies required yet may yield important information for prognosis. Our results clearly support that there is a statistically significant correlation of anaplasia grading with high-risk histologic features: optic nerve invasion (p=<0.0001), choroidal invasion (p=<0.0001), and anterior segment invasion (p=0.04). Our analysis also demonstrated increased mitotic activity as the severity of anaplasia increased. In medulloblastomas, patients with diffuse severe anaplasia had worse clinical outcomes compared to those with focal severe anaplasia and there were no significant differences between diffuse and focal groups with moderate anaplasia.9 Our study reflected similar findings for diffuse severe versus focal severe anaplasia with regard to development of metastasis. We did, however, note a difference in the moderate anaplasia group. None of the focal moderate cases developed metastasis, while some in the diffuse moderate group did.
Our study did contain a few cases in which the anaplasia grade did not coincide with the presence or absence of high-risk features. For example, there were cases of mild anaplasia that had optic nerve and/or choroidal invasion and there were cases of severe anaplasia that did not have any histologic high-risk features. Increasing grades of anaplasia were associated with decreased survival and increased probability of metastasis. Mild anaplasia was not adversely related to prognosis although patients with moderate and severe anaplasia had significantly worse outcomes. We found that anaplasia grading may be a valuable feature that can aid in the identification of patients at high-risk of metastasis and death. This observation was highlighted in two patients in our series who had no evidence of high-risk histologic features but still proceeded to develop distant metastasis. Both of these two cases were found to have severe anaplasia (Cases 6 and 7 in Table 2). For both cases, the calottes were processed and carefully examined for any high-risk features and were found to have none. There are many factors that contribute to cancer metastases that may be unrelated to local tissue invasion. These factors include selective pressures in the tumor microenvironment, tumor stem cell properties, epithelial to mesenchymal transformation, extracellular matrix remodeling, and end organ specific properties among others.34 It is possible that highly anaplastic retinoblastoma cells may invade tumor vasculature prior to or without invasion of adjacent tissue. There is evidence that there are tumor-related modifications in nuclear organization leading to nuclear changes in size, shape, and chromatin texture35 as seen in anaplasia and related to compromised gene regulation and expression during the onset and progression of cancer.35, 36 These alterations may develop independent of local tissue invasion.
Among the 11 cases with metastasis listed in Table 2, there were 6 that did not receive therapy despite the presence of high-risk histologic features. Four of them were older cases (case numbers 1-3 and 9, diagnosed between the years 1947-1975) when post-enucleation chemotherapy was not yet widely adopted. The other 2 cases were more recent (case numbers 4 and 10, diagnosed in 1993 and 1999 respectively). Case 4 had non-massive choroidal invasion combined with non-postlaminar optic nerve invasion and Case 10 had isolated massive choroidal invasion. These patients did not receive adjuvant chemotherapy probably because the definitions of “massive choroidal invasion” and “non-massive choroidal plus non-postlaminar optic nerve invasion” had not yet been clearly defined as high-risk histologic features at the time of diagnosis.
Our results showed a variable association between anaplasia grade and level of differentiation. The terms differentiation and anaplasia are sometimes used interchangeably. We note that that these are two separate, albeit related, concepts. Well-differentiated tumors are composed of cells resembling mature normal cells of the tissue of origin, whereas poorly differentiated tumors have primitive-appearing, unspecialized cells. Anaplasia literally means “to form backward” implying the reversion from a high to lower level of differentiation of normal cells during tumorigenesis. However, there is substantial evidence that cancers arise from cancer stem cells in specialized tissues. Well-differentiated malignant tumors may evolve from maturation or specialization of undifferentiated cells as they proliferate, whereas undifferentiated malignant tumors derive from proliferation without maturation of cancer stem cells. Lack of differentiation therefore results from failure to differentiate, rather than as a consequence of dedifferentiation. Many cells lose their capacity to replicate as they become specialized but in neoplasia, replicative capability may be retained along with specialization.6 We found that in retinoblastomas, as is in medulloblastomas, rosette formation or differentiation may be seen even with severe anaplasia.9 The differentiating potential of neuroblastic tumor cells is related to the morphogenesis of the area of the central nervous system in which they originate.8 The presumed cell of origin is the retinoblast, which is derived from the neuroepithelium of the primitive optic cup, and has the capability of differentiating into neuroblastic or photosensory cells. Recent evidence suggests that the cell of origin for poorly differentiated retinoblastoma is the primitive retinal progenitor cell or retinoblast and for well-differentiated retinoblastoma is the cone precursor cell.37,38
The degree of differentiation in retinoblastoma is determined by the development of rosettes and fleurettes, which has been confirmed by electron microscopy and tissue culture.39-41 The least differentiated is the neuroblastic Homer Wright rosette, which can also be observed in other embryonal central nervous system tumors such as pineoblastoma and medulloblastoma. The Flexner-Wintersteiner rosette and the more differentiated fleurette are two formations mimicking the cytological and architectural features of photosensory rod and cone cells, which are evidence of further differentiation. It is debatable whether differentiation in retinoblastoma is related to prognosis, as studies have shown conflicting results.42, 43 Poorly differentiated tumors have been shown to be associated with high-risk features, especially massive choroidal invasion.44
The association of anaplasia with age at enucleation was also analyzed. Ninety-five percent of cases of retinoblastoma are diagnosed before age 5 and 66% of cases occur before age 2. Older age at the time of diagnosis is usually associated with more advanced disease and a poorer prognosis. Previous studies have advocated that the older the child, the longer the retinoblastoma has presumably been present and the more undifferentiated the retinoblastoma becomes.16, 45 Our present study showed a positive correlation of increasing severity of anaplasia with older age. Retinoblastomas that have differentiated may undergo additional mutational events over a period of time, thus resulting in higher grade cellular anaplasia. This may explain the increased grade of anaplasia that correlated with increased patient age in our study.
Though criteria for grading anaplasia has been described in standard textbooks,6 it remains largely subjective resulting in inter-observer variability. We employed objective measures of nuclear morphometry in an attempt to validate the categorization determined by ophthalmic pathologists through microscopic evaluation. Nuclear morphometric analysis revealed that there were no striking differences in the nuclei of retinocytoma and tumors with mild anaplasia, as they both can possess small, round, and bland nuclei. The characterizing feature that distinguishes retinocytoma from a mildly anaplastic retinocytoma is the greater amount of cytoplasm and the presence of fleurettes denoting photoreceptor differentiation in retinocytoma. Similarly, nuclear size in itself is not discriminatory between moderate and severe anaplasia. However, size does appear to be a reliable parameter for dividing retinoblastoma tumors into 2 groups: lower grades of anaplasia (retinocytoma and mild) and higher grades (moderate and severe). Hyperchromaticity did not prove useful in separating mild, moderate, and severe anaplasia. These findings are similar to what was observed in medulloblastomas.9 The features that were most significant in differentiating each of the anaplasia grades were those related to nuclear shape. We found the degree of nuclear pleomorphism, indicated by elongation and angularity, consistently increasing as anaplasia was deemed to be more severe.
Our study demonstrates the utility of grading for anaplasia in distinguishing retinoblastoma patients at increased risk of developing metastasis especially in patients without identifiable high-risk histologic features. We recommend including the description of anaplasia grade as part of the histopathologic assessment and closer monitoring as well as consideration of adjuvant therapy for patients whose enucleated eyes for retinoblastoma exhibit severe anaplasia. The enucleated eyes that we evaluated had no treatment prior to enucleation and our findings do not necessarily translate to enucleated eyes with prior treatment, as chemotherapy may mask histologic and cytologic features. We recognize that the small number of cases with metastasis is a limitation of this study. Further investigations with larger cohorts and more definitive criteria for anaplasia are needed to validate these findings.
Supplementary Material
Supplemental Table 1: Correlation of Grade of Anaplasia and Degree of Optic Nerve Invasion in 266 Patients with Retinoblastoma
Supplemental Table 2. Correlation of Grade of Anaplasia and Degree of Choroidal Invasion in 266 Patients with Retinoblastoma
Supplemental Table 3. Correlation of Grade of Anaplasia and Anterior Chamber Invasion in 266 Patients with Retinoblastoma
Supplemental Table 4. One-Way Analysis of Variance (ANOVA) and Multiple Comparison Test (MCT) Results for Nuclear Morphometric Analysis in 47 Cases of Retinoblastoma
Acknowledgments
Funding/Support: This study was funded in part by grants from the National Institutes of Health National Eye Institute P30EY006360, National Institutes of Health K25CA181503, and an unrestricted departmental grant from Research to Prevent Blindness Inc, New York, New York.
The authors would like to thank Dr. Daniel Brat and Dr. Merete Williams of the Winship Cancer Institute, Emory University – Pathology Core Lab for their assistance with the nuclear morphometry analysis.
Biography
Pia R. Mendoza, MD finished her Ophthalmology residency at St. Luke's Medical Center, Quezon City, Philippines. She completed a fellowship in Ophthalmic Pathology at the David G. Cogan Laboratory, Massachusetts Eye and Ear Infirmary in Boston, Massachusetts and a second fellowship in Ophthalmic Pathology and Oncology at the L.F. Montgomery Laboratory, Emory Eye Center in Atlanta, Georgia.

Footnotes
Financial Disclosures: National Institutes of Health CA192153 (HEG, pending); Clearside Biomedical, Atlanta, Georgia (HEG, patent submitted)
Contributions of Authors: Design of the study (PRM, HEG, CSS); Conduct of the study (PRM, HEG, CSS); Collection, management, analysis, and interpretation of the data (PRM, HEG, CSS, GBH, JRW, MJL, QZ, JK); Preparation, review, or approval of the manuscript (PRM, HEG, CSS, GBH, JRW, MJL, QZ, JK)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975-2004. Br J Ophthalmol. 2009;93(1):21–23. doi: 10.1136/bjo.2008.138750. [DOI] [PubMed] [Google Scholar]
- 2.Shields CL, Shields JA, Baez KA, Cater JR, De Potter PV. Optic nerve invasion of retinoblastoma: metastatic potential and clinical risk factors. Cancer. 1994;73(3):692–698. doi: 10.1002/1097-0142(19940201)73:3<692::aid-cncr2820730331>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Shields CL, Shields JA, Baez KA, Cater JR, De Potter PV. Choroidal invasion of retinoblastoma: metastatic potential and clinical risk factors. Br J Ophthalmol. 1993;77(9):544–548. doi: 10.1136/bjo.77.9.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haik BG, Dunleavy SA, Cooke C, et al. Retinoblastoma with anterior chamber extension. Ophthalmology. 1987;94(4):367–370. doi: 10.1016/s0161-6420(87)33437-2. [DOI] [PubMed] [Google Scholar]
- 5.Kaliki S, Shields CL, Shah SU, Eagle RC, Jr, Shields JA, Leahey A. Postenucleation adjuvant chemotherapy with vincristine, etoposide, and carboplatin for the treatment of high-risk retinoblastoma. Arch Ophthalmol. 2011;129(11):1422–1427. doi: 10.1001/archophthalmol.2011.289. [DOI] [PubMed] [Google Scholar]
- 6.Kumar V, Abbas AK, Aster J, editors. Robbins Basic Pathology. 9th. Philadelphia: Saunders Elsevier; 2013. pp. 176–177. [Google Scholar]
- 7.Schmitt HP. Rapid anaplastic transformation of gliomas in childhood. Neuropediatrics. 1983;14(3):137–143. doi: 10.1055/s-2008-1059566. [DOI] [PubMed] [Google Scholar]
- 8.Rubinstein LJ. Embryonal central neuroepithelial tumors and their differentiating potential: a cytogenetic view of a complex neuro-oncological problem. J Neurosurg. 1985;62(6):795–805. doi: 10.3171/jns.1985.62.6.0795. [DOI] [PubMed] [Google Scholar]
- 9.Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer. 2002;94(2):552–560. doi: 10.1002/cncr.10189. [DOI] [PubMed] [Google Scholar]
- 10.Shields CL, Ghassemi F, Tuncer S, Thangappan A, Shields JA. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthal mology. 2008;115(12):2253–2258. doi: 10.1016/j.ophtha.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Jijelava KP, Grossniklaus HE. Diffuse anterior retinoblastoma: a review. Saudi J Ophthalmol. 2013;27(3):135–139. doi: 10.1016/j.sjopt.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthal Otolaryngol. 1963;67:164–172. [PubMed] [Google Scholar]
- 13.Murphree AL. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41–53. doi: 10.1016/j.ohc.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Sastre X, Chantada GL, Doz F, et al. International Retinoblastoma Staging Working Group. proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med. 2009;133(8):1199–1202. doi: 10.5858/133.8.1199. [DOI] [PubMed] [Google Scholar]
- 15.Edge SB, Byrd DR, Compton CC, et al., editors. Retinoblastoma AJCC Cancer Staging Manual. 7th. New York: Springer; 2010. pp. 560–568. [Google Scholar]
- 16.Eagle RC., Jr High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med. 2009;133(8):1203–1209. doi: 10.5858/133.8.1203. [DOI] [PubMed] [Google Scholar]
- 17.Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23(4):291–299. [PubMed] [Google Scholar]
- 18.Frangi AF, Niessen W, Vincken KL, Viergever MA. Multiscale vessel enhancement filtering. Lecture Notes in Computer Science. 1998;1496:130–137. [Google Scholar]
- 19.Hancock ER, Kittler J. Adaptive estimation of hysteresis thresholds. Computer Vision and Pattern Recognition: Proceedings IEEE Computer Society Conference. 1991:196–201. [Google Scholar]
- 20.Rushlow DE, Mol BM, Kennett JY, et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 2013;14(4):327–334. doi: 10.1016/S1470-2045(13)70045-7. [DOI] [PubMed] [Google Scholar]
- 21.Grossniklaus HE. Retinoblastoma: fifty years of progress The LXXI Edward Jackson Memorial Lecture. Am J Ophthalmol. 2014 doi: 10.1016/j.ajo.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gatta G, Capocaccia R, Coleman MP, Ries LA, Berrino F. Childhood cancer survival in Europe and the United States. Cancer. 2002;95(8):1767–1772. doi: 10.1002/cncr.10833. [DOI] [PubMed] [Google Scholar]
- 23.Abramson DH, Niksarli K, Ellsworth RM, Servodidio CA. Changing trends in the management of retinoblastoma: 1951-1965 vs 1966-1980. J Pediatr Ophthal mol Strabismus. 1994;31(1):32–37. doi: 10.3928/0191-3913-19940101-07. [DOI] [PubMed] [Google Scholar]
- 24.Chawla B, Sharma S, Sen S, et al. Correlation between clinical features, magnetic resonance imaging, and histopathologic findings in retinoblastoma: a prospective study. Ophthalmology. 2012;119(4):850–856. doi: 10.1016/j.ophtha.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 25.Wilson MW, Qaddoumi I, Billups C, Haik BG, Rodriguez-Galindo C. A clinicopathological correlation of 67 eyes primarily enucleated for advanced intraocular retinoblastoma. Br J Ophthalmol. 2011;95(4):553–558. doi: 10.1136/bjo.2009.177444. [DOI] [PubMed] [Google Scholar]
- 26.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68(4):820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 28.Rootman J, Ellsworth RM, Hofbauer J, Kitchen D. Orbital extension of retinoblastoma: a clinicopathological study. Can J Ophthalmol. 1978;13(2):72–80. [PubMed] [Google Scholar]
- 29.Kopelman JE, McLean IW, Rosenberg SH. Multivariate analysis of risk factors for metastasis in retinoblastoma treated by enucleation. Ophthalmology. 1987;94(4):371–377. doi: 10.1016/s0161-6420(87)33436-0. [DOI] [PubMed] [Google Scholar]
- 30.Messmer EP, Heinrich T, Hopping W, de Sutter E, Havers W, Sauerwein W. Risk factors for metastases in patients with retinoblastoma. Ophthalmology. 1991;98(2):136–141. doi: 10.1016/s0161-6420(91)32325-x. [DOI] [PubMed] [Google Scholar]
- 31.Finger PT, Harbour JW, Karcioglu ZA. Risk factors for metastasis in retinoblastoma. Surv Ophthalmol. 2002;47(1):1–16. doi: 10.1016/s0039-6257(01)00279-x. [DOI] [PubMed] [Google Scholar]
- 32.Yan J, Zhang H, Li Y. Establishment of the relationship between tumor size and range of histological involvement to evaluate the rationality of current retinoblastoma management. PLoS one. 2013;8:e80484. doi: 10.1371/journal.pone.0080484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karcioglu ZA, Huaman AM. Angiogenesis in retinoblastoma. Middle East J Ophthal mol. 1996;4(1):18–25. [Google Scholar]
- 34.Chiang AC, Massague J. Molecular basis of metastasis. New Engl J Med. 2008;359(26):2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4(9):677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- 36.Stein GS, Montecino M, van Wijnen AJ, Stein JL, Lian JB. Nuclear structure-gene expression interrelationships: implications for aberrant gene expression in cancer. Cancer Res. 2000;60(8):2067–2076. [PubMed] [Google Scholar]
- 37.Kapatai G, Brundler MA, Jenkinson H, et al. Gene expression profiling ideintifies different sub-types of retinoblastoma. Br J Cancer. 2013;109(2):512–525. doi: 10.1038/bjc.2013.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu XL, Fang Y, Lee TC, et al. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137(6):1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ts'o MO, Zimmerman LE, Fine BS. The nature of retinoblastoma. I. Photoreceptor differentiation: a clinical and histopathologic study. Am J Ophthal mol. 1970;69(3):339–349. doi: 10.1016/0002-9394(70)92263-4. [DOI] [PubMed] [Google Scholar]
- 40.Ts'o MO, Fine BS, Zimmerman LE. The nature of retinoblastoma. II. Photoreceptor differentiation: an electron microscopic study. Am J Ophthalmol. 1970;69(3):350–359. doi: 10.1016/0002-9394(70)92264-6. [DOI] [PubMed] [Google Scholar]
- 41.Bogenmann E, Mark C. Routine growth and differentiation of primary retinoblastoma cells in culture. J Natl Cancer Inst. 1983;70(1):95–104. [PubMed] [Google Scholar]
- 42.Sevel D, Rohm GF, Sealy R. Clinical significance of the fleurette in retinoblastoma. Br J Ophthalmol. 1974;58(7):687–693. doi: 10.1136/bjo.58.7.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mashiah M, Barishak YR. Photoreceptor differentiation in retinoblastomas and its significance in prognosis. Br J Ophthalmol. 1977;61(6):417–422. doi: 10.1136/bjo.61.6.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kashyap S, Meel R, Pushker N, et al. Clinical predictors of histopathology in retinoblastoma. Pediatr Blood Cancer. 2012;58(3):356–61. doi: 10.1002/pbc.23239. [DOI] [PubMed] [Google Scholar]
- 45.Carbajal UM. Observations on retinoblastoma. Am J Ophthalmol. 1958;45(3):391–402. doi: 10.1016/0002-9394(58)90821-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Correlation of Grade of Anaplasia and Degree of Optic Nerve Invasion in 266 Patients with Retinoblastoma
Supplemental Table 2. Correlation of Grade of Anaplasia and Degree of Choroidal Invasion in 266 Patients with Retinoblastoma
Supplemental Table 3. Correlation of Grade of Anaplasia and Anterior Chamber Invasion in 266 Patients with Retinoblastoma
Supplemental Table 4. One-Way Analysis of Variance (ANOVA) and Multiple Comparison Test (MCT) Results for Nuclear Morphometric Analysis in 47 Cases of Retinoblastoma