

Abstract
Increasing evidence suggests that the pathogenesis of neuropathic pain is mediated through activation of microglia in the spinal cord. Hydrogen sulfide attenuates microglial activation and central nervous system inflammation; however, the role of hydrogen sulfide in neuropathic pain is unclear. In this study, we examined the effects of hydrogen sulfide breathing on neuropathic pain in mice. C57BL/6J mice were subjected to chronic constriction injury (CCI) of the sciatic nerve. After CCI, mice breathed air alone or air mixed with hydrogen sulfide at 40 ppm for 8 h on 7 consecutive days. The expression levels of inflammatory cytokines including interleukin 6 (IL-6) were measured in the spinal cord. Effects of hydrogen sulfide on IL-6-induced activation of microglia were examined in primary rat microglia. Mice that breathed air alone exhibited the neuropathic pain behavior including mechanical allodynia and thermal hyperalgesia and increased mRNA levels of IL-6 and chemokine CC motif ligand 2 (CCL2) after CCI. Inhaled hydrogen sulfide prevented the neuropathic pain behavior and attenuated the upregulation of inflammatory cytokines. Sodium sulfide inhibited IL-6-induced activation of primary microglia. These results suggest that inhaled hydrogen sulfide prevents the development of neuropathic pain in mice possibly via inhibition of the activation of microglia in the spinal cord.
Keywords: Neuropathic pain, Hydrogen sulfide, Microglia, Inflammatory cytokine
1. Introduction
Chronic pain affects about 30% of the population and is estimated to cost $650 billion a year in health-care costs and lost productivity in the United States [1]. The estimate of the annual cost of chronic pain was greater than the annual costs of heart disease ($309 billion), cancer ($243 billion), and diabetes ($188 billion) [2]. Neuropathic pain is a common chronic pain condition [3] that is caused by peripheral nerve injury and is characterized by long-lasting exaggerated pain behavior such as allodynia and hyperalgesia [4]. Although neuropathic pain is known as a particularly unpleasant type of pain [3], the management of patients with neuropathic pain is challenging because of the multiplicity of mechanisms involved in neuropathic pain conditions [5]. Precise pathophysiological mechanisms of neuropathic pain are still unclear, however, a number of studies have suggested that microglial activation and inflammatory cytokines in the spinal cord play important roles in the development and maintenance of neuropathic pain [6,7]. To date, management of neuropathic pain is aimed only at reducing symptoms; however, current drugs have limited efficacy and dose-limiting toxic effects [8]. Several clinical trials of drugs for neuropathic pain have reported negative results despite encouraging results from preclinical and early clinical studies [9]. Therefore, additional therapeutic strategies are urgently needed.
Hydrogen sulfide is a colorless, flammable and water-soluble gas with the characteristic odor of rotten eggs typically found in sulfur hot spring and sewer [10]. Recently, hydrogen sulfide was rediscovered as an endogenously produced signaling molecule along with nitric oxide and carbon monoxide [11]. An abundance of experimental evidence suggests that hydrogen sulfide plays a prominent role in physiology and pathophysiology [12]. We have previously reported that inhaled hydrogen sulfide prevents neurodegeneration in a mouse model of Parkinson’s disease induced by neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [13]. The protective effects of inhaled hydrogen sulfide in the mouse model of Parkinson’s disease were associated with inhibition of glial activation and upregulation of antioxidant and detoxification proteins in the brain. Furthermore, we have recently reported that breathing hydrogen sulfide prevents the systemic inflammation induced by lipopolysaccharide (LPS) and improves survival rate in mice [14]. Based on these findings, we hypothesized that breathing hydrogen sulfide prevents the development of neuropathic pain behavior via inhibiting microglial activation and neuroinflammation in the spinal cord. Here we report that inhaled hydrogen sulfide prevents the neuropathic pain behavior induced by chronic constriction injury of the sciatic nerve in mice.
2. Materials and methods
2.1. Animals
After approval by the Massachusetts General Hospital Subcommittee on Research Animal Care, we studied 2–3 month-old male C57BL/6J wild-type (WT) mice (The Jackson Laboratory, Bar Harbor, ME) and 3–4 month-old male Sprague-Dawley rats (Charles River, Wilmington, MA).
2.2. Surgical procedure
Chronic constriction injury (CCI) of the sciatic nerve was performed in mice as previously described [15,16]. Briefly, after instrumentation under anesthesia, one side of the common sciatic nerve was exposed and two loose ligatures (4-0 chromic gut) were made around the dissected nerve.
2.3. Hydrogen sulfide inhalation
Mice breathed air alone or air mixed with hydrogen sulfide at 40 ppm for 8 h each day for 7 days starting immediately after the CCI operation in custom made chambers, as previously described [13]. We chose the dose of H2S inhalation based on our recent study in which H2S inhalation at 40 ppm prevented neurodegeneration in a murine model of Parkinson’s disease induced by MPTP [13].
2.4. Behavioral test of neuropathic pain
2.4.1. Mechanical allodynia
Mechanical allodynia was assessed by using the von Frey filament test as previously described [16,17]. To carry out this test, mice were placed in a cage in which the bottom is made of gauze. This set-up allowed an experimenter to touch the mid-plantar surface of mice’s hind-paw from the bottom of the cage. An experimenter applied von Frey filaments in ascending order of bending force to the mid-plantar surface of the mice’s hind-paw. A von Frey filament was applied perpendicular to the skin and depressed slowly until it bent. A threshold force of response was defined as the first filament in the series that evoked at least one clear paw-withdrawal out of five applications. Each of these five stimuli was applied to slightly different areas of the mid-plantar surface with 2–3 s intervals.
2.4.2. Thermal hyperalgesia
Thermal hyperalgesia to radiant heat was assessed by using a foot-withdrawal test as previously described [16,18]. Mice were placed in plastic boxes on a glass plate. The radiant heat source was applied from a projection bulb placed directly under the planter surface of the mice’s hind-paw. The foot-withdrawal latency was defined as the time elapsed from the onset of radiant heat stimulation to withdrawal of mice’s hind-paw. The radiant source was adjusted to result in base-line latencies of 12 s and a cut-off time of 20 s was preset to prevent possible tissue damage. Three test trials with 5 min interval were performed and scores from each trial were averaged to yield the mean foot-withdrawal latency.
2.5. Measurements of gene expression in the mice spinal cord after peripheral nerve injury
The lumber parts of spinal cord were obtained from mice at 2 days after sham operation or CCI with or without hydrogen sulfide breathing. RNA was extracted from spinal cord using the RNAspin Mini kit (GE Healthcare, Piscataway, NJ) and cDNA was synthesized using moloney murine leukemia virus reverse transcriptase (M-MLV RT) (Promega, Madison, WI). The mRNA expressions of interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), chemokine CC motif ligand 2 (CCL2), activating transcription factor 3 (ATF3), integrin alpha M (ITGAM), glial fibrillary acidic protein (GFAP), and 18S ri-bosomal RNA were measured by real-time PCR using Realplex 2 system (Eppendorf North America, Westbury, NY). The primer sequences are listed in Table 1.
Table 1.
List of primer sequences for real-time PCR.
| IL-6 for mice | Forward | 5′-CCGGAGAGGAGACTTCACAGA-3′ |
| IL-6 for mice | Reverse | 5′-GGTCTGGGCCATAGAACTGA-3′ |
| TNF-α for mice | Forward | 5′-CAGCCTCTTCTCATTCCTGC-3′ |
| TNF-α for mice | Reverse | 5′-GGTCTGGGCCATAGAACTGA-3′ |
| CCL-2 for mice | Forward | 5′-ATGCAGGTCCCTGTCATGCTTC-3′ |
| CCL-2 for mice | Reverse | 5′-ACTCATTGGGATCATCTTGCTGG-3′ |
| ATF3 for mice | Forward | 5′-CAGTTACCGTCAACAACAGACCC-3′ |
| ATF3 for mice | Reverse | 5′-CTTTCTGCAGGCACTCTGTCTTC-3′ |
| ITGAM for mice | Forward | 5′-CCATGACCTTCCAAGAGAATGC-3′ |
| ITGAM for mice | Reverse | 5′-ACCGGCTTGTGCTGTAGTC-3′ |
| GFAP for mice | Forward | 5′-ACCAGCTTACGGCCAACAG-3′ |
| GFAP for mice | Reverse | 5′-CCAGCGATTCAACCTTTCTCT-3′ |
| 18S for mice | Forward | 5′-CGGCTACCACATCCAAGGAA-3′ |
| 18S for mice | Reverse | 5′-GCTGGAATTACCGCGGCT-3′ |
| IL-6 for rat | TaqMan Rn00561420_m1 (Life Technologies, Grand Island, NY) | |
| TNF-α for rat | TaqMan Rn99999017_m1 (Life Technologies) | |
| CCL-2 for rat | TaqMan Rn00580555_m1 (Life Technologies) | |
| 18S for rat | TaqMan Hs 99999901_s1 (Life Technologies) |
IL-6, interleukin 6; TNF-α, tumor necrosis factor α; CCL2, chemokine CC motif ligand 2; ATF3, Activating transcription factor 3; ITGAM, integrin alpha M; GFAP, glial fibrillary acidic protein.
2.6. Activation of primary microglia and measurements of gene expression
Primary mixed glial cells were cultured from 0 to 2 days old Sprague Dawley rat pups as previously described [19]. Cerebral cortices were dissected, minced and digested. The cells were seeded in poly-D-lysine-coated 75-cm2 T flasks with Dulbecco’s Modified Eagle’s medium containing 20% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cell cultures were con-fluent in 10–14 days, and then the flasks were shaken for an hour in an orbital shaker at 218 rpm in 36.5 °C to dissociate the microg-lia. Then, the media from the flasks were extracted. Cells in the extracted media were seeded into poly-D-lysine-coated six-well plates (2 ×10 5 cells/cm2). Cells were incubated for 7 days and then serum starved with 0.1% FBS. Vehicle, or sodium sulfide (Na2S) at 50, or 200 μM was administered to the media 1 min after the addition of 50 ng/ml recombinant rat IL-6 (Peprotech, Rocky Hill, NJ). We chose the dose of Na2S based on a recent study by Whiteman and colleagues [20]. After the incubation at 37 °C for 6 h, cells were washed thoroughly. The mRNA expressions of IL-6, TNF-a, CCL2 and 18S ribosomal RNA were measured by real-time PCR using Realplex 2 system (Eppendorf North America). The primer sequences are listed in Table 1.
2.7. Statistical analysis
All data are expressed as mean ± SD. Data were analyzed using one-way ANOVA or two-way ANOVA with a Bonferroni post hoc test. GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA) was used for statistical analyses. P values less than 0.05 were considered statistically significant.
3. Results
3.1. Inhaled hydrogen sulfide prevents the neuropathic pain behavior after peripheral nerve injury
To examine the effects of inhaled hydrogen sulfide on the neuropathic pain behavior induced by CCI, mice breathed air alone or air mixed with hydrogen sulfide at 40 ppm for 8 h each day starting immediately after the CCI operation on day 0. The hydrogen sulfide breathing session was performed 7 consecutive days from day 0 through postoperative day 6. Behavioral experiments were performed before CCI operation (day 0) and 1, 3, 5, 7, 9, and 14 days after CCI operation. In mice that breathed air alone, peripheral nerve injury resulted in the neuropathic pain behavior including mechanical allodynia and thermal hyperalgesia on the ipsilateral (operated) side (Fig. 1A and B). No significant mechanical hypersensitivity or thermal hyperalgesia was observed on the contralateral side (non-operated, data not shown). In contrast, breathing hydrogen sulfide for 8 h daily for 7 days significantly attenuated both mechanical allodynia (Fig. 1A) and thermal hyperalgesia (Fig. 1B). These observations demonstrate that inhaled hydrogen sulfide prevents the neuropathic pain behavior induced by peripheral nerve injury.
Fig. 1.
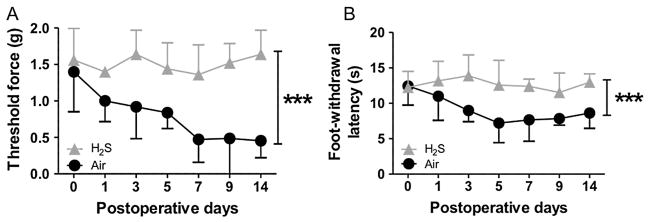
Effects of inhaled hydrogen sulfide on neuropathic pain behavior in mice. Mechanical allodynia (A) and thermal hyperalgesia (B) were attenuated by hydrogen sulfide breathing at 40 ppm for 8 h (H2S) on 7 consecutive days. H2S, mice that breathed hydrogen sulfide at 40 ppm mixed in air after CCI. Air, mice that breathed air alone after CCI. N = 5 in each group. ***P < 0.001 versus Air. Data were analyzed using repeated measures two-way ANOVA.
3.2. Inhaled hydrogen sulfide prevents microglial activation in the spinal cord after peripheral nerve injury
To elucidate the mechanisms responsible for the beneficial effect of inhaled hydrogen sulfide on neuropathic pain, we examined the expression levels of glial activation in the spinal cord. The mRNA expression of ITGAM (marker of microglial activation) was increased by CCI in mice that breathe air alone (Fig. 2A). In contrast, inhaled hydrogen sulfide at 40 ppm for 8 h for 7 days prevented the upregulation of ITGAM. Although the expression level of GFAP (marker of astrocyte activation) (Fig.2B) was not affected by CCI operation, inhaled hydrogen sulfide attenuated the mRNA expression of GFAP (Fig. 2B). These results suggest that inhaled hydrogen sulfide prevents microglial activation induced by peripheral nerve injury in the spinal cord.
Fig. 2.

Relative gene expression levels of glial activation (A and B), inflammatory cytokines (C-E) and activating transcription factor 3 (ATF3) (F) in the spinal cord at 2 days after chronic constriction injury (CCI) of the sciatic nerve. Sham, mice that were subjected to sham operation without sciatic nerve ligations. Air, mice that breathed air alone after CCI. H2S, mice that breathed hydrogen sulfide at 40 ppm for 8 h on 2 consecutive days after CCI. ITGAM, integrin alpha M; GFAP, glial fibrillary acidic protein; IL-6, interleukin 6; CCL2, chemokine CC motif ligand 2; TNF-α, tumor necrosis factor α. N = 8 in each group. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were analyzed using one-way ANOVA with a Bonferroni post hoc test.
3.3. Inhaled hydrogen sulfide attenuates the inflammatory cytokines in the spinal cord after peripheral nerve injury
To examine whether inhaled hydrogen sulfide prevents the inflammation induced by CCI operation, we examined the expression levels of inflammatory cytokines in the spinal cord. The mRNA expression of inflammatory cytokines including IL-6, TNF-α, and CCL2 were increased at 2 days after CCI in mice that breathe air alone (Fig. 2C–E). Inhaled hydrogen sulfide prevented the upregulation of IL-6 and CCL2 (Fig. 2C and D). These results suggest that inhaled hydrogen sulfide attenuates inflammation in the spinal cord after peripheral nerve injury.
3.4. Inhaled hydrogen sulfide attenuates the neuronal damage after peripheral nerve injury
ATF3 has been reported as a maker of the neuronal damage after peripheral nerve injury [21]. To examine whether inhaled hydrogen sulfide attenuates the neuronal damage after CCI operation, we measured the expression level of ATF3 in the spinal cord. The ATF3 mRNA was markedly increased in mice that breathed air alone after CCI. Inhaled hydrogen sulfide blocked the upregulation of ATF3 after CCI in the spinal cord (Fig. 2F).
3.5. The inflammatory cytokine production from activated microglia was prevented by a hydrogen sulfide donor
To elucidate whether hydrogen sulfide prevents the production of inflammatory cytokines from activated microglia, we examined the effects of sodium sulfide, hydrogen sulfide donor, in cultured microglia. Stimulation of microglia with IL-6 upregulated inflammatory cytokines including IL-6, CCL2, and TNF-α (Fig. 3). In contrast, administration of 50 or 200 μM sodium sulfide significantly blocked the IL-6-evoked upregulation of IL-6, CCL2, and TNF-α. Taken together, these observations suggest that hydrogen sulfide prevents the production of inflammatory cytokines in activated microglia.
Fig. 3.
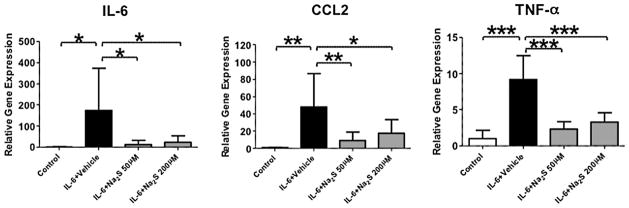
Interleukin 6 (IL-6) induced expression levels of inflammatory cytokines in the primary microglial cells. The microglia was incubated with or without IL-6 (Control) for 6 h. Vehicle (IL-6 + Vehicle), sodium sulfide (Na2S) at 50 μM (IL-6 + Na2S 50 μM), or 200 μM (IL-6 + Na2S 200 μM) was administered to the media 1 min after the addition of IL-6. CCL2, chemokine CC motif ligand 2; TNF-α, tumor necrosis factor α. N = 7–8 in each group. *P < 0.05, **P < 0.01, and ***P < 0.001. Data were analyzed using one-way ANOVA with a Bonferroni post hoc test.
4. Discussion
The current study revealed that breathing hydrogen sulfide at 40 ppm for 8 h on 7 consecutive days prevented the development of mechanical allodynia and thermal hyperalgesia that are the hallmark-symptoms of CCI-induced neuropathic pain. Inhaled hydrogen sulfide prevented microglial activation and inhibited the upregulation of inflammatory cytokines in the spinal cord after CCI. The protective effects of breathing hydrogen sulfide were associated with suppression of ATF3 in the spinal cord. We also observed that sodium sulfide, a hydrogen sulfide donor, attenuated the increased levels of inflammatory cytokines in the cultured microglia. Taken together, these observations suggest that inhaled hydrogen sulfide confers protective effects on neuropathic pain after peripheral nerve injury possibly via inhibiting microglial activation and inflammation in the spinal cord.
Increasing evidence demonstrates that microglia contributes to the development of neuropathic pain after peripheral nerve injury [6,7]. Recent data have shown that the inhibition of microglial activation prevents development of neuropathic pain behavior in animal models of neuropathic pain [22,23]. The chemokine CCL2 (also known as monocyte chemoattractant protein 1 (MCP1)) was reported to act as a key mediator of microglial activation in neuropathic pain [6,7,23]. Although CCL2 is not normally expressed in healthy conditions, it is dramatically upregulated in the spinal cord neurons and dorsal root ganglion after peripheral nerve injury [24]. Thacker and colleagues demonstrated that intrathecal administration of CCL2 leads to activation of spinal-microglia and mechanical allodynia [25]. The authors also reported that intrathical administration of CCL2 combined with anti-CCL2 antibody prevents both microglial activation and neuropathic pain behavior. Similarly, we observed that inhaled hydrogen sulfide prevented the increased expression of CCL2 as well as microglial activation in the spinal cord after peripheral nerve injury, leading to the attenuation of neuropathic pain behavior.
The activated microglia is known to release inflammatory cytokines and prostaglandins after peripheral nerve injury [6,7], and these inflammatory responses in the spinal cord play important roles in the development and maintenance of neuropathic pain status [6,23]. The inflammatory cytokine IL-6 has been implicated as a key mediator in the development of neuropathic pain behavior in both rodents and humans [26–28]. For example, DeLeo and colleagues reported that intrathecal administration of IL-6 in the absence of nerve injury produces mechanical allodynia in rats [26]. Further, Arruda and colleagues demonstrated that intrathecal administration of anti-IL-6 antibody inhibits the development of mechanical allodynia after L5 spinal nerve transection [29]. The relation between IL-6 and neuropathic pain behavior seems to be consistent with our results that inhibition of IL-6 in the spinal cord by hydrogen sulfide breathing resulted in preventing the development of neuropathic pain behavior. To further explore the mechanisms of the protective effects of hydrogen sulfide, we examined the effects of sodium sulfide in the primary microglial cell culture. We observed a robust increase of IL-6, and TNF-α in IL-6-evoked activated microglia. Administration of sodium sulfide strongly inhibited the upregulation of these inflammatory cytokines. These observations suggest that inhaled hydrogen sulfide confers protective effects on the neuropathic pain via preventing the inflammatory cytokine production from activated microglia.
ATF3 is a member of the activating transcription factor/cAMP-responsive element binding protein (ATF/CREB) family of transcription factors and is induced by various stress signals [30]. A number of studies have demonstrated that peripheral nerve injury induces the expression of ATF3 in the dorsal root ganglia (DRG) and spinal cord neurons [4,21,31]. Although ATF3 has been recognized as a marker of neuronal injury [21], its precise role is incompletely understood. Recent studies demonstrated that ATF3 is also associated with the establishment of long-lasting pain conditions [31,32]. The relationship between the increased level of IL-6 and upregulation of ATF3 in the spinal cord has been reported after CCI [4]. Similarly, we observed the correlation of the increased level of IL-6 and ATF3 in the spinal cord after peripheral nerve injury, and the upregulation of IL-6 and ATF3 were completely blocked by hydrogen sulfide breathing. Taken together, these observations suggest that inhaled hydrogen sulfide prevents neuropathic pain by inhibiting not only the inflammatory cytokine production but also the upregulation of ATF3 in the spinal cord.
Although the current study was not designed to examine the molecular mechanisms responsible for the beneficial effects of inhaled hydrogen sulfide, it has been reported that hydrogen sulfide prevents macrophage activation via inhibition of nuclear factor kappa B (NF-κB)-dependent signaling [33]. It is likely that inhaled hydrogen sulfide inhibited microglial activation via inhibition of NF-κB in our study thus preventing the development of neuropathic pain.
Concentrations of H2S produced by H2S inhalation and administration of various H2S donors are poorly defined. Inhalation of low concentrations of H2S only modestly increases blood H2S levels (<1 μM) in rats and mice [14,34]. Administration of Na2S to cell culture medium only transiently increases sulfide levels that return to the baseline levels within 30 min according to Whitfield and colleagues [35]. Therefore, both inhaled H2S at 40 ppm and administration of Na2S at 50–200 μM in cell culture appear to have negligible impact on H2S levels in circulating blood and culture medium, respectively. It is widely believed that H2S is quickly metabolized in circulation and in culture medium and at least some of it is taken up by tissue or cells. Nonetheless, the identity of the sulfide metabolites is currently unknown. Further studies are warranted to better characterize the metabolic fate of H2S, either breathed or administered as H2S donor compounds, in vivo and in vitro.
Balneotherapy in sulfur containing hot water has been widely used for the treatment of chronic pain associated with a number of conditions including osteoarthritis and fibromyalgia [36–38]. While the mechanisms responsible for the beneficial effects of balneotherapy are poorly understood, it has been reported that immersion in sulfur-rich hot water increases serum cysteine levels [39] while decreasing plasma levels of inflammatory cytokines and reactive oxygen species [40]. Based on the current observations, it is tempting to speculate that at least part of the beneficial effects of balneotherapy may be mediated by increased sulfide levels caused by absorption or inhalation of hydrogen sulfide contained in sulfur hot spring.
In conclusion, the current study revealed the robust protective effects of inhaled hydrogen sulfide, which prevented neuropathic pain behavior after peripheral nerve injury in mice. The protective effects of breathing hydrogen sulfide were associated with the prevention of microglial activation as well as attenuation of the upregulation of inflammatory cytokines in the spinal cord. Our observations also demonstrate that hydrogen sulfide donor markedly prevented the production of inflammatory cytokines from activated microglia. The ability of hydrogen sulfide to attenuate the neuropathic pain behavior in mice, if extrapolated to human beings, is clinically relevant and may serve as the experimental basis for future studies in which the effects of hydrogen sulfide donor compounds against neuropathic pain are examined.
Acknowledgments
This work was supported by postdoctoral fellowships from the American Heart Association’s Founders Affiliate 13POST14530016 and from the Massachusetts General Hospital Tosteson Fund for Medical Discovery to KK and R01 grant from the NHLBI HL101930 to FI.
References
- 1.Gilron I, Jensen TS, Dickenson AH. Combination pharmacotherapy for management of chronic pain: from bench to bedside. Lancet Neurol. 2013;12:1084–1095. doi: 10.1016/S1474-4422(13)70193-5. [DOI] [PubMed] [Google Scholar]
- 2.Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13:715–724. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Smith BH, Lee J, Price C, Baranowski AP. Neuropathic pain: a pathway for care developed by the British Pain Society. Br J Anaesth. 2013;111:73–79. doi: 10.1093/bja/aet206. [DOI] [PubMed] [Google Scholar]
- 4.Latrmolire A, Mauborgne A, Masson J, Bourgoin S, Kayser V, Hamon M, et al. Differential implication of proinflammatory cytokine interleukin-6 in the development of cephalic versus extracephalic neuropathic pain in rats. J Neurosci. 2008;28:8489–8501. doi: 10.1523/JNEUROSCI.2552-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attal N. Neuropathic pain: mechanisms, therapeutic approach, and interpretation of clinical trials. Continuum (Minneap Minn) 2012;18:161–175. doi: 10.1212/01.CON.0000411564.41709.2d. [DOI] [PubMed] [Google Scholar]
- 6.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 7.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attal N, Cruccu G, Baron R, Haanpapa M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17:1113–e88. doi: 10.1111/j.1468-1331.2010.02999.x. [DOI] [PubMed] [Google Scholar]
- 9.Baron R, Farster M, Binder A. Subgrouping of patients with neuropathic pain according to pain-related sensory abnormalities: a first step to a stratified treatment approach. Lancet Neurol. 2012;11:999–1005. doi: 10.1016/S1474-4422(12)70189-8. [DOI] [PubMed] [Google Scholar]
- 10.Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Bench-to-bedside review: hydrogen sulfide – the third gaseous transmitter: applications for critical care. Crit Care. 2009;13:213. doi: 10.1186/cc7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, et al. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98:235–244. doi: 10.1161/01.RES.0000200739.90811.9f. [DOI] [PubMed] [Google Scholar]
- 12.Predmore BL, Lefer DJ, Gojon G. Hydrogen sulfide in biochemistry and medicine. Antioxid Redox Signal. 2012;17:119–140. doi: 10.1089/ars.2012.4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kida K, Yamada M, Tokuda K, Marutani E, Kakinohana M, Kaneki M, et al. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid Redox Signal. 2010;15:343–352. doi: 10.1089/ars.2010.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokuda K, Kida K, Marutani E, Crimi E, Bougaki M, Khatri A, et al. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid Redox Signal. 2012;17:11–21. doi: 10.1089/ars.2011.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 16.Lim G, Wang S, Zhang Y, Tian Y, Mao J. Spinal leptin contributes to the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2009;119:295–304. doi: 10.1172/JCI36785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 18.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 19.Arai K, Lo EH. An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci. 2009;29:4351–4355. doi: 10.1523/JNEUROSCI.0035-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2010;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, et al. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol Cell Neurosci. 2000;15:170–182. doi: 10.1006/mcne.1999.0814. [DOI] [PubMed] [Google Scholar]
- 22.Watkins LR, Maier SF. GLIA: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- 23.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 25.Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LDF, et al. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 26.DeLeo JA, Colburn RW, Nichols MICH, Malhotra AMIT. Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. J Interferon Cytokine Res. 1996;16:695–700. doi: 10.1089/jir.1996.16.695. [DOI] [PubMed] [Google Scholar]
- 27.Oka T, Oka K, Hosoi M, Hori T. Intracerebroventricular injection of interleukin-6 induces thermal hyperalgesia in rats. Brain Res. 1995;692:123–128. doi: 10.1016/0006-8993(95)00691-i. [DOI] [PubMed] [Google Scholar]
- 28.Geiss A, Varadi E, Steinbach K, Bauer HW, Anton F. Psychoneuro-immunological correlates of persisting sciatic pain in patients who underwent discectomy. Neurosci Lett. 1997;237:65–68. doi: 10.1016/s0304-3940(97)00810-0. [DOI] [PubMed] [Google Scholar]
- 29.Arruda JL, Sweitzer S, Rutkowski MD, DeLeo JA. Intrathecal anti-IL-6 antibody and IgG attenuates peripheral nerve injury-induced mechanical allodynia in the rat: possible immune modulation in neuropathic pain. Brain Res. 2000;879:216–225. doi: 10.1016/s0006-8993(00)02807-9. [DOI] [PubMed] [Google Scholar]
- 30.Hai TSON, Wolford CC, Chang AYS. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 2010;15:1–11. doi: 10.3727/105221610x12819686555015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obata K, Yamanaka H, Fukuoka T, Yi D, Tokunaga A, Hashimoto N, et al. Contribution of injured and uninjured dorsal root ganglion neurons to pain behavior and the changes in gene expression following chronic constriction injury of the sciatic nerve in rats. Pain. 2003;101:65–77. doi: 10.1016/s0304-3959(02)00296-8. [DOI] [PubMed] [Google Scholar]
- 32.Shortland PJ, Baytug B, Krzyzanowska A, McMahon SB, Priestley JV, Averill S. ATF3 expression in L4 dorsal root ganglion neurons after L5 spinal nerve transection. Eur J Neurosci. 2006;23:365–373. doi: 10.1111/j.1460-9568.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- 33.Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2009;12:1147–1154. doi: 10.1089/ars.2009.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 36.Kovacs C, Pecze M, Tihanyi AG, Kovacs L, Balogh SA, Bender T. The effect of sulphurous water in patients with osteoarthritis of hand. Double-blind, randomized, controlled follow-up study. Clin Rheumatol. 2012;31:1437–1442. doi: 10.1007/s10067-012-2026-0. [DOI] [PubMed] [Google Scholar]
- 37.Verhagen AP. Balneotherapy for osteoarthritis. Cochrane Database Syst Rev. 2008;(4):CD006864. doi: 10.1002/14651858.CD006864. [DOI] [PubMed] [Google Scholar]
- 38.Baranowsky J, Klose P, Musial F, Haeuser W, Dobos G, Langhorst J. Qualitative systemic review of randomized controlled trials on complementary and alternative medicine treatments in fibromyalgia. Rheumatol Int. 2009;30:1–21. doi: 10.1007/s00296-009-0977-5. [DOI] [PubMed] [Google Scholar]
- 39.Bagnato G, De Filippis LG, Morgante S, Morgante ML, Farina G, Caliri A, et al. Clinical improvement and serum amino acid levels after mud-bath therapy. Int J Clin Pharmacol Res. 2004;24:39–47. [PubMed] [Google Scholar]
- 40.Fioravanti A, Cantarini L, Guidelli GM, Galeazzi M. Mechanisms of action of spa therapies in rheumatic diseases: what scientific evidence is there? Rheumatol Int. 2011;31:1–8. doi: 10.1007/s00296-010-1628-6. [DOI] [PubMed] [Google Scholar]