

Abstract
Mitochondriopathies are a group of clinically heterogeneous genetic diseases caused by defects in mitochondrial metabolism, bioenergetic efficiency, and/or signaling functions. The large majority of proteins involved in mitochondrial function are encoded by nuclear genes, with many yet to be associated with human disease. We performed exome sequencing on a young girl with a suspected mitochondrial myopathy that manifested as progressive muscle weakness, hypotonia, seizures, poor weight gain, and lactic acidosis. She was compound heterozygous for two frameshift mutations, p. Asn112HisfsX29 and p. Leu659AlafsX4, in the PNPLA8 gene, which encodes mitochondrial calcium independent phospholipase A2γ (iPLA2γ). Western blot analysis of affected muscle displayed the absence of PNPLA8 protein. iPLA2s are critical mediators of a variety of cellular processes including growth, metabolism, and lipid second messenger generation, exerting their functions through catalyzing the cleavage of the acyl groups in glycerophospholipids. The clinical presentation, muscle histology and the mitochondrial ultrastructural abnormalities of this proband are highly reminiscent of Pnpla8 null mice. Although other iPLA2–related diseases have been identified, namely infantile neuroaxonal dystrophy and neutral lipid storage disease with myopathy, this is the first report of PNPLA8-related disease in a human. We suggest PNPLA8 join the increasing list of human genes involved in lipid metabolism associated with neuromuscular diseases due to mitochondrial dysfunction.
Keywords: PNPLA8, mitochondrial dysfunction, phospholipase, dystonia
The myriad important roles assumed by mitochondria in cellular function, including energy production, metabolism, signaling pathways and the regulation of apoptosis, are reflected in the clinical diversity of human mitochondrial diseases. Significant pleiotropy exists for these disorders, which frequently present with neurologic and myopathic phenotypes, often involving multiple organ systems. The organs most reliant on mitochondrial energy production are most susceptible to such defects, such as the extraocular muscles and pancreas. Typical clinical features often include, but are not limited to, myopathy, diabetes mellitus, liver failure, ataxia, seizures, encephalopathy, sensorineural deafness and bone marrow failure (DiMauro and Schon, 2003). Given this complexity, the extensive display of locus and allelic heterogeneity present in mitochondriopathies is not surprising, with over 1,300 nuclear genes implicated in mitochondrial function and 265 disease genes identified to date (Pagliarini, et al., 2008). Mitochondrial diseases may be caused by maternally inherited or sporadic defects of the mitochondrial genome (mtDNA) or by defects in nuclear genes, which may be inherited in an autosomal recessive, dominant or X-linked manner (Thorburn and Dahl, 2001). Collectively, mitochondrial diseases are a significant source of morbidity and mortality, with a conservative estimate for prevalence being 1 in 5000 live births (Skladal, et al., 2003). The diverse clinical presentation of mitochondrial disease in conjunction with the large number of both nuclear and mitochondrial gene disease targets make molecular diagnoses of mitochondrial diseases challenging. With the growing use of whole exome sequencing (WES), definitive genetic diagnoses of mitochondriopathies are now possible, with important implications for prognosis and clinical management.
Herein, we report the use of exome sequencing to identify a novel human mitochondrial disease in patient CMH193, a 7 year old female who was evaluated for a suspected mitochondrial myopathy with dystonia, abnormal gait and abnormal muscle biopsy. This patient was a term baby born to a G4P4 mother by spontaneous vaginal delivery following an uneventful pregnancy, weighing 3.1 kg (25th percentile). She was asymptomatic in the first year, meeting all developmental milestones; however, the parents noted toe-walking. At age 2, she was noted to have a positive Gowers sign, and by the age of 3 years could no longer walk. Independent sitting was restricted to 10 minutes due to fatigue. Her fine motor skills declined and speech became dysarthric. She had proximal muscle weakness which was more pronounced proximally, tight heel cords, subtle rightward deviation of the tongue, and generalized mildly increased tone. These symptoms have progressed over the past several years, with increased proximal muscle weakness and dysmetria. Hypotonicity progressed to spasticity of different opposing muscle groups. She recently developed complex partial seizures associated with recurrent left temporal lobe spikes on EEG, and slow wave discharges exacerbated by sleep. Treatment with a multivitamin containing iron, Carbidopa-Levodopa and leucovorin initially improved symptoms. Supplementation with CoQ10 increased appetite without subsequent weight gain, and persistence of a very lean body habitus. In addition, she developed significant lactic acidosis, requiring bicarbonate supplementation. Relevant positive family history includes a maternal history of migraines and renal stones, a paternal grandmother with early stroke (age 45), and multiple family members with late onset cardiac problems. Complete blood count, metabolic panel, AST, ALT, liver function, creatine phosphokinase have been performed multiple times and are normal, with the exception of a consistently elevated anion gap (17 mmol/L) since age 5. Karyotype, microarray, mtDNA sequence analysis for tRNAs, and mtDNA depletion studies were performed at age four and were normal. Neurotransmitter studies in cerebrospinal fluid performed at the age of 6 showed deficiency of 5-methyltetrahydrofolate. At age 7, elevated lactic and pyruvic acid was demonstrated in blood on multiple occasions (lactic acid 3.8-5.6; n=0.7-2.1 mmol/L and pyruvic acid 0.18-0.32; n=0.08-0.16 mmol/L) with ratios of 17.5-21.1 (n=<20); plasma acylcarnitine profile was normal. Initial MRI brain scan at 33 months of age showed subtle increased signal intensity involving the medial globus pallidus bilaterally which was no longer visualized two years later at the second study, suggesting maturation of myelination. MR spectroscopy at 33 months showed metabolites within the normal range including choline, creatine and NAA. CT of the head was also unremarkable. Accurate cognitive assessment of this patient is limited due to frequent seizures, level of fatigue, and oral motor dystonia, however she attends Elementary school and receives physical therapy, occupational therapy, and speech therapy.
Exome sequencing was performed on patient CMH193 and her two healthy parents (CMH466 & CMH467) following informed consent to participate in a study approved by the Institutional Review Board (IRB) of Children's Mercy Hospital (CMH). DNA was prepared utilizing the KAPA Biosystems library preparation kit (KAPA Biosystems, Woburn, MA) followed by Illumina TruSeqExome enrichment (Illumina, San Diego, CA). Enrichment efficiency was verified using quantitative real-time PCR. Samples were sequenced on an Illumina HiSeq 2000 instrument with TruSeq v3 reagents, yielding paired end 100 nucleotide reads. Alignment and variant calling were performed as previously reported (Bell, et al., 2011; Saunders, et al., 2012). Briefly, gapped alignment to reference sequences (Hg19 and GRCH37) was performed with GSNAP and the GATK. Nucleotide variants were genotyped and annotated utilizing RUNES (Saunders, et al., 2012). Variant analysis for all samples was completed utilizing Viking (Saunders, et al., 2012). The proband was sequenced to a depth of 7.7 gigabases resulting in median target coverage of 135×, which identified ∼170,000 nucleotide variants. The mitochondrial genome was represented at an average depth of 1835×. Variants were filtered to 1% minor allele frequency in an internal database of 1913 samples, then prioritized by ACMG categorization, OMIM identity and phenotypic assessment. No diagnostic genotypes were found in previously reported disease genes, however the patient was compound heterozygous for two frameshift variants, c.334_337delAATT (p.Asn112HisfsX29) and c.1975_1976delAG (p.Leu659AlafsX4) in PNPLA8 (MIM# 612123; nomenclature based on NM_001256011.1; +1 as the A of the ATG initiation codon in the reference sequence; initiation codon as codon 1), which encodes iPLA2γ. The two variants were confirmed by Sanger sequencing and submitted to the LOVD database (http://databases.lovd.nl/shared/transcripts/16455). Segregation analysis confirmed that the variants were inherited from carrier parents, consistent with an autosomal recessive inheritance pattern. In addition, genotyping of two healthy siblings revealed they were negative for both variants. Both PNPLA8 variants were absent from the NHLBI Exome Sequencing Project (EVS; http://evs.gs.washington.edu/EVS/) and an internal variant database of 1913 samples. Truncating PNPLA8 variants were absent from the internal dataset and rare in the EVS, and homozygous truncating variants were absent.
Mammalian iPLA2s serve critical roles in cellular signaling, growth, lipid homeostasis, second messenger generation and ion channel function (Mancuso, et al., 2000) by catalyzing the cleavage of acyl groups from glycerophosholipids resulting in the generation of free fatty acids and lysolipids. Released polyunsaturated fatty acids can be further metabolized by a variety of cycloxygenases, lipoxygenases and P450 enzymes generating potent lipid second messengers of signal transduction. Previous studies using both pharmacologic and genetic approaches have demonstrated the prominent roles of iPLA2s in neurotransmitter release, long-term potentiation, and cognition (Mancuso, et al., 2009; Wolf, et al., 1995). In mitochondria, the products of iPLA2s and their downstream metabolites play key roles in mitochondrial bioenergetics, signaling and the opening of the mitochondrial permeability transition pore (Moon, et al., 2012a). Of the 9 known PNPLA family members, four have previously been identified that differentially regulate various aspects of mitochondrial bioenergetics and signaling, including PNPLA2 (MIM# 609059), PNPLA3 (MIM# 609567), PNPLA6 (MIM# 603197) and PNPLA9 (MIM# 603604). Variants in three of these genes have been reported in association with human disease: PNPLA2, which encodes an adipose triglyceride lipase, is associated with Neutral Lipid Storage Disease with Myopathy, (MIM# 610717) (Fischer, et al., 2007; Janssen, et al., 2013; Reilich, et al., 2011), PNLPA6 defects cause a broad spectrum of neurodegenerative diseases including Boucher-Neuhäuser and Gordon Holmes syndromes (MIM#s 215470, 612020) (Deik, et al., 2014; Synofzik, et al., 2014a; Synofzik, et al., 2014b; Topaloglu, et al., 2014; Wortmann, et al., 2014; Yoon, et al., 2013.) PNPLA9 is associated with Infantile Neuronal Axonal Dystrophy (MIM# 603604) (Illingworth, et al., 2014; Khateeb, et al., 2006; Morgan, et al., 2006; Salih, et al., 2013; Tonelli, et al., 2010).
PNPLA8, the predominant phospholipase in mammalian mitochondria, has been extensively characterized using genetic loss of function and gain of function mouse models in conjunction with enantio selective mechanism-based inhibition (Elimam, et al., 2013; Mancuso, et al., 2007a; Mancuso, et al., 2000; Mancuso, et al., 2009; Mancuso, et al., 2007b; Moon, et al., 2012a; Moon, et al., 2012b; Yan, et al., 2005). The alterations in mitochondrial ultrastructure and function in the Pnpla8 null mouse model bears a striking resemblance to CMH193. The Pnpla8 -/- mouse has mitochondrial dysfunction, impaired learning, decreased exercise endurance, enhanced insulin sensitivity, a thin body habitus (from energy dissipation by mitochondrial uncoupling), and cold intolerance (Mancuso, et al., 2009).
In addition to the similarities in phenotypes, there are unique morphologic changes observed in mitochondria of the Pnpla8 -/- mouse, which closely match those observed in the right quadriceps muscle of CMH193 at 4 years of age (Fig. 1). Specifically, mitochondria from CMH193 exhibit marked disarray of cristae and globular dense osmiophilic inclusions by EM analysis. In addition, subsarcolemmal aggregates of abnormal mitochondria with concentric lamellar membranes and dense round or oval, strongly osmiophilic inclusions were observed, suggestive of degenerating mitochondria (Fig. 1b, c). No large aggregates of mitochondria were observed. In addition, variable fiber size was found with isolated small atrophic fibers of all subtypes observed, but with no ragged red fibers (Fig. 1a). Interestingly, there was a significant increase in secondary lysosomes containing residual bodies that appeared to be derived from mitochondria, which raised the possibility of lipofuscinosis. However, no pathogenic variants were found in lipofuscinosis genes. Test results supporting mitochondrial dysfunction in CMH193 include an elevated anion gap and moderately elevated lactate acid levels in blood requiring bicarbonate supplementation. In the absence of an elevated lactate: pyruvate ratio, these results suggest mitochondrial dysfunction rather than an electron transport chain defect. Consistent with this concept, oxidative enzyme reactions (SDH and COX) showed normal activity in muscle (Supp. Figure S1), and electron transport chain function was not affected (Supp. Table S1). In addition, CSF neurotransmitter studies in CMH193 showed decreased 5-methyltetrahydrofolate, and secondary CSF folate deficiency is seen in many disorders of mitochondrial function (Garcia-Cazorla, et al., 2008). The genotype observed in CMH193 (two frameshift variants) was expected to generate a null phenotype, secondary to a truncated or degraded product. To confirm this, Western blotting against iPLA2γ protein was performed on a muscle biopsy from the patient as well as three de-identified samples with known unrelated diagnoses (CPT2 deficiency, Duchenne muscular dystrophy, and pulmonary hypoplasia; Fig. 2). As predicted, Western blotting of tissue from the patient exhibited a dramatic decrease in multiple immunoreactive iPLA2γ bands known to be absent in the Pnpla8 -/- mouse. In addition, a novel 110 KD band was present in “control” patients, but absent in CMH193. Due to the surprising coordinate disappearance of both the known iPLA2γ bands and the novel immunoreactive 110 kDa band in CMH193, further studies were undertaken to determine the identity of the latter. Protein was extracted from a cadaveric gastrocnemius muscle at 10hr post mortem and purified by sequential DEAE anion exchange and FPLC Mono Q chromatographies. The resultant highly purified 110 kDa protein was electrophoresed on SDS PAGE, visualized by silver staining, and the 110 kDa band was excised from the gel and subjected to trypsinolysis. Resultant peptides were identified by LC-MS/MS using an LTQ-Orbitrap. The results demonstrated the 110 kD band to be heat shock protein family member 4 (HSPA4) (Supp. Table S2). Furthermore, the 110 kDa band did not contain any peptides corresponding to those present in iPLA2γ, either by traditional LC-MS/MS, or through the enhanced sensitivity provided by multiple reaction monitoring of known transitions of previously identified peptides in iPLA2γ (PNPLA8). Collectively, these results indicate that the “immunoreactivity” of the 110 kDa protein detected by Western blotting was due to a non-specific interaction of HSPA4 with the antibody. Previously, we reported the co-chromatography of a 63kDa iPLA2γ isoform and another member of the heat shock protein family that was distinct from HSPA4 (Yan, et al., 2005). We speculate that the concomitant disappearance of iPLA2γ and the 110 kDa band is likely due to the absence of a transcriptional activator that may be derived from intronic DNA in the PNPLA8 gene, as has previously been described for other protein-chaperone systems (Moabbi, et al., 2012).
Figure 1. Comparison of Histological and EM findings in PNPLA8-affected tissues from human and mice.
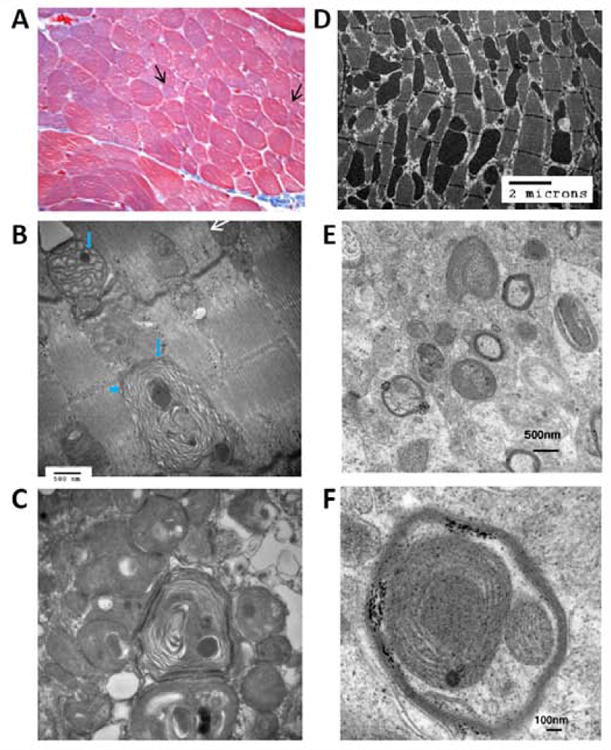
Panels (A)-(C) show paraffin-embedded muscle biopsy from patient CMH193. Trichromestaining (A) (magnification 400 ×) revealed variability in fiber size with isolated smallatrophic fibers (arrows). Normal fibers average 35 microns. ATPase stains (not shown) revealed that atrophic fibers were type 1 and 2 fibers. Ragged red fibers were not found. Oxidative enzyme reactions (SDH and COX) were normal and no large aggregates of mitochondria were demonstrable.
(B) EM photograph of muscle biopsy from CMH193 at 48000 magnification. Mitochondria show abnormal concentric disarray of internal cristae (arrow head) and globular dense osmiophilic inclusions (short arrows). White arrow shows a mitochondrion close to normal. (C) EM photograph of muscle biopsy from CMH193 at 36000 magnification, exhibiting subsarcolemmal aggregates of abnormal mitochondria with concentric lamellar membranes and dense round to oval osmiophilic inclusions. D) Electron micrograph of myocardium from the Pnpla8 -/- mouse; E) Electron micrograph of the cerebellum from Pnpla8 -/- mouse exhibiting dysmorphic mitochondria; F) High power view of the cerebellum from the Pnpla8 -/- mouse.
Figure 2. Near absence of PNPLA8 protein in CMH193.
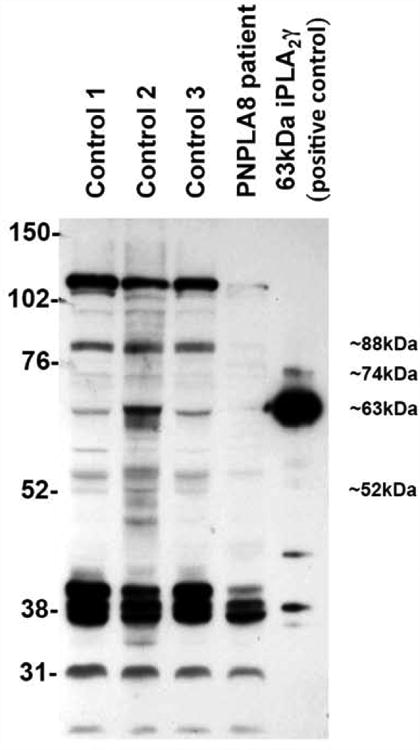
Western blot showing dramatic decrease of the 88, 63, and 52kD PNPLA8 protein (NP_001242940.1) in the muscle from CMH193, compared to muscle from three control samples with known non-mitochondrial diagnoses.
Thus, PNPLA8 may be added to an increasing list of genes involved in lipid metabolism associated with neurodegenerative diseases (Boukhris, et al., 2013; Martin, et al., 2013; Schuurs-Hoeijmakers, et al., 2012; Tesson, et al., 2012). Other iPLA2-specific disorders include PLA2G6, associated with infantile neuroaxonal dystrophy 1, PNPLA2, associated with neutral lipid storage disease with myopathy and PNPLA6, associated with Boucher-Neuhäuser and Gordon Holmes syndromes (Deik, et al., 2014; Synofzik, et al., 2014a; Synofzik, et al., 2014b; Topaloglu, et al., 2014; Wortmann, et al., 2014) as well as spastic ataxia and hereditary spastic paraplegia with or without motor neuropathy (Synofzik, et al., 2014a; Yoon, et al., 2013). In the view of this case report, and given that mitochondrial disorders are clinically heterogeneous diseases, there appears to be a variable spectrum in iPLA2-related disorders. While patients with variants in PNPLA2 share a recognizable phenotype characterized by slowly progressive adult onset proximal muscle weakness affecting the upper and lower limbs, distal muscle weakness is also present. In addition, half of the affected patients show cardiomyopathic changes as adults (typically around 40 years of age). Other variable features include diabetes mellitus, hepatic steatosis, hypertriglyceridemia, and possibly sensorineural hearing loss (Reilich, et al., 2011). Elevated serum transaminases and creatine are clinical markers of PNPLA2 disease, but were not found in CMH193.
Children with infantile neuroaxonal dystrophy have a severe progressive psychomotor disorder with typical onset within the first 24 months of life and rapid progression of hypotonia, hyperreflexia, tetraparesis leading to loss of ambulation within 5 years, pyramidal signs and seizures. Skin biopsies showed axonal spheroids. Although brain iron accumulation is variable, MRI studies have demonstrated areas of increased intensity, early degenerative processes, and cerebellar cortical atrophy. Death usually occurs prior to age ten (Morgan, et al., 2006). PLA2G6 variants were predicted to result in an almost complete lack of enzyme activity, although gain of function could not be excluded, as missense variants have been reported in families with adult-onset dystonia-parkinsonism (PARK14; MIM# 612953) and PLA2G6 protein is active as a tetramer (Morgan, et al., 2006). As reported in our patient, PLA2G6 affected patients have dystonia (Tonelli, et al., 2010). The biochemical activity of the respiratory chain complexes I-IV were normal in all patients with iPLA2 variants examined thus far. Moreover, histopathological analysis of skeletal muscle in iPLA2-related diseases revealed some similar findings (Khateeb, et al., 2006; Reilich, et al., 2011). In PLA2G6-related diseases, the degeneration of the mitochondrial inner membrane is well characterized and proposed to be the primary pathogenic mechanism (Illingworth, et al., 2014). This may also be the case with PNPLA8-related disease, where proteins involved in electron transport and ATP synthesis located in the inner membranes would be secondarily affected by altered membrane homeostasis but not the primary pathogenic mechanism.
Boucher-Neuhäuser and Gordon Holmes syndromes are defined by early-onset ataxia and hypogonadism plus chorioretinal dystrophy (Boucher-Neuhäuser syndrome) or brisk reflexes (Gordon Holmes syndrome), associated with variants in PNPLA6 (Synofzik, et al., 2014a; Synofzik, et al., 2014b; Topaloglu, et al., 2014). In addition, PNPLA6 pathogenic variants are also associated with spastic ataxia and hereditary spastic paraplegia, suggesting a broad spectrum of neurodegenerative PNPLA6-related diseases (Synofzik, et al., 2014a; Yoon, et al., 2013). PNPLA6 catalyzes the hydrolysis of membrane phosphatidylcholine into fatty acids and lysophosphatidylcholines; defects may lead to disturbance of membrane homeostasis at the synaptic junctions in a variety of neuronal networks. PNPLA6 shares 41% amino acid sequence identity with the Drosophila ‘Swiss Cheese’ (Sws) protein, and has been implicated in the regulating interactions between neurons and glia in the developing fly brain (Beck, et al., 2011). Secondly, it may affect cellular signaling in the nervous system by impacting biosynthesis of acetylcholine or other neurotransmitters (van Tienhoven, et al., 2002). Although there is an intriguingly widening range of phenotypes and variable severity associated with alteration in these genes, it is also possible that in addition to the loss of the PNPLA8 protein, the absence of the unknown chaperone detected by immunoblotting (110-kD band) could contribute to the pathogenesis in our PNPLA8 patient.
In summary, a causal link between the proband's phenotype and PNPLA8 variantsis supported by the parallel alterations in neuromuscular dysfunction, cognitive deficits and the striking similarities in dysmorphic and degenerating mitochondria present in the Pnpla8 -/- mouse. It appears that many metabolic and genetic factors may account for the clinical heterogeneity of iPLA2-related diseases. The quantitative importance of phospholipids in the nervous system makes the brain and peripheral nerves a critical target for dysfunction in cellular growth, signaling and lipid homeostasis (Lamari, et al., 2013; Mancuso, et al., 2009; Wortmann, et al., 2014). It remains to be seen whether other (milder) variants in this gene result in a different iPLA2γ-related phenotype and whether the 110 kD heat shock protein can indeed provide additional insights into the chemical biology of iPLA2γ mediated mitochondrial dysfunction and its downstream sequelae.
Supplementary Material
Acknowledgments
We thank the family for their participation. We thank Dr. Lili Miles for the EM studies. This work was supported by the Clare Giannini Fund and RO1 HL118639.
Grant sponsors: Clare Giannini Fund; NIH RO1 HL118639
Footnotes
Conflict of interest: Authors state that they have no conflicts of interest.
References
- Beck G, Sugiura Y, Shinzawa K, Kato S, Setou M, Tsujimoto Y, Sakoda S, Sumi-Akamaru H. Neuroaxonal dystrophy in calcium-independent phospholipase A2beta deficiency results from insufficient remodeling and degeneration of mitochondrial and presynaptic membranes. J Neurosci. 2011;31(31):11411–20. doi: 10.1523/JNEUROSCI.0345-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhris A, Schule R, Loureiro JL, Lourenco CM, Mundwiller E, Gonzalez MA, Charles P, Gauthier J, Rekik I, Acosta Lebrigio RF, et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am J Hum Genet. 2013;93(1):118–23. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deik A, Johannes B, Rucker JC, Sanchez E, Brodie SE, Deegan E, Landy K, Kajiwara Y, Scelsa S, Saunders-Pullman R, et al. Compound heterozygous PNPLA6 mutations cause Boucher-Neuhauser syndrome with late-onset ataxia. J Neurol. 2014 doi: 10.1007/s00415-014-7516-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348(26):2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Elimam H, Papillon J, Takano T, Cybulsky AV. Complement-mediated activation of calcium-independent phospholipase A2gamma: role of protein kinases and phosphorylation. J Biol Chem. 2013;288(6):3871–85. doi: 10.1074/jbc.M112.396614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Lefevre C, Morava E, Mussini JM, Laforet P, Negre-Salvayre A, Lathrop M, Salvayre R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39(1):28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- Garcia-Cazorla A, Quadros EV, Nascimento A, Garcia-Silva MT, Briones P, Montoya J, Ormazabal A, Artuch R, Sequeira JM, Blau N, et al. Mitochondrial diseases associated with cerebral folate deficiency. Neurology. 2008;70(16):1360–2. doi: 10.1212/01.wnl.0000309223.98616.e4. [DOI] [PubMed] [Google Scholar]
- Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R, Hardy C, McDonald F, Childs AM, Stewart B, et al. PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab. 2014;112(2):183–9. doi: 10.1016/j.ymgme.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen MC, van Engelen B, Kapusta L, Lammens M, van Dijk M, Fischer J, van der Graaf M, Wevers RA, Fahrleitner M, Zimmermann R, et al. Symptomatic lipid storage in carriers for the PNPLA2 gene. Eur J Hum Genet. 2013;21(8):807–15. doi: 10.1038/ejhg.2012.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. 2006;79(5):942–8. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamari F, Mochel F, Sedel F, Saudubray JM. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Dis. 2013;36(3):411–25. doi: 10.1007/s10545-012-9509-7. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Han X, Jenkins CM, Lehman JJ, Sambandam N, Sims HF, Yang J, Yan W, Yang K, Green K, et al. Dramatic accumulation of triglycerides and precipitation of cardiac hemodynamic dysfunction during brief caloric restriction in transgenic myocardium expressing human calcium-independent phospholipase A2gamma. J Biol Chem. 2007a;282(12):9216–27. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A(2) J Biol Chem. 2000;275(14):9937–45. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, et al. Genetic ablation of calcium-independent phospholipase A2{gamma} leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J Biol Chem. 2009;284(51):35632–44. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, et al. Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem. 2007b;282(48):34611–22. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Schule R, Smets K, Rastetter A, Boukhris A, Loureiro JL, Gonzalez MA, Mundwiller E, Deconinck T, Wessner M, et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am J Hum Genet. 2013;92(2):238–44. doi: 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moabbi AM, Agarwal N, El Kaderi B, Ansari A. Role for gene looping in intron-mediated enhancement of transcription. Proc Natl Acad Sci U S A. 2012;109(22):8505–10. doi: 10.1073/pnas.1112400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SH, Jenkins CM, Kiebish MA, Sims HF, Mancuso DJ, Gross RW. Genetic ablation of calcium-independent phospholipase A(2)gamma (iPLA(2)gamma) attenuates calcium-induced opening of the mitochondrial permeability transition pore and resultant cytochrome c release. J Biol Chem. 2012a;287(35):29837–50. doi: 10.1074/jbc.M112.373654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SH, Jenkins CM, Liu X, Guan S, Mancuso DJ, Gross RW. Activation of mitochondrial calcium-independent phospholipase A2gamma (iPLA2gamma) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J Biol Chem. 2012b;287(18):14880–95. doi: 10.1074/jbc.M111.336776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38(7):752–4. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134(1):112–23. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilich P, Horvath R, Krause S, Schramm N, Turnbull DM, Trenell M, Hollingsworth KG, Gorman GS, Hans VH, Reimann J, et al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J Neurol. 2011;258(11):1987–97. doi: 10.1007/s00415-011-6055-4. [DOI] [PubMed] [Google Scholar]
- Salih MA, Mundwiller E, Khan AO, AlDrees A, Elmalik SA, Hassan HH, Al-Owain M, Alkhalidi HM, Katona I, Kabiraj MM, et al. New findings in a global approach to dissect the whole phenotype of PLA2G6 gene mutations. PLoS One. 2013;8(10):e76831. doi: 10.1371/journal.pone.0076831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, Andraws N, Patterson ML, Krivohlavek LA, Fellis J, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4(154):154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, Swinkels ME, Gilissen C, Willemsen MA, Holvoet M, Steehouwer M, Veltman JA, de Vries BB, et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012;91(6):1122–7. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain : a journal of neurology. 2003;126(Pt 8):1905–12. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, Hannequin D, Strom TM, Prokisch H, Kernstock C, et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014a;137(Pt 1):69–77. doi: 10.1093/brain/awt326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Kernstock C, Haack TB, Schols L. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher-Neuhauser syndrome due to PNPLA6 mutations. J Neurol Neurosurg Psychiatry. 2014b doi: 10.1136/jnnp-2014-307793. [DOI] [PubMed] [Google Scholar]
- Tesson C, Nawara M, Salih MA, Rossignol R, Zaki MS, Al Balwi M, Schule R, Mignot C, Obre E, Bouhouche A, et al. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet. 2012;91(6):1051–64. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn DR, Dahl HH. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. American journal of medical genetics. 2001;106(1):102–14. doi: 10.1002/ajmg.1380. [DOI] [PubMed] [Google Scholar]
- Tonelli A, Romaniello R, Grasso R, Cavallini A, Righini A, Bresolin N, Borgatti R, Bassi MT. Novel splice-site mutations and a large intragenic deletion in PLA2G6 associated with a severe and rapidly progressive form of infantile neuroaxonal dystrophy. Clin Genet. 2010;78(5):432–40. doi: 10.1111/j.1399-0004.2010.01417.x. [DOI] [PubMed] [Google Scholar]
- Topaloglu AK, Lomniczi A, Kretzschmar D, Dissen GA, Kotan LD, McArdle CA, Koc AF, Hamel BC, Guclu M, Papatya ED, et al. Loss-of-Function Mutations in PNPLA6 Encoding Neuropathy Target Esterase Underlie Pubertal Failure and Neurological Deficits in Gordon Holmes Syndrome. J Clin Endocrinol Metab. 2014;99(10):E2067–75. doi: 10.1210/jc.2014-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tienhoven M, Atkins J, Li Y, Glynn P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem. 2002;277(23):20942–8. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Izumi Y, Zorumski CF, Gross RW. Long-term potentiation requires activation of calcium-independent phospholipase A2. FEBS Lett. 1995;377(3):358–62. doi: 10.1016/0014-5793(95)01371-7. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Espeel M, Almeida L, Reimer A, Bosboom D, Roels F, de Brouwer AP, Wevers RA. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J Inherit Metab Dis. 2014 doi: 10.1007/s10545-014-9759-7. [DOI] [PubMed] [Google Scholar]
- Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2gamma: identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signaling. J Biol Chem. 2005;280(29):26669–79. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- Yoon G, Baskin B, Tarnopolsky M, Boycott KM, Geraghty MT, Sell E, Goobie S, Meschino W, Banwell B, Ray PN. Autosomal recessive hereditary spastic paraplegia-clinical and genetic characteristics of a well-defined cohort. Neurogenetics. 2013;14(3-4):181–8. doi: 10.1007/s10048-013-0366-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.