

Abstract
Aims: Typical 2-Cys peroxiredoxins (2-Cys Prxs) are Cys peroxidases that undergo inactivation by hyperoxidation of the catalytic Cys, a modification reversed by ATP-dependent reduction by sulfiredoxin (Srx). Such an attribute is thought to provide regulation of 2-Cys Prxs functions. The initial steps of the Srx catalytic mechanism lead to a Prx/Srx thiolsulfinate intermediate that must be reduced to regenerate Srx. In Saccharomyces cerevisiae Srx, the thiolsulfinate is resolved by an extra Cys (Cys48) that is absent in mammalian, plant, and cyanobacteria Srxs (1-Cys Srxs). We have addressed the mechanism of reduction of 1-Cys Srxs using S. cerevisiae Srx mutants lacking Cys48 as a model. Results: We have tested the recycling of Srx by glutathione (GSH) by a combination of in vitro steady-state and single-turnover kinetic analyses, using enzymatic coupled assays, Prx fluorescence, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and reverse-phase chromatography coupled to mass spectrometry. We demonstrate that GSH reacts directly with the thiolsulfinate intermediate, by following saturation kinetics with an apparent dissociation constant of 34 μM, while producing S-glutathionylated Srx as a catalytic intermediate which is efficiently reduced by the glutaredoxin/glutathione reductase system. Total cellular depletion of GSH impacted the recycling of Srx, confirming in vivo that GSH is the physiologic reducer of 1-Cys Srx. Innovation: Our study suggests that GSH binds to the thiolsulfinate complex, thus allowing non-rate limiting reduction. Such a structural recognition of GSH enables an efficient catalytic reduction, even at very low GSH cellular levels. Conclusion: This study provides both in vitro and in vivo evidence of the role of GSH as the primary reducer of 1-Cys Srxs. Antioxid. Redox Signal. 22, 731–743.
Introduction
Mechanisms of H2O2-dependent signaling generally rely on thiol chemistry and involve specific protein sensors such as peroxiredoxins (19). Typical 2-Cys peroxiredoxins (Prx) constitute an important class of thiol peroxidases that are structured as obligate head-to-tail dimers, with each monomer carrying two Cys residues that are named peroxidatic (CP) and resolving (CR) Cys. Their peroxidatic cycle involves a reaction of CP with H2O2, which leads to the formation of a reactive sulfenic CP–SOH intermediate (6). The fast kinetics of this reaction, in the 105–107 M−1s−1 range, explain the high H2O2-reducing efficiency of these enzymes as peroxidases and their unique ability to detect very low levels of H2O2 (4, 7, 10, 26, 39). The nascent sulfenic acid then condensates with the CR of the symmetrical subunit to form an intermolecular disulfide, which is then reduced by thioredoxin (Trx), thus completing the cycle (41).
Innovation.
Typical 2-Cys peroxiredoxins (Prx) are thiol peroxidases regulated by a sulfinic redox switch reverted by the sulfiredoxin (Srx). Unlike yeast Srx, which is recycled by thioredoxin, a mutant of yeast Srx lacking the resolving cysteine residue operates via a distinct mechanism and is efficiently reduced by glutathione both in vivo and in vitro. This supports glutathione as the in vivo reducer of the 1-Cys Srx existing in mammals, plants, and cyanobacteria. Improved understanding of the crosstalk between Prx/Srx and the main cellular redox systems should contribute to elucidating the role of the Prx/Srx couple in antioxidant resistance, cell signaling, and aging.
Typical 2-Cys peroxiredoxins carry the unique ability to exit the catalytic cycle by H2O2-mediated hyperoxidation of Cys CP–SOH to the sulfinic acid form (Prx–SO2), thereby becoming inactive. Hence, the CP–SOH sulfenic acid is poised to condense with CR to a disulfide or to react with H2O2 to form a sulfinic acid (32, 40). Hyperoxidation is reversed by ATP-dependent reduction by sulfiredoxin (Srx) (1). The physiological significance of the inactivation of Prxs by hyperoxidation is not intuitive. The observation that this attribute and the presence of an Srx gene are both exclusive to eukaryotes, with some exception in cyanobacteria, has led to a suggestion that it might entail a regulation of Prx functions (2, 31, 40, 41). The so-called “floodgate model” was hence proposed, which posits that during H2O2 signaling, Prx must be inactivated for allowing the oxidant to reach its regulatory targets unhampered (40). This model is now supported by the feedback regulation of corticosterone biosynthesis in the mouse adrenal cortex by an H2O2 signaling pathway involving Prx3 and Srx (18). Hyperoxidation also stabilizes Prx into decameric and higher-order oligomeric forms that are capable of preventing aggregation of heat-denatured model substrates (12, 37). In yeast, the major cytosolic Prx Tsa1 accumulates in the hyperoxidized form during replicative aging, and elevating Srx activity counteracts this hyperoxidation and extends lifespan (23). Prx hyperoxidation has also been shown to undergo circadian oscillations, a phenomenon conserved across kingdoms (28, 29).
Srx catalyzes the ATP-dependent reduction of Prx–SO2 to Prx–SOH by a unique enzymatic chemistry (Fig. 1A). Studies both on the human enzyme from the Lowther's group and on the S. cerevisiae enzyme from our group support a mechanism by which the sulfinic moiety of the Prx–SO2 substrate is first activated by slow, rate-limiting formation of an anhydride bond with the γ-phosphate of ATP (35), leading to a phosphoryl sulfinic intermediate (oxidation state of Prx CP+II) (15, 35). The sulfinyl sulfur (S=O) is then reduced by attack by the Srx catalytic Cys (Cys84 for S. cerevisiae), resulting in a thiolsulfinate intermediate Prx–SO–S–Srx (oxidation state of Prx CP +I) (16, 34). For the S. cerevisiae Srx, the cycle is completed by (i) the attack of the thiolsulfinate by a second Srx Cys residue at position 48, leading to formation of an oxidized form of Srx with an intramolecular disulfide Cys48–Cys84 as catalytic intermediate, and (ii) the reduction of this intermediate by Trx (36) (Fig. 1A). This mechanism has been described in vitro but has not yet been verified in vivo.
FIG. 1.

The possible recycling pathways of the Srx catalytic mechanism. (A) Srx catalyzes the reduction of Prx hyperoxidized on the peroxidatic Cys CP by (i) ATP-dependent phosphate transfer on the sulfinic acid group allowing activation of the Prx–SO2 substrate, (ii) formation of a covalent Prx–SO–S–Srx thiolsulfinate complex involving the catalytic Cys of Srx, and (iii) recycling of Srx by formation of an intramolecular disulfide bond between the catalytic C84 and C48 for two-Cys Srxs, or attack of an external thiol reducer RSH for 1-Cys Srxs. This attack could, a priori, occur on the sulfinyl sulfur (SO) or the sulfenyl sulfur (S) of the thiolsulfinate bond (See “Kinetic competency of the glutathionylated Srx intermediate product” section). (B) For wild-type Prx, the product released is the sulfenic acid form, which is resolved by formation of intermolecular disulfide bond with Cys CR, which is subsequently reduced by Trx. (C) If a mutant lacking Cys CR is used (C171A Prx), the reactive sulfenic acid will evolve as a mixed disulfide with the external thiol reducer added. R, thiol reducer.
The resolving Srx Cys48 is located in an extra sequence that is lacking in mammalian and plant Srxs (1), which indicates that the latter, or “1-Cys Srx” utilizes a mechanism for the reduction of the thiolsulfinate that is different from the one proposed for the yeast enzyme, or 2-Cys Srx (Fig. 1A). Specifically, the nature of the thiol-reducing system and the chemical details of this mechanism are unknown. Initial experiments using S. cerevisiae Srx C48A mutant indicated that the rate of formation of the thiolsulfinate intermediate is unaffected, but that recycling by Trx becomes slow and rate limiting for the overall reaction (36). A reasonable alternative reducer for 1-Cys Srx is glutathione (GSH), given its thiol-reduction property and its high cellular concentration (i.e., 1–10 mM).
We have investigated the recycling mechanism of 1-Cys Srx, using S. cerevisiae Srx mutants lacking the resolving Cys48 as a model. Since the kinetics of reduction will eventually determine whether GSH can act as 1-Cys Srx reducing system, we performed a kinetic analysis in the presence of a large range of GSH concentrations. These analyses were performed under steady-state and single-turnover conditions to get insight into the rate of the individual steps of the process. To establish the recycling mechanism, the products resulting from GSH attack were characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), reverse-phase chromatography, and mass spectrometry. Finally, we have monitored the recycling of wild-type and C48S Srxs in S. cerevisiae strains lacking either the Trx or GSH pathways. Our study supports the conclusion that GSH is the reducing system for 1-Cys Srx, and confirms the role of Trx as the reducing system for 2-Cys Srx.
Results
GSH is not rate limiting when coupled with Grx for reduction of SrxC84 in steady-state conditions
S. cerevisiae Srx has a catalytic Cys (Cys84), a resolving Cys (Cys48) and Cys106 that has no known function. To investigate the reduction mechanism of 1-Cys Srxs, we used an Srx mutant retaining only Cys84 (SrxC84) in which both Cys48 and Cys106 were substituted to Ala. Substitution of Cys106 was necessary to avoid the formation of unwanted side products in vitro (34). These two substitutions did not affect the rate of thiolsulfinate formation, which thus allowed the study of the subsequent steps of the Srx catalytic mechanism (36). The kinetics of the SrxC84 reaction were compared in the steady state under multiple enzymatic cycle conditions, using Trx/NTR (Trx, NADPH Trx reductase, and excess NADPH), GSH/GR (GSH, glutathione reductase, and excess NADPH), or GSH/Grx/GR (GSH, glutaredoxin, glutathione reductase, and excess NADPH)(Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/ars) as recycling systems. We previously reported that Trx can efficiently reduce the Srx Cys48–Cys84 disulfide in vitro, and that in the presence of the Trx/NTR system, reaction of wild-type Srx is only limited by the ATP-dependent activation of the Prx–SO2 substrate at a rate constant of 1–2 min−1 at pH 7 (36). In contrast, recycling of SrxC84 by the Trx/NTR system appeared to be rate limiting with a rate constant of 0.11 and 0.06 min−1, for 50 and 100 μM Trx, respectively (not shown). In the presence of the GSH/Grx/GR system, however, the observed steady-state rate constant increased with the GSH concentration until a plateau value of 1.2–1.4 min−1 for concentrations>2 mM (Fig. 2), indicating that under these conditions, the recycling process of SrxC84 is not rate limiting with respect to the ATP-dependent step. In the presence of only GSH and GR, the kinetics of the reaction were much slower and linearly dependent on the GSH concentration, indicating that Grx is required for an efficient recycling of SrxC84 by the GSH/GR coupled system. These results were obtained with a Prx mutant lacking the resolving Cys171 (Fig. 2). Similar results were obtained with wild-type Prx (Supplementary Fig. S2), which indicates that Prx Cys171 is not involved in the Srx-recycling process. Furthermore, to validate the use of S. cerevisiae SrxC84 for the study of 1-Cys-Srx recycling, we performed an experiment similar to the one shown in Figure 2 using recombinant Srx and Prx1 from mouse. The data indicate that mouse Srx shows similar kinetics to S. cerevisiae SrxC84 (Supplementary Fig. S3 and Supplementary Materials and Methods): under steady-state conditions, recycling of mouse Srx by the GSH/GR coupled assay is rate limiting (maximum rate constant of 0.1 min−1), while in the presence of Grx, the observed rate constant increases with GSH concentration until 0.8 min−1. This latter value is similar to the rate constant measured for the first step of the reaction (0.9 min−1, data not shown), which corresponds to the formation of the thiolsulfinate species measured under single-turnover conditions. This shows that, as with S. cerevisiae SrxC84, the recycling process by the GSH/Grx/GR coupled assay is not rate limiting.
FIG. 2.

SrxC84 is recycled by the GSH/Grx/GR system in the steady state. The reaction of 50 μM C171A Prx–SO2 with 10 μM SrxC84 in the presence of 1 mM MgCl2 was followed using the GSH/GR (0.5 μM GR and 200 μM NADPH, circles) or GSH/Grx/GR (50 μM Grx, 0.5 μM GR, and 200 μM NADPH, squares) coupled assays. The assay was started by the addition of 1 mM ATP. Initial rate measurements were carried out at 30°C in buffer TK by following the decrease of the absorbance at 340 nm due to the oxidation of NADPH. The observed steady-state rate constant kss was calculated as the ratio between the rate measured and Srx concentration.
GSH reduces the Prx–SO–S–Srx thiolsulfinate intermediate by saturation kinetics
We next explored the kinetics of the recycling of SrxC84 by GSH under single-turnover conditions, in the absence of the GSH/GR coupled system to limit the reaction at the step of thiolsulfinate reduction by GSH (Fig. 1A). We monitored by SDS-PAGE the formation of the reaction products after ATP addition. We previously showed by LC-MS that the thiolsulfinate species is the major species among Prx-Srx complexes that form by incubating equal concentrations of C171A Prx–SO2 and SrxC84, ATP and Mg2+ in the absence of added reducer, up to 1 min of incubation (34) (Supplementary Fig. S4). In the absence of GSH (Fig. 3A, left panel), formation of two electrophoretic bands was observed in addition to monomeric Srx and Prx, which correspond to the thiolsulfinate intermediate and to a disulfide-linked Srx dimer that forms due to the reactivity of extra monomeric SrxC84 with the thiolsulfinate, although this reaction is slow, as similarly shown for the recycling reaction with Trx (see previous section). In the presence of 5 mM GSH, formation of the disulfide-linked Srx dimer was prevented, and the accumulation of the Prx-Srx species was significantly dampened, which supports the notion that GSH reacts directly with the thiolsulfinate intermediate at a rate at least as fast as formation of the thiolsulfinate (Fig. 3A, right panel). As shown in the “Kinetic competency of the glutathionylated Srx intermediate product” section, this attack releases Prx–SOH and Prx–SSG that can react with SrxC84 to give Prx–S–S–Srx, with both reactions being favored by specific complex formation between Prx and Srx. This implies that in this experiment, the Prx-Srx band contains a fraction of disulfide species in addition to the thiolsulfinate.
FIG. 3.

SrxC84 is recycled by GSH in single-turnover conditions. The SrxC84-catalyzed reaction was followed in the presence of GSH by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of the reaction products (A) and by fluorescence kinetics (B–D). (A) Equimolar concentrations of C171A Prx–SO2 and SrxC84 (30 μM) were incubated for the indicated time in the presence of 1 mM MgCl2, in buffer TK at 30°C, in the presence or absence of GSH (5 mM). The reaction was started by the addition of 1 mM ATP final (third lane: control without ATP) and quenched by acidification before NEM treatment and SDS-PAGE analysis in nonreducing conditions. First/second lanes: SrxC84/Prx–SO2 alone incubated with ATP and MgCl2 for 1 min (left panel) with ATP, MgCl2, and GSH for 1 min (right panel). (B–D) Final concentrations of 40 μM Prx–SO2, 1 mM ATP, 1 mM MgCl2, and variable GSH were rapidly mixed with 5 μM SrxC84 in buffer TK at 30°C in a rapid kinetics spectrofluorometer. The blank-corrected progress curves collected for GSH 25, 50, 75, 100, 150, 250, 500, and 2500 μM (B, top to bottom) were analyzed using a first-order kinetic model to deduce the rate constant kobs (circles) (C) and amplitudes (squares) (D) of the process.
To obtain quantitative data on the kinetics of reduction by GSH, we studied the reaction kinetics of SrxC84 in the presence of increasing GSH concentrations. Here, we used wild-type Prx, which allowed monitoring reduction of the sulfinic acid through the decrease of Prx intrinsic tryptophan fluorescence caused by formation of the CP-CR disulfide bond (35). If, indeed, one assumes that the GSH attack on the Prx–SO–S–Srx is on the sulfenyl sulfur atom (the –S– of the Srx catalytic Cys) (17, 27), the reaction should release the Prx–SOH species, followed by the rapid condensation of the latter with the CR residue into a disulfide-linked Prx homodimer (Fig. 1A, B). We first verified that the disulfide-linked Prx homodimer is not reduced by GSH on the time scale of the Srx-catalyzed reaction (data not shown). As shown in Figure 3B, decreasing fluorescence time courses were obtained in the presence of GSH, thus attesting the formation of the disulfide-linked Prx dimer. Since the phosphotransfer step is slow and determines the rate of all subsequent steps, this multistep mechanism could be modeled as a simple first-order process: time courses were best described by first-order, single exponential kinetics characterized by (i) a rate constant kobs that slightly increased with the concentration of GSH, reaching a plateau of 1.4 min−1 for concentrations >150 μM (Fig. 3C), and unexpectedly (ii) signal amplitudes that followed a hyperbolic dependence on the GSH concentration, with a characteristic apparent KGSH constant of 34±7 μM (Fig. 3D). This result suggests that GSH binds to the thiolsulfinate complex.
Kinetic competency of the glutathionylated Srx intermediate product
If as earlier assumed, GSH selectively reduces the thiolsulfinate species by an attack on the Srx sulfenyl sulfur of the thiolsulfinate bond (Fig. 1A), a glutathionylated Srx species should be released as a catalytic intermediate. We thus sought this intermediate among the products formed during the reduction of C171A Prx–SO2 by SrxC84, under single-turnover conditions, that is, in the presence of GSH and the absence of GR. Chromatographic analysis of the reaction mixture before initiation of the reaction identified Srx (peak b) and Prx–SO2 (peak e) (Fig. 4A, B and Table 1). Upon ATP addition, three additional peaks appeared, one corresponding to the previously identified Prx/Srx complexes (peak c) (34), and the other two with masses compatible with glutathionylated SrxC84 (Srx–S–SG, peak a, Table 1) and glutathionylated Prx (Prx–S–SG, peak d, Table 1). The latter should form upon a reaction between the released Prx–SOH and GSH (Fig. 1C), and its presence, thus indicates that GSH, indeed, reacts on the Srx sulfenyl sulfur within the thiolsulfinate intermediate (Fig. 1A). An oxidized form of glutathionylated Prx (Prx–S–SG +16 Da) (estimated by mass spectrometry as amounting to less than 30% of peak d) was also present, which could have formed as a result of either the attack of GSH on the Prx sulfinyl sulfur (Fig. 1A) leading to Prx–SO–SG or the oxidation of the glutathionylated Prx species during the analysis. Addition of DTT reduced glutathionylated SrxC84 back to reduced SrxC84 (not shown), thus confirming that peak a corresponds to a mixed disulfide between SrxC84 Cys84 and GSH.
FIG. 4.
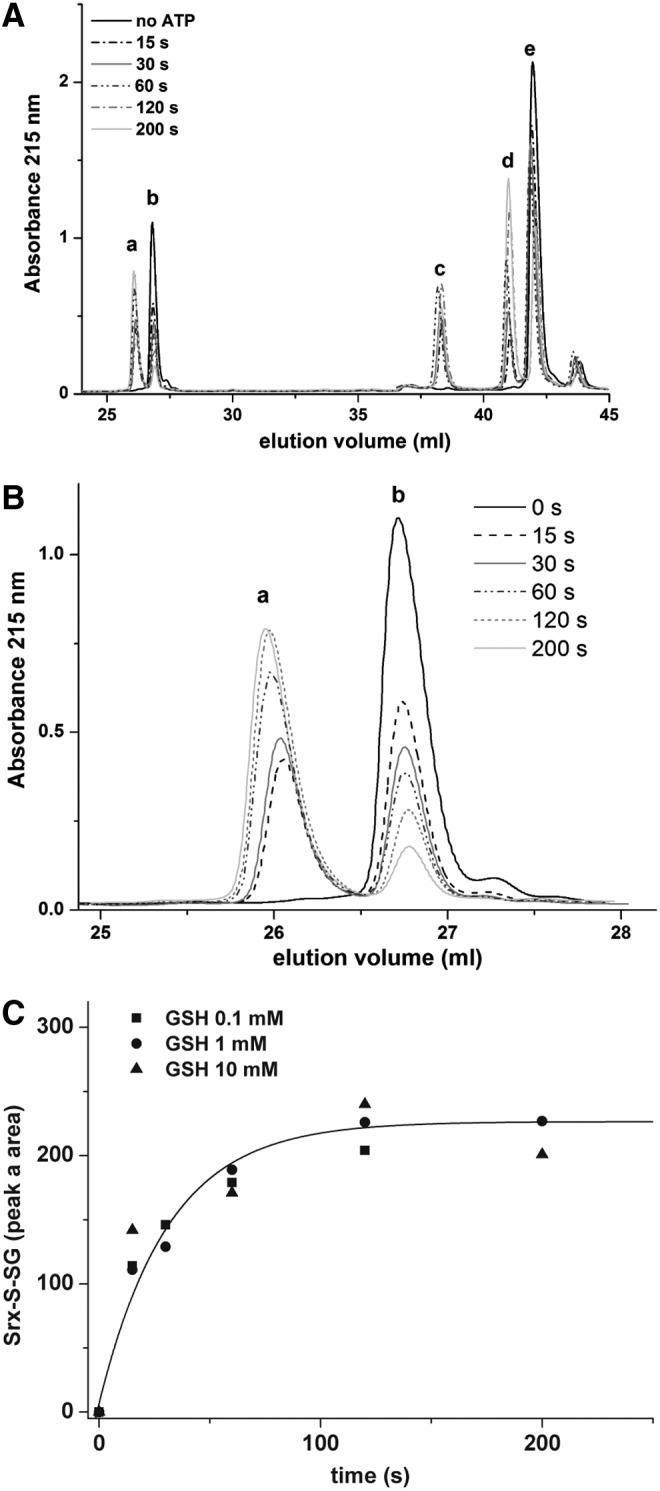
Glutathionylated SrxC84 is a catalytic intermediate. The reaction was conducted as in Figure 3A and quenched by acidification before analysis by reverse-phase chromatography. (A) Chromatograms were obtained before and at 15 s, 30 s, 60 s, 120 s, and 200 s after ATP addition, in the presence of 1 mM GSH. Masses of the products eluted in each peak are in Table 1. (B) A zoom of the peaks a and b shown in (A). (C) Evolution of the area of peak a (S-glutathionylated SrxC84) plotted as a function of time for GSH 0.1 mM (squares), 1 mM (circles), and 10 mM (triangles) and analyzed using a first-order mono-exponential kinetic model (solid line shown for GSH 1 mM).
Table 1.
Theoretical and Observed Masses of the Species Observed in Reverse-Phase Chromatographic Analysis of SrxC84-Catalyzed Reaction in the Presence of Glutathione
| Peak | Observed massa, Da | Speciesb(theoretical mass, Da) |
|---|---|---|
| a | 13963.7 | Srx–S–SG (13964.0) |
| b | 13658.5 | Srx reduced (13658.7) |
| c | 37261.5 (∼50%) | Prx–SO–S–Srx (37262.5) |
| 37246.4 (∼50%) | Prx–S–S–Srx (37246.5) | |
| d | 23895.3 (>70%) | Prx–S–SG (23895.1) |
| 23911.8 (<30%) | Prx–S–SG+O (23911.1) | |
| e | 23621.8 | Prx–SO2 (23621.8) |
The estimated amount of each species found under peaks c and d is indicated in parentheses.
Species observed in the chromatographic analysis from Figure 4 using C171A Prx–SO2 substrate and SrxC84.
We next established the kinetics of evolution of peak a area (glutathionylated Srx) obtained in the presence of 1 mM GSH (Fig. 4A, B) and compared them with those obtained in the presence of 0.1 and 10 mM GSH (Fig. 4C). As in Figure 3B, this multistep mechanism could be modeled as a simple first-order process because the phosphotransfer step is very slow and determines the rate of all subsequent steps. The observed kinetics were very similar and could be described by a single exponential process characterized by comparable rate constants of 2.2±0.4 min−1 (0.1 mM GSH), 1.9±0.4 min−1 (1 mM GSH), and 3.6±1.0 min−1 (10 mM GSH).
These data indicate that glutathionylated SrxC84 is produced at a rate compatible with the rate of thiolsulfinate formation, as expected for a catalytic intermediate. A similar result was obtained using wild-type Prx–SO2 as substrate (data not shown).
Determination of the rate constant for the attack of the thiolsulfinate by GSH
Since the first step of the Srx catalytic mechanism is rate limiting for the overall reaction with a slow rate constant of 1–2 min−1, the intrinsic rate of reaction of GSH on the thiolsulfinate intermediate cannot be measured by following the overall reaction. To probe this step individually, we first accumulated the thiolsulfinate intermediate by incubating C171A Prx–SO2 and SrxC84 for 1 min in the presence of ATP, and then monitored the disappearance of this intermediate on addition of 5 mM GSH by SDS-PAGE (Figs. 3A and 5A). At this concentration, GSH is saturating for binding to the thiolsulfinate complex, as shown in Figure 3D, thus allowing to observe the breakdown of the thiolsulfinate/GSH complex, which can be modeled as a simple first-order process. Densitometric quantification of the thiolsulfinate intermediate over time allowed modeling the kinetics of thiolsulfinate reduction by GSH as a single exponential law, characterized by a rate constant of 7±1 min−1 at 5 mM GSH (Fig. 5B). Secondary formation of Prx–S–S–Srx product explains why the band does not completely disappear on the SDS-PAGE. In similar experiments, we monitored glutathionylated Srx by reverse-phase chromatography, which confirmed that a lower limit for the intrinsic rate of GSH reduction of thiolsulfinate was of the same order of magnitude for 200 μM and 1 mM GSH (data not shown).
FIG. 5.
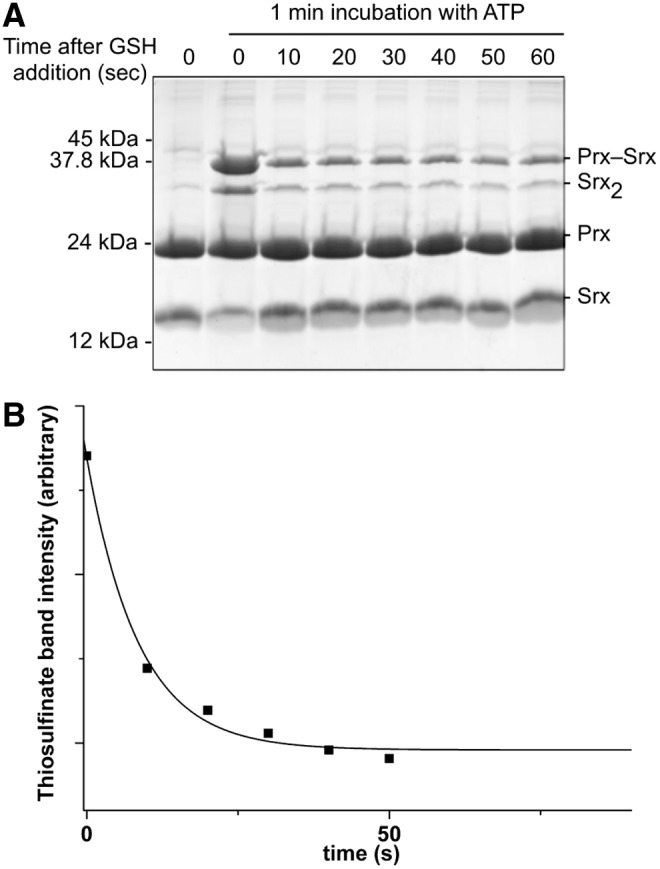
Kinetics of the attack of GSH on the thiolsulfinate intermediate. (A) The reaction was conducted in the same conditions as in Figure 3A except that GSH (5 mM) was added after 1 min of incubation after ATP addition to accumulate the thiolsulfinate intermediate. The reaction was quenched by acidification and analyzed by nonreducing SDS-PAGE. (B) The amount of thiolsulfinate species was measured as a function of time by densitometry of the corresponding Coomassie-stained band (squares). The time course was analyzed as a first-order monoexponential model (solid line).
In vivo recycling of 1-Cys and 2-Cys Srxs
We next sought to evaluate the role of GSH in Srx recycling in vivo. For this purpose, we inspected in yeast cells exposed to H2O2 the redox interaction between Srx and its Prx substrate Tsa1, taken as a token of in vivo Srx catalytic cycling (Fig. 6). We used a strain lacking both SRX1 and GSH1 (ΔsrxΔgsh1) and carrying a one-copy plasmid expressing either HA-tagged Srx (HA-Srx) or HA-C48S Srx. Deletion of GSH1, which encodes γ-glutamylcysteine synthase, the rate-limiting step enzyme of the GSH biosynthesis pathway, allows to control cellular GSH levels by growth in the absence (GSH depletion) or presence of defined GSH amounts (20). Cells were first exposed to 100 μM H2O2 during 30 min to induce Srx expression, and then challenged with 500 μM H2O2 to cause full Prx sulfinylation (1). In the absence of GSH depletion (100 μM of GSH added to cultures), three Srx-containing bands were observed with both wild-type and C48S Srx, one at 15 kDa, which represents monomeric Srx, and the other two at 40 and 60 kDa, which represent redox-linked Srx-Tsa1 complexes of different stoichiometry, as indicated by their β-mercaptoethanol (β-ME) sensitivity (Fig. 6A), and by their total absence in lysates of an HA-Srx-expressing Δtsa1 strain (1). Based on the Srx catalytic mechanism, we propose that the 40-kDa band is the Prx–SO–S–Srx thiolsulfinate intermediate, and the 60 kDa band is Srx attached by a thiolsulfinate bond to a disulfide-linked Prx dimer. The latter complex is a reaction intermediate in which one of the CP of a Prx dimer is still attached by a thiolsulfinate to Srx, while the other has just been released on thiolsulfinate reduction in the form of CP–SOH, and condenses into a disulfide with the CR of the other subunit of the Prx dimer (Fig. 7). It should be noted that compared with the 40 and 60 kDa bands formed by the wild-type enzyme, which both appeared at 5 min after H2O2 exposure and started to disappear after 60, and 30 min, respectively, those formed with Srx C48S only started to disappear after 60 min, as a result of slower recycling. GSH depletion slowed down reduction of wild-type enzyme as the result of the global redox imbalance of the strain, but prevented formation of the C48S Srx 60 kDa band, indicating that in this case the Srx-catalyzed reaction was aborted after formation of the 40 kDa thiolsulfinate by defective reduction of the latter. To further demonstrate the role of GSH in 1-Cys-Srx recycling, the extracts expressing Srx C48S were blotted against anti-Prx-SO2/3 antibody under reducing conditions to monitor Prx-SO2 reduction. The results (Supplementary Fig. S5) show that for Srx C48S under conditions of GSH depletion, the reduction of PrxSO2 is significantly delayed, as it starts at 60 min after H2O2 treatment, while in the presence of GSH, reduction begins after 5 min and reaches near completion by 120 min.
FIG. 6.

In vivo analysis of the role of the Trx and GSH pathways in Srx redox status. (A) Δgsh1Δsrx1 strain expressing pRS316-HA2-Srx (left panel) or pRS316-HA2-SrxC48S (right panel), grown to an OD600nm of 0.4 in selective minimal medium (SD URA−) supplemented with GSH (100 μM), were treated for 30 min with 100 μM H2O2 to induce SRX1. After 30 min, cells were treated with 500 μM H2O2 to hyperoxidize Prx and lysed using the TCA-protocol after 5, 15, 30, 45, 60, and 120 min. Normalized total protein extracts were immunoblotted with the anti-HA monoclonal antibody after nonreducing 15% SDS-PAGE. The last lane is the same as after 30 min of treatment except that precipitated protein pellet was dissolved in the presence of 10% β-mercaptoethanol (β-ME). (B) Δgsh1Δsrx1 strains expressing pRS316-HA2-Srx (left panel) or pRS316-HA2-Srx C48S (right panel) grown in selective minimal medium (SD URA−) lacking GSH for 10 divisions were analyzed in the same conditions as in (A). NSB: indicates a protein reacting nonspecifically with the antibody. (C) Δsrx1 (left panel) or Δsrx1Δtrx1Δtrx2 strain (right panel) expressing pRS316-HA2-Srx were grown, and total proteins extracts were immunoblotted against the anti-HA monoclonal antibody in the same conditions as in (A).
FIG. 7.
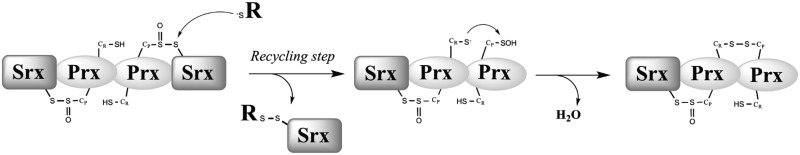
Mechanism of formation of the Srx-Prx2 species proposed as a hallmark of the recycling process. Within the hyperoxidized Prx–SO2 dimer, the first steps of the Srx catalytic mechanism lead to the formation of the thiolsulfinate intermediate. If recycling occurs on one of the Prx monomers, the formation of the Prx sulfenic acid form will result rapidly in a disulfide Prx dimer, with one Prx monomer covalently linked to an Srx molecule by a thiolsulfinate bond, thus leading to a ∼60 kDa species. R, thiol reducer.
We also evaluated the role of the Trx system in 2-Cys Srx recycling in vivo by a similar approach, using a ΔsrxΔtrx1Δtrx2 strain. In this case, the Cys48 Srx residue resolves the Prx–SO–S–Srx thiolsulfinate with formation of an Srx intramolecular Cys48–Cys84 disulfide. In wild-type cells, monomeric Srx was present as a single band throughout the H2O2 time-course analysis (Fig. 6A, C). However, in ΔsrxΔtrx1Δtrx2 cells, an extra HA-immunoreactive band that migrated slightly faster than monomeric Srx was present, which corresponds to the Cys48–Cys84 intramolecular disulfide intermediate of Srx previously identified in vitro (36). This species appeared at 15 min after H2O2 exposure, increased in intensity till 1 h, and then disappeared (Fig. 6C, right panel). These in vivo data thus confirm the prominent role of Trx in Srx recycling previously shown in vitro. They also indicate that in the absence of cytosolic Trxs, recycling still occurs, albeit inefficiently, by the probable engagement of GSH. The latter assumption is supported by in vitro data of the kinetics of the reduction of Cys48–Cys84 disulfide Srx by the GSH/Grx/GR coupled system, which, although much less efficient than the Trx/NTR system, appeared not to be rate limiting relative to the Srx sulfinyl reductase kinetics (36).
Discussion
Catalytic mechanism of 1-Cys Srx recycling in vitro
Reduction of the Prx sulfinic acid by Srx leads to the formation of a thiolsulfinate as a reaction intermediate for both the S. cerevisiae and mammalian enzymes (16, 34). In the S. cerevisiae Srx, the thiolsulfinate is resolved by a second Cys residue. However, in mammals, plants, and cyanobacteria, the resolving Cys residue is absent, which suggests that the thiolsulfinate intermediate is reduced by an external thiol molecule. A comparison of the steady-state kinetics of SrxC84 with the Trx or GSH systems (Supplementary Fig. S1) showed that only the latter is able to recycle SrxC84 at a rate that reaches a nonlimiting value with regard to the formation of the thiolsulfinate intermediate. Furthermore, our data indicated that GSH efficiently reduces the thiolsulfinate intermediate only when Grx is present. Since neither Trx nor Grx (not shown) can efficiently recycle SrxC84, due likely to steric constraints on access to the Prx–SO–S–Srx thiolsulfinate bond (Fig. 8), GSH must operate as the direct reducer of the thiolsulfinate. Accordingly, the kinetics of the GSH-dependent reaction measured under single-turnover conditions showed that even at concentrations as low as 150 μM, GSH reacts with the thiolsulfinate intermediate at a rate that is only limited by thiolsulfinate formation process (Fig. 3C). Therefore, as soon as it is formed, the thiolsulfinate is attacked by GSH and does not accumulate (Figs. 3 and 4).
FIG. 8.

Identification of potential access channels in human Prx1/Srx disulfide-linked complex. The structure of the complex formed between human Prx1 dimer (blue/yellow) and Srx (red) (pdb code 2RII) (14), linked by a disulfide bond between Srx catalytic Cys and Prx1 Cys CP was used as a model of the thiolsulfinate intermediate to search for tunnels (grey) by the program CAVER (33). The center of gravity of the sulfur atoms of the disulfide bond (spheres) was used as a starting point in the calculation. Three potential access channels that could direct GSH attack on the thiolsulfinate bond are identified. Only one Srx molecule is shown for clarity. The scheme was prepared with PyMOL 0.99 (www.pymol.org). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Mass analysis of the reaction products formed on reduction of the thiolsulfinate by GSH revealed that the major species released is S-glutathionylated SrxC84. In line with the selectivity of thiol attack on thiolsulfinates (17, 27), this result supports the notion that GSH preferentially reacts with the Prx–SO–S–Srx intermediate on the sulfenyl sulfur of SrxC84, releasing S-glutathionylated SrxC84 and Prx–SOH as products (Fig. 1A). For wild-type Prx, a disulfide-linked dimer rapidly forms, as attested by the decrease in Prx fluorescence during the reaction (Fig. 1B). For C171A Prx, the reactive sulfenic species reacts with an additional GSH to form a Prx–S–SG intermediate, which is the predominant species observed in mass analysis of peak d (Figs. 1C and 4). Reactivity of this species with SrxC84 likely explains the formation of the Prx–S–S–Srx disulfide found under peak c (30), along with the thiolsulfinate intermediate. Importantly, kinetic monitoring of Srx–S–SG release by reverse-phase chromatography establishes the role of S-glutathionylated Srx as a catalytic intermediate.
The steady-state kinetics presented in Figure 2 show that in the absence of Grx, the recycling process is rate limiting for the overall Srx reaction. This strongly suggests that the reduction of SrxS–SG by a second GSH molecule is slow and largely rate limiting (Supplementary Fig. S1). Such results are consistent with previous studies performed on mammalian or plant Srx using GSH alone as reducer and explain the low Srx activity reported (11, 13). By contrast, the process is catalyzed by Grx, which will efficiently deglutathionylate SrxS–SG. In the presence of Grx, for GSH below 1 mM, after the glutathionyl exchange from SrxC84 to Grx, the rate-limiting process would likely be associated with reduction of glutathionylated Grx by a second GSH molecule, assuming that Grx follows a monothiol mechanism (9). For GSH concentration superior to 1 mM, the SrxC84 catalysis assumes the rate-limiting process, explaining why the steady-state rate reaches a plateau of 1.2 min−1.
Determinants of the efficiency of SrxC84 recycling by GSH
The kinetics of the SrxC84-catalyzed reaction monitored in single turnover using GSH as a reducer revealed unexpected features: While the rate constant of the reaction was only barely influenced by the GSH concentration and remained close to the value of thiolsulfinate formation (1.4 min−1), the amplitude of the signal followed a saturation profile. This suggests that a process of binding of the GSH molecule on the thiolsulfinate complex occurs before reduction and controls the reaction for GSH concentrations below ∼250 μM. At saturating GSH concentrations, since the Prx–SO2 substrate was used in excess in this experiment, the amplitude of the reaction reached a limit corresponding to the SrxC84 concentration. With a deduced apparent binding constant of 34 μM, this means that GSH saturation is reached for concentrations largely submillimolar and suggests that in vivo, for 1-Cys Srxs, the rate of thiolsulfinate reduction will not depend on GSH, unless its concentration drops drastically down to micromolar concentrations. This is consistent with the observation that the intrinsic rate of GSH attack on the thiolsulfinate was not increased at 5 mM of GSH compared with 200 μM.
The fact that GSH could bind to the thiolsulfinate complex raises the question as to whether the reaction is catalyzed within the active site, through GSH deprotonation, thiolate alignment relative to the thiolsulfinate bond, or stabilization of the transition state. The second-order rate constant for GSH attack on the thiolsulfinate intermediate (ratio of intrinsic rate of attack to the apparent binding constant) is 3.4 103 M−1s−1 for GSH at pH 7, which corresponds to 2.8 105 M−1s−1 for the GS− thiolate (which is the reactive form of GSH), considering an apparent pKa of 8.9 (21) for GSH thiol group. This latter value is similar to the one measured at 4.7 105 M−1s−1 for the reduction of a chemical thiolsulfinate model by Cys thiolate (27), suggesting that the thiol apparent pKa of GSH is either not or only weakly altered within the active site. The efficiency of Prx–SO–S–Srx reduction by GSH would thus mainly rely on (i) the intrinsic reactivity of the thiolsulfinate bond with thiolate; (ii) the high GSH local concentration imparted by a specific recognition mode, allowing efficient reduction of the thiolsulfinate at physiological GSH concentrations; and potentially (iii) an optimized orientation of the thiolate relative to the thiolsulfinate bond.
From a structural standpoint, the thiolsulfinate complex likely participates in the recognition of GSH via both the flanking Prx and Srx moieties. To test this hypothesis, we looked for potential access paths for GSH within the crystal structure of the human disulfide-linked Prx1/Srx complex (pdb access 2RII) (14), a good model for the 1-Cys Srx thiolsulfinate complex, using the program CAVER (33) (Fig. 8). The identified potential tunnels suggest that GSH binding probably occurs at the interface between Srx and one Prx monomer or dimer. This observation is reminiscent of the studies that have reported the existence of a deglutathionylation activity for Srx, and which suggested specific interactions between Srx and glutathionylated Prx, protein tyrosine phosphatase 1B, and actin (8, 30). In addition, the interaction between Srx and the protein S100A4, which plays a key role in regulating nonmuscle myosin IIA activity, is strongly enhanced when S100A4 is glutathionylated (3). Finally, the observed interaction between GSH and the S. cerevisiae Prx/Srx complex (this work) suggests that this feature appeared early in evolution.
Srx catalytic cycle in vivo
In vitro kinetic data provided evidence for the existence of distinct recycling pathways for 2-Cys vs. 1-Cys Srx ((36) and this work). We first confirmed in vivo the mechanism proposed for the S. cerevisiae 2-Cys Srx in vitro, which was suggested to proceed through an intramolecular disulfide Srx form that is efficiently reduced by Trx (Fig. 6C). We also compared the Srx redox species formed in vivo by 2-Cys (wild-type) and 1-Cys (C48S) Srx, which revealed the formation of two catalytic complexes (Fig. 6), both of which contain Srx and Prx moieties. We propose that the complex that migrates at 60 kDa corresponds to an Srx-Prx2 complex and represents a hallmark for the reduction process of the thiolsulfinate intermediate occurring within the dimeric Prx unit (Fig. 7). For 2-Cys Srx, this intramolecular process is fast and does not depend on an external reducer (36). For 1-Cys Srx, this complex was also present in wild-type cells, but was almost totally absent in cells totally depleted of GSH (Δgsh1 grown for 10 divisions without GSH supplementation) (Fig. 6), which indicates that (i) it is formed as a result of GSH reduction of 1-Cys Srx-Prx thiolsulfinate intermediate; (ii) Trx is not able to efficiently reduce this intermediate, in accordance with in vitro kinetic data. The presence of the 60 kDa band in cells that were not totally depleted of GSH [Δgsh1 grown for 7 divisions without GSH supplementation, total cellular concentration of about 100 μM GSH (20)] (not shown) further indicates that low amounts of GSH are sufficient for the recycling of 1-Cys Srx in vivo, which is also in full agreement with the kinetic studies. Our study thus highlights the pertinence to combine both in vivo and in vitro kinetic characterization of the processes involved in redox signaling pathways.
Conclusion
Here, we have addressed the question of which of the two thiol-redox control pathways assists Srx sulfinyl reductase activity. In S. cerevisiae, which carries a 2-Cys enzyme, this function is primarily devoted to Trx, which is consistent with a previous work (22, 36). We have also shown that GSH can compensate for the lack of Trx, albeit inefficiently. For other organisms possessing 1-Cys Srx, our study provides evidence that GSH acts as the reducer of the Prx/Srx thiolsulfinate intermediate in vivo. The catalytic efficiency of reduction of this intermediate is secured even in case of GSH depletion by a mechanism of binding of GSH to the Prx/Srx complex.
Materials and Methods
Materials
KCl, MgCl2, EDTA, ATP, trichloroacetic acid (TCA), and acetonitrile were purchased from Merck. Dithiothreitol (DTT) and Tris were from Euromedex. NADPH was obtained from Roche. Glutathione (GSH), N-ethylmaleimide (NEM), and glutathione reductase (GR) were from Sigma-Aldrich. Trifluoroacetic acid (TFA) and H2O2 were obtained from Acros Organics. SDS was obtained from AppliChem GmbH. Monoclonal anti-HA antibody and ammonium acetate were obtained from Sigma-Aldrich.
Preparation of recombinant proteins
Recombinant thioredoxin1 (Trx), NADPH Trx reductase (NTR) from Escherichia coli, wild-type and C171A Tsa1 (Prx) and wild-type and C48A-C106A Srxs (SrxC84) from S. cerevisiae were prepared following the experimental procedures previously described (34). Recombinant glutaredoxin1 (Grx) from E. coli was obtained by cloning the grx1 open reading frame amplified by PCR using the high-fidelity DNA polymerase pfu (Thermo Scientific), into the pET20b+ vector (Novagen), between the Nde1 and Sac1 restriction sites (sequence of oligonucleotides not shown). Grx was produced in E. coli and purified following the same experimental procedures as previously described for Trx (24, 25). Preparation of hyperoxidized Prx–SO2 was performed as previously described for both wild-type and C171A Prx (34).
Yeast strains and growth conditions
The S. cerevisiae strains used are derived from YPH98. The isogenic derivative Δsrx1 of strain yAD1-1C (MATa, ura3-52, lys2-801amber, ade2-101ochre, trp1-Δ1, leu2-Δ1, and his3-Δ200) was described (13). The Δsrx1Δtrx1Δtrx2 strain was made by replacing the coding region of SRX1, TRX1, and TRX2 ORFs by HIS3, TRP1, and KAN. Y252 Δgsh1 was described (38). The isogenic strain Δgsh1Δsrx1 was obtained by replacing SRX1 ORF by KAN. Cells carrying the pRS316-HA2-Srx (wild-type or C48S) plasmid (1) were grown at 30°C in minimal medium SD (0.67% yeast nitrogen base without amino acids, 2% glucose) complemented with adenine, amino acids, and GSH as appropriate. For experiments in GSH-depleted cells, Δgsh1 cultures were grown for ten divisions in SD medium lacking GSH.
SDS-PAGE analyses
Kinetic analysis of the species formed during the Srx-catalyzed reaction was followed by incubating C171A Prx–SO2 and SrxC84 (30 μM each), 1 mM ATP, and 1 mM MgCl2 in buffer TK (50 mM Tris-HCl, 100 mM KCl, pH 7, close to physiological pH), in the presence or absence of 5 mM GSH as indicated. The reaction was stopped by TCA (20%); precipitated proteins were washed in acetone, air dried, and dissolved in 100 mM Tris-HCl pH 8, 10 mM EDTA, and 1% SDS containing 50 mM NEM at 30°C for 1 h. Proteins were loaded onto 15% nonreducing SDS-PAGE gel, followed by Coomassie Blue staining. Species quantification was done by densitometry (ImageJ).
Redox Western blots
For in vivo Srx redox-state analysis, we first added H2O2 (100 μM) to an early-log-phase culture (OD600 nm=0.4) to induce SRX1. After 30 min, cells were treated with 500 μM H2O2 to hyperoxidize Prx. We collected cells corresponding to 5 OD600 of culture at the indicated time. Lysates were prepared by the TCA lysis protocol (5). Precipitated proteins were solubilized in buffer containing Tris-HCl pH 8 (100 mM), SDS (1%), EDTA (1 mM), and NEM (50 mM) at 30°C for 1h (cysteine-trapping method). Extracts were analyzed by nonreducing 15% SDS–PAGE. Srx was immunodetected with anti-HA monoclonal antibody.
Steady-state kinetics
The reduction of Prx–SO2 by SrxC84 was measured in the steady state by using the following enzymatic coupled assays: Trx/NTR (variable Trx, 0.5 μM NTR, and 200 μM NADPH), GSH/GR (Variable GSH, 0.5 μM GR, and 200 μM NADPH), and GSH/Grx/GR (variable GSH, 50 μM Grx, 0.5 μM GR, and 200 μM NADPH) (Supplementary Fig. S1). The assay contained 50 μM of C171A Prx–SO2 in excess to SrxC84 (10 μM), 1 mM MgCl2 and was started by the addition of 1 mM ATP. Initial rate measurements were carried out at 30°C in buffer TK on a UV mc2 spectrophotometer (Safas) by following the decrease of the absorbance at 340 nm due to the oxidation of NADPH. We confirmed that the recycling system was not in itself rate limiting, as doubling of the GR concentration produced no change in the measured rate. A blank measurement recorded in the absence of SrxC84 was systematically deduced from the assay to account for the nonspecific oxidation of NTR or GR. The observed steady-state rate constant kss was calculated as the ratio between the rate measured and Srx concentration (Supplementary Fig. S6). A stoichiometry of 2 mol of NADPH per mol of C171A Prx–SO2 was used in rate calculations to account for the oxidation of 2 mol GSH in reduction of the glutathionylated Srx product and of the Prx sulfenic intermediate. When wild-type Prx–SO2 was used in the Trx/NTR assay, a stoichiometry of 2 mol of NADPH per mol of Prx–SO2 was used in rate calculations to account for the oxidation of 1 mol Trx in SrxC84 recycling, and of 1 mol Trx per mol Prx–SO2 in Prx own catalytic cycle.
Stopped-flow kinetics
Recycling of SrxC84 by GSH was followed on a stopped-flow apparatus by monitoring the quenching of fluorescence intensity of wild-type Prx on going from the oxidized Prx–SO2 to the disulfide form (Fig. 1) (35). The decrease of the emission fluorescence intensity was recorded at 30°C on an SX18MV-R stopped-flow apparatus fitted for fluorescence measurements, with excitation wavelength set at 295 nm, and emitted light collected above 320 nm using a cutoff filter. One syringe contained oxidized Prx–SO2 (40 μM), ATP (1 mM), MgCl2 (1 mM), and GSH at variable concentrations as indicated, in buffer TK. The other syringe contained the SrxC84 (5 μM) in buffer TK (final concentrations after mixing). Equal volumes of each syringe were rapidly mixed to start the reaction. An average of at least three runs was recorded for each concentration of GSH. For each condition, the data were corrected from the corresponding blank time course recorded in the absence of reducer. Rate constants kobs, were obtained by fitting fluorescence traces against a single exponential model by nonlinear regression analysis. The amplitude of the monoexponential process was proportional to the Srx concentration, which is limiting relative to the substrate and cofactors (Supplementary Fig. S7).
Reverse-phase chromatography
To monitor the formation of products of the reaction in the presence of GSH, reaction mixtures containing 30 μM SrxC84, 30 μM C171A Prx–SO2, 1 mM ATP, and 1 mM MgCl2 were incubated in the presence of 0.1, 1, and 10 mM GSH in TK buffer at 30°C. Aliquots were quenched by using TFA 0.1% (final concentration) and were loaded onto a Vydac 208TP52 (C8) column, 2.1×250 mm, 5 μm (PerkinElmer Life Sciences), equilibrated in H2O plus 0.1% TFA, coupled to the ÄKTA explorer system (Amersham Biosciences). Proteins were eluted from the column using a linear gradient from 30% to 80% of B (0.1%TFA in acetonitrile), at a flow rate of 1 ml.min−1. Fractions eluted were collected and stored at −20°C until mass spectrometry (MS) analysis. For the reduction of the glutathionylated SrxC84 product, the sample was lyophilized, redissolved in 50 mM AcNH4 pH 6.8, in the presence of 10 mM DTT (30 min, RT). Before MS analysis, the sample was diluted twice in ACN/H2O/formic acid (50/50/1).
Mass spectrometry
Electrospray ionization mass spectrometry measurements were performed on a MicrOTOF-Q instrument (Bruker Daltonics). Samples were directly injected with a syringe pump in the mass spectrometer at a flow rate of 6 μl/min. The nebulization gas pressure was 0.4 bar, drying gas flow was 4 L/min, source temperature was 190°C, and the capillary and end plate voltage was set to 4500 V and 500 V, respectively. The acquisition range was 500–3000 m/z. For the measurements, an external calibration standard was used (Tuning Mix solution). Collected data were processed via Bruker Data Analysis software (version 4.0). Deconvoluted mass spectra were prepared using the Maximum Entropy Charge Deconvolution module.
Supplementary Material
Abbreviations Used
- 1-Cys Srx
Srx possessing only one Cys as catalytic residue and no resolving Cys
- β-ME
β mercaptoethanol
- CP, CR
Prx peroxidatic and resolving cysteines
- CP–SOH
Prx peroxidatic cysteine oxidized as a sulfenic acid
- GR
glutathione reductase
- Grx
glutaredoxin
- GSH, GSSG
reduced and oxidized glutathione
- HA-Srx
N-terminus tagged S. cerevisiae Srx
- kobs
observed presteady-state rate constant
- kss
steady-state rate constant
- LC-MS
mass spectrometry coupled to liquid chromatography
- NTR
NADPH thioredoxin reductase
- Prx
typical 2-cysteine peroxiredoxin
- Prx–SO2
hyperoxidized Prx with Cys CP oxidized as a sulfinic acid
- Prx–SOH
Prx oxidized with peroxidatic Cys CP oxidized as a sulfenic acid
- Prx–SO–S–Srx
catalytic thiolsulfinate intermediate of the reduction of Prx–SO2 by Srx
- Prx–S–SG
Prx glutathionylated on the peroxidatic cysteine CP
- Prx–S–S–Srx
disulfide complex linking Prx and Srx catalytic cysteines
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- Srx
sulfiredoxin
- SrxC84
S. cerevisiae C48A-C106A Srx
- Srx–S–SG
Srx glutathionylated on the catalytic cysteine
- Trx
thioredoxin
- Tsa1
major cytosolic typical 2-cysteine peroxiredoxin in S. cerevisiae
Acknowledgments
This work was supported by the CNRS, the University of Lorraine, the Fédération de Recherche “Bioingénierie Moléculaire, Cellulaire et Thérapeutique,” local funds from the Région Lorraine and the Ligue contre le Cancer, and ANR and INCA grants to MBT. Mass spectrometry was performed at the “Service commun de spectrométrie de masse et des techniques chromatographiques couplées” from the Université de Lorraine. The authors thank Prs. K. Weissman, and S. Boschi-Muller for helpful discussions, and A. Kriznik, J. Charbonnel and G. Palais for their very efficient technical help.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Biteau B, Labarre J, and Toledano MB. ATP-dependent reduction of cysteine–sulphinic acid by S. cerevisiae sulphiredoxin. Nature 425: 980–984, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Boileau C, Eme L, Brochier-Armanet C, Janicki A, Zhang C-C, and Latifi A. A eukaryotic-like sulfiredoxin involved in oxidative stress responses and in the reduction of the sulfinic form of 2-Cys peroxiredoxin in the cyanobacterium Anabaena PCC 7120. New Phytol 191: 1108–1118, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Bowers RR, Manevich Y, Townsend DM, and Tew KD. Sulfiredoxin redox-sensitive interaction with S100A4 and non-muscle myosin IIA regulates cancer cell motility. Biochemistry 51: 7740–7754, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Autreaux B. and Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Delaunay A, Isnard A-D, and Toledano MB. H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J 19: 5157–5166, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis HR. and Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry 36: 13349–13356, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, and Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol 24: 434–450, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, and Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res 66: 6800–6806, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gallogly MM, Starke DW, and Mieyal JJ. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid Redox Signal 11: 1059–1081, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hall A, Parsonage D, Poole LB, and Karplus PA. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J Mol Biol 402: 194–209, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iglesias-Baena I, Barranco-Medina S, Lázaro-Payo A, López-Jaramillo FJ, Sevilla F, and Lázaro J-J. Characterization of plant sulfiredoxin and role of sulphinic form of 2-Cys peroxiredoxin. J Exp Bot 61: 1509–1521, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, and Lee SY. Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 117: 625–635, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Jeong W, Park SJ, Chang T-S, Lee D-Y, and Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem 281: 14400–14407, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Jönsson TJ, Johnson LC, and Lowther WT. Structure of the sulphiredoxin–peroxiredoxin complex reveals an essential repair embrace. Nature 451: 98–101, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jönsson TJ, Murray MS, Johnson LC, and Lowther WT. Reduction of cysteine sulfinic acid in peroxiredoxin by sulfiredoxin proceeds directly through a sulfinic phosphoryl ester intermediate. J Biol Chem 283: 23846–23851, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jönsson TJ, Tsang AW, Lowther WT, and Furdui CM. Identification of intact protein thiosulfinate intermediate in the reduction of cysteine sulfinic acid in peroxiredoxin by human sulfiredoxin. J Biol Chem 283: 22890–22894, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kice JL. and Liu C-CA. Reactivity of nucleophiles toward and the site of nucleophilic attack on phenyl benzenethiolsulfinate. J Org Chem 44: 1918–1923, 1979 [Google Scholar]
- 18.Kil IS, Lee SK, Ryu KW, Woo HA, Hu M-C, Bae SH, and Rhee SG. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol Cell 46: 584–594, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Klomsiri C, Karplus PA, and Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal 14: 1065–1077, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar C, Igbaria A, D'Autreaux B, Planson A-G, Junot C, Godat E, Bachhawat AK, Delaunay-Moisan A, and Toledano MB. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J 30: 2044–2056, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchal S. and Branlant G. Evidence for the chemical activation of essential cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from streptococcus mutans. Biochemistry 38: 12950–12958, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Moan NL, Clement G, Maout SL, Tacnet F, and Toledano MB. The Saccharomyces cerevisiae proteome of oxidized protein thiols. Contrasted functions for the thioredoxin and glutathione pathways. J Biol Chem 281: 10420–10430, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Molin M, Yang J, Hanzén S, Toledano MB, Labarre J, and Nyström T. Life span extension and H2O2 resistance elicited by caloric restriction require the peroxiredoxin Tsa1 in Saccharomyces cerevisiae. Mol Cell 43: 823–833, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Mössner E, Huber-Wunderlich M, and Glockshuber R. Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Protein Sci Publ Protein Soc 7: 1233–1244, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulrooney SB. Application of a single-plasmid vector for mutagenesis and high-level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Protein Expr Purif 9: 372–378, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Nagy P, Karton A, Betz A, Peskin AV, Pace P, O'Reilly RJ, Hampton MB, Radom L, and Winterbourn CC. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen Peroxide. A kinetic and computational study. J Biol Chem 286: 18048–18055, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagy P, Lemma K, and Ashby MT. Reactive sulfur species: kinetics and mechanisms of the reaction of cysteine thiosulfinate ester with cysteine to give cysteine sulfenic acid. J Org Chem 72: 8838–8846, 2007 [DOI] [PubMed] [Google Scholar]
- 28.O'Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, Reddy AB, and Millar AJ. Circadian rhythms persist without transcription in a eukaryote. Nature 469: 554–558, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Neill JS. and Reddy AB. Circadian clocks in human red blood cells. Nature 469: 498–503, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JW, Mieyal JJ, Rhee SG, and Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J Biol Chem 284: 23364–23374, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pascual MB, Mata-Cabana A, Florencio FJ, Lindahl M, and Cejudo FJ. Overoxidation of 2-Cys Peroxiredoxin in Prokaryotes. Cyanobacterial 2-Cys peroxiredoxins sensitive to oxidative stress. J Biol Chem 285: 34485–34492, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, and Winterbourn CC. Hyperoxidation of Peroxiredoxins 2 and 3. Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem 288: 14170–14177, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrek M, Otyepka M, Banás P, Kosinová P, Koca J, and Damborský J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics 7: 316, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roussel X, Béchade G, Kriznik A, Van Dorsselaer A, Sanglier-Cianferani S, Branlant G, and Rahuel-Clermont S. Evidence for the formation of a covalent thiosulfinate intermediate with peroxiredoxin in the catalytic mechanism of sulfiredoxin. J Biol Chem 283: 22371–22382, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Roussel X, Boukhenouna S, Rahuel-Clermont S, and Branlant G. The rate-limiting step of sulfiredoxin is associated with the transfer of the γ-phosphate of ATP to the sulfinic acid of overoxidized typical 2-Cys peroxiredoxins. FEBS Lett 585: 574–578, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Roussel X, Kriznik A, Richard C, Rahuel-Clermont S, and Branlant G. Catalytic Mechanism of sulfiredoxin from Saccharomyces cerevisiae passes through an oxidized disulfide sulfiredoxin intermediate that is reduced by thioredoxin. J Biol Chem 284: 33048–33055, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saccoccia F, Di Micco P, Boumis G, Brunori M, Koutris I, Miele AE, Morea V, Sriratana P, Williams DL, Bellelli A, and Angelucci F. Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Struct Lond Engl 199320: 429–439, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spector D, Labarre J, and Toledano MB. A genetic investigation of the essential role of glutathione. Mutations in the Proline biosynthesis pathway are the only suppressor of glutathione auxotrophy in yeast. J Biol Chem 276: 7011–7016, 2001 [DOI] [PubMed] [Google Scholar]
- 39.Tavender TJ, Springate JJ, and Bulleid NJ. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J 29: 4185–4197, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood ZA, Poole LB, and Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300: 650–653, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Wood ZA, Schröder E, Robin Harris J, and Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci 28: 32–40, 2003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.