

Abstract
Hypertrophy increases the risk of heart failure and arrhythmia. Prevention or reversal of the maladaptive hypertrophic phenotype has thus been proposed to treat heart failure. Chronic β-adrenergic receptor (β-AR) stimulation induces cardiomyocyte hypertrophy by elevating 3′, 5′-cyclic adenosine monophosphate (cAMP) levels and activating downstream effectors such protein kinase A (PKA). Conversely, hydrolysis of cAMP by phosphodiesterases (PDEs) spatiotemporally restricts cAMP signaling. Here, we demonstrate that PDE4, but not PDE3, is critical in regulating cardiomyocyte hypertrophy, and may represent a potential target for preventing maladaptive hypertrophy. We identify a sequence within the upstream conserved region 1 of PDE4D, termed UCR1C, as a novel activator of PDE4 long isoforms. UCR1C activates PDE4 in complex with A-Kinase anchoring protein (AKAP)-Lbc resulting in decreased PKA signaling facilitated by AKAP-Lbc. Expression of UCR1C in cardiomyocytes inhibits hypertrophy in response to chronic β-AR stimulation. This effect is partially due to inhibition of nuclear PKA activity, which decreases phosphorylation of the transcription factor cAMP response element-binding protein (CREB). In conclusion, PDE4 activation by UCR1C attenuates cardiomyocyte hypertrophy by specifically inhibiting nuclear PKA activity.
Keywords: Phosphodiesterase 4 (PDE4) activation; A-Kinase Anchoring Protein (AKAP)-Lbc; compartmentalized signaling; 3′, 5′-cyclic monophosphate (cAMP); protein kinase A (PKA); cardiomyocyte hypertrophy
1. Introduction
Cardiac hypertrophy, characterized by increased myocyte size, occurs in response to pressure or volume stress. Under physiological conditions, this response is initially beneficial due to a compensatory decrease in wall stress and increase in cardiac output. However, in pathological cases, prolonged hypertrophic stimuli induce maladaptive changes, which significantly increase the risk of cardiac failure and arrhythmia independent of the underlying cause of hypertrophy [1, 2]. Prevention or reversal of maladaptive hypertrophy has thus been proposed as a desirable therapeutic intervention [3]. It is well-established that multiple pathological hypertrophic pathways converge on a set of transcriptional regulators such as CREB, nuclear factor of activated T cells (NFAT), and myocyte enhancer factor 2 (MEF2), which promote initiation of a developmental gene-reprogramming paradigm, often termed fetal gene response [4–7]. These “fetal” cardiac genes, whose activation accompanies cardiac hypertrophy, are thus called hypertrophic fetal genes.
Localized regulation and integration of cAMP signaling is important for proper cardiac function and perturbation of this signaling leads to heart failure. Upon chronic β-AR stimulation, cardiomyocyte hypertrophy is induced via elevated cAMP and activation of its downstream effectors, including PKA and Epac [8–10]. Mice lacking the PKA catalytic subunit Cβ (PKA-Cβ) are resistant to hypertrophy induction [11]. The first visualization of cAMP microdomains in cardiomyocytes by fluorescent probes for cAMP demonstrated the presence of compartmental cAMP [12]. The subcellular distribution of cAMP signaling components, including adenylyl cyclases (ACs), PDEs, PKA and Epac, is a well-appreciated mechanism to spatiotemporally regulate cAMP signaling in cardiomyocytes [10, 13, 14]. However many of the molecular components and cAMP-signaling networks remain to be defined. PDEs control cAMP signaling in a temporal and spatial manner by degrading cAMP. There are 11 PDE families with more than 100 PDE enzyme variants with distinct kinetic and regulatory properties [15]. Among those, PDE3 and PDE4 contribute up to 90% of cAMP-hydrolyzing activity in cardiomyocytes [16]. Specific inhibition of PDE3 or PDE4 potentiates cAMP production in response to β-AR stimulation of cardiomyocytes [17]. The importance of PDE4 in the myocardium is highlighted by the observation that PDE4D ablation results in age-related hypertrophy, which predisposes mice to development of heart failure [18, 19]. Additionally, abnormal PDE4 expression and activity are observed in hypertrophied and failing hearts [20]. Thus, we hypothesized that PDE4 inhibits hypertrophy and that PDE4 activation might be used as a potential therapeutic strategy for heart diseases
There are 4 separate genes encoding over 20 PDE4 isoforms, each possessing an N-terminal regulatory domain and a C-terminal catalytic domain. Alignment of the N-termini of PDE4 long isoforms identified two conserved regions: Upstream Conserved Region 1 and 2 (UCR1 and UCR2). UCR2 itself inhibits PDE activity, which is counteracted by UCR1 [21]. Antiserum against UCR2 activates PDE4. Phosphorylation through multiple kinases including PKA, MAPK-activated protein kinase 2 (MK2), AMP-activated protein kinase (AMPK) and extracellular signal-regulated kinase (ERK) modulates PDE4 activity [21, 22]. For example, PKA phosphorylates a Ser in the UCR1 of PDE4 long isoforms (equivalent to Ser54 of human PDE4D3) and increases catalytic activity via the UCR1-UCR2 module [23]. In contrast, ERK phosphorylation of a Ser in the catalytic domain inhibits PDE4 activity (equivalent to Ser579 of human PDE4D3 [24, 25]. Moreover, molecules or proteins that bind UCR1 or UCR2 also modulate PDE activity [10, 26]. For example, phosphatidic acid binds to the UCR1 region of PDE4D3 and stimulates its activity [27]. Thus UCR1 and UCR2 are critical domains that regulate PDE4 function.
Compartmentalized cAMP signaling is often facilitated by a family of structurally distinct scaffold proteins called AKAPs. All members of the AKAP family possess a conserved PKA anchoring domain as well as binding sites for additional signaling components, including PDEs [28–30]. Importantly, AKAPs target signaling complexes to distinct subcellular locations, thereby generating signaling specificity [31–33]. AKAPs perform a critical role in mediating the effects of neurohumoral stimulation of the heart by integrating PKA activity with additional enzymes [34–36]. Previously, we and others have demonstrated a critical role for AKAP-Lbc in hypertrophy induction [37–39]. This regulation occurs possibly due to AKAP-Lbc-facilitated PKA-mediated phosphorylation of its substrates. PKA phosphorylates AKAP-Lbc at Ser 1565 to inhibit its Rho-GEF activity [40, 41], and Ser 2737 to reduce PKD binding to AKAP-Lbc, promoting protein kinase C (PKC)-mediated protein kinase D (PKD) activation [42]. Moreover, we showed that AKAP-Lbc facilitates PKA-mediated phosphorylation and inhibition of tyrosine phosphatase Shp2 under hypertrophic conditions stimulated by isoproterenol (ISO) [37]. Additionally, AKAP-Lbc modulates ERK signaling through PKA phosphorylation of kinase suppressor of Ras-1 (KSR-1) [43]. Despite this increased understanding of AKAP-Lbc in regulating PKA signaling, the role of AKAP-Lbc-associated PDE4 in this regulation remains to be determined.
Given the central importance of PKA in AKAP-Lbc signaling and PDE4 in controlling cAMP levels in the heart, we examined the role of specific PDE4 activation in the AKAP-Lbc complex and the resulting effect on hypertrophic PKA signaling. Here, we first demonstrate that PDE4, but not PDE3, plays a critical role in preventing cardiac myocyte hypertrophy. We also identify a short fragment derived from the conserved region of PDE4D long isoforms, termed UCR1C, which functions as a specific activator of PDE4 long isoforms. Expression of UCR1C in neonatal rat ventricular myocytes (NRVMs) inhibits ISO-induced hypertrophy suggesting that PDE4 activation may represent a novel approach to prevent hypertrophy. This inhibition of hypertrophy is due in part to reduced nuclear PKA activity. Finally, we demonstrate that UCR1C activates PDE4 associated with AKAP-Lbc, which accounts for the reduced nuclear PKA activity.
2. Materials and Methods
2.1. Chemicals and antibodies
GIBCO cell culture media, serum, and antibiotics were obtained from Invitrogen. All chemicals were obtained from Sigma: a general PDE inhibitor IBMX (Cat# I5879); a specific PDE4 inhibitor rolipram (Cat# R6520); a specific PDE3 inhibitor cilostamide (Cat# C7971); β-AR agonist isoproterenol hydrochloride (ISO, Cat# I6504); PKA inhibitor fragment 14–22, myristoylated trifluoroacetate salt (PKI, Cat# P9115); forskolin (FSK, F6886); H89 dihydrochloride hydrate (H89, B1427), vehicle control dimethyl sulfoxide (DMSO, Cat# D4540). Antibodies used were as follows: anti-α-actinin (clone BM-75.2, Sigma, Cat# A5044), anti-Flag (M2, Sigma, Cat# F1804), anti-VSV-G (Sigma, Cat# V4888,), anti-V5-agarose affinity gel (V5-10, Sigma, Cat# A7345), anti-Myc (4A6, EMD Millipore, Cat# 05-724), anti-V5 (Invitrogen, Cat# R96025), anti-GFP (Clontech, Cat# 632375 and Cat# 632592), anti-phospho-CREB at Ser133 (87G3, cell signaling, Cat# 9198), anti-CREB (48H2, cell signaling, Cat#3955). AKAP-Lbc polyclonal antibody VO96 were previously described [44].
2.2. Cell culture, transfection and co-immunoprecipitation
Human embryonic kidney (HEK) 293T cells were cultured in DMEM plus 10% (v/v) FBS and penicillin (10 I.U.)/streptomycin (100 μg/ml). Cells were transfected with using calcium phosphate method and lysed in the phosphate buffer with protease inhibitor and phosphatase inhibitor cocktail [39]. AKAP-Lbc siRNAs (Dharmacon, Cat# 4390824) and scramble control siRNA (Dharmacon, Cat# 4390843) (10 nM of each) were transfected into HEK293T cells using Dharmafect 1 (GE Healthcare, Cat# T-2001). Seventy-two hours later, cells were stimulated with adenylyl cyclase activator FSK for 20 min and then lysates harvested. Cleared lysates were incubated with indicated antibodies and Protein A/G-agarose (Millipore) for 2–16 h at 4°C with rocking. Immunoprecipitants were washed in 1ml lysis buffer for 5 times, and then analyzed by Western blot or in vitro cAMP-PDE assays.
2.3. Molecular Cloning
A fragment encoding UCR1 (amino acid 17-136) or UCR1C (amino acid 80-136) of human PDE4D3 was generated by PCR using GFP-PDE4D3 as a template. Forward and reverse oligonucleotides containing restriction sites were used (5′KpnI-UCR1: 5′GCGAATTCCTTTGATGTGGACAATGGCACATC3′; 5′EcoRV-UCR1C: 5′GCGAATTCCATACACGGAGATGACTTGATTGTGAC3′; 3′EcoRI-UCR1:AAAGGTACCTCATCAGGCCTCCTCTGTTATGGTGG3′). The amplified fragments were clone into KpnI/EcoRI (for Cerulean-UCR1) and EcoRV/EcoRI (for Cerulean-UCR1C) sites of FLAG-Cerulean vector digested with restriction enzymes EcoRI (NEB Cat# R0101S)/KpnI (NEB Cat#R0142) (for cerulean-UCR1), and EcoRI/EcoRV (NEB Cat# R0195S) (for Cerulean-UCR1C) respectively. Using QuickChange II site-directed mutagenesis kit (Cat# 200523) from Agilent Technology. UCR1C R/A mutants were generated using complimentary primers (5′GTCTTGGCCAGTCTGGCAACTGTAGCAAACAACTTTGCTGCA3′). Cerulean was replace with RFP to produce FLAG-RFP vectors of control, UCR1C, and UCR1C R/A. RFP was amplified from TagRFP vector (Evrogen Cat #FP141) using primer set (5′ primer: 5′GACAAGCTTATGGTGTCTAAGGGCGAAGAG3′; 3′ primer: 5′GATGAATTCGCTCTAGATCCGGTGGATCCC3′), and inserted into corresponding Cerulean vectors digested with HindIII and EcoRI. The clones were all confirmed by sequencing.
2.3. Western blot analysis
Equal amount of cell lysates were fractionated on polyacrylamide gels and transferred to nitrocellulose membranes. After 1 hour blocking with 10% (w/v) milk or 5% (w/v) BSA, membranes were incubated with primary antibodies overnight at 4°C, followed by washing of the membranes, and then incubation with the appropriate secondary mouse or rabbit antibodies for 1 hour before imaging using ECL reagents.
2.4. Neonatal rat ventricular myocytes (NRVMs) preparation, transfection and immunochemistry
Primary NRVMs were prepared as described [45]. Freshly isolated myocytes were then transfected with indicated plasmids either by electroporation using amaxa rat cardiomyocyte-neonatal nucleofector kit (Lonza, Cat# VPE-1002) or by Jetprime (Polyplus transfection, Cat# 114). The electroporated NRVMs were cultured in plating medium (70% DMEM, 15% Medium 1999, 10% horse serum (HS), 5% FBS and 1x penicillin/streptomycin) on chamber slides (Lab-Tec, Cat# 177437) coated with 1% gelatin (Sigma, Cat# 1393). The following day cells were changed to maintenance medium (80% DMEM, 20% medium 199 and 1X penicillin/streptomycin) for treatment. For transfection using Jetprime, freshly isolated NRVMs were cultured in plating medium for overnight. The following day, the medium was changed to maintenance medium plus 4% horse serum before transfection (according to the manufacturer’s protocol). Twenty-four hours after transfection, cells were treated with isoproterenol (ISO, 10 μM) for 48 hours to induce hypertrophy. Cells were then fixed in 3.7% paraformaldehyde (Fisher, Cat# S74337) for 10 min, permeabilized in 1% Triton X-100 (Fisher, Cat# BP151) for 10 min, then immunostained with the specific cardiomyocyte marker α-actinin. Size of α-actinin and Cerulean double positive myocytes was quantified using ImageJ. For measuring CREB phosphorylation, cells were treated with vehicle (DMSO) or ISO for 20 min before fixation, permeabilization and subsequent immuno-staining with antibody against pCREB (Ser133). The intensity of CREB phosphorylation was analyzed by MetaMorph software.
2.5. in vitro cAMP-PDE assay
PDE activity was measured as described [46] with the following modifications. Either cell lysate or immunoprecipitates from transfected cells were incubated with 100,000 cpm 3H-cAMP and 2 μM cAMP at 30°C for 5–20 min. The reaction was stopped by addition of boiling water. The phosphodiesterase hydrolysis product, 5′AMP, was further broken down to adenosine by snake venom. Adenosine was separated from other compounds using an ionic exchange resin. The activity was calculated using the formula (Counts-background)/(SA*min*ml), SA is equal to CPM in 1μl of total/(pmol of cAMP in reaction). The specific activity is defined as PDE activity/PDE expression in each IP, while the total activity is defined as PDE activity/AKAP-Lbc expression in each IP
2.6. Fluorescence resonance energy transfer (FRET)
HEK293T cells were transfected using the calcium phosphate method and allowed to grow for 12–24 hours before plating onto sterilized glass coverslips coated with 0.2% gelatin. Immediately prior to imaging, cells were washed once with pre-warmed PBS, and placed in L-15 imaging medium supplemented with 5% FBS at 37°C. FSK or H89 (a specific PKA inhibitor) were added as indicated. Cells were imaged on Zeiss total internal reflectance fluorescent (TIRF) microscope. The FRET sensor AKAR2.2 and nls-AKAR2.2 utilized CFP and Citrine with a 405 nm laser line for excitation of fluorescence. Fluorescence emission was collected for CFP between 450 and 490 nm while emission for YFP was collected between 520 and 590 nm. The ratio of fluorescence recorded using these wavelength bands was normalized to the average of baseline ratios before stimulation and plotted against time to illustrate changes of PKA activity. RFP is an indicator of UCR1C expression, and only RFP-positive cells were chosen for analysis. Quantification of recorded time-series was performed using Metamorph.
2.7. Statistical analyses
All data are shown as mean ± standard error of the mean (s.e.m.). Significance was determined using an unpaired two-tailed test using InStat or student T-test. A p-value less than 0.05 was considered significant (*), a p-value less than 0.01 was considered very significant (**), a p-value less than 0.001 was considered extremely significant (***); while a p-value larger than 0.05 was considered not significant (n.s.).
3. Results
3.1. PDE4, but not PDE3, prevents ISO-induced cardiomyocyte hypertrophy
Chronic β-AR stimulation by ISO increased cellular size of NRVMs, which was reduced by the specific PKA inhibitor myristoylated PKI 5-24 peptide (Fig. 1). This ISO-induced, PKA-mediated cardiomyocyte hypertrophy was thus used as an in vitro cellular model to study hypertrophic cAMP-PKA signaling. The general PDE inhibitor IBMX induced cardiomyocyte hypertrophy (Fig. 1), suggesting an active role for PDEs in preventing hypertrophy. PDE3 and PDE4 contribute to the majority of PDE activity in cardiomyocytes [47]. Specific PDE4 inhibition of PDE4 by its specific inhibitor rolipram increased cardiomyocyte size. In contrast, PDE3 inhibition by its specific inhibitor cilostamide, did not affect cardiomyocyte size (Fig 1). These results indicated the critical and primary role of PDE4 in actively preventing cardiomyocyte hypertrophy. This observation is consistent with studies of PDE4D-deficient mice, which developed progressive cardiomyopathy, accelerated heart failure and arrhythmias [18].
Figure 1. PDE4 but not PDE3 actively prevents hypertrophy.

Freshly isolated NRVMs were treated with PDE inhibitors [a general PDE inhibitor IBMX (10 μM), a specific PDE4 inhibitor rolipram (7.5 μM), and a specific PDE3 inhibitor cilostamide (1 μM)], vehicle control DMSO, a β-AR agonist ISO (10 μM), and ISO plus a cell permeable PKA inhibitor PKI (1 μM). After 48 hours treatment, cells were fixed, permeabilized, and immunostained for the cardiomyocyte marker α-actinin. Representative images are shown in 1A. The sizes of NRVMs were quantified using NIH Image J software and plotted in 1B. Data are expressed as mean ± s.e.m.; the number of cells counted is indicated. Student’s t-Tests were performed for statistical analysis. A p-value <0.001 was highly significant (***) while a p-value > 0.05 was not significant (n.s.).
3.2. UCR1C peptide specifically activates PDE4 long isoforms
PDE4 activity is regulated through phosphorylation by kinases such as PKA, ERK, AMPK and MK. However, PDE4 activity is also modulated through protein:protein interactions [10]. Several regulators of PDE4 activity, including phosphatidic acid (PA), phosphatidylserine (PS), X-associated protein 2 (XAP2), and Deleted in Schizophrenia 1 (DISC1), bind the conserved N-terminal regulatory domains UCR1 or UCR2 [27, 48–50]. Although the detailed mechanisms are unknown, it is clear that PDE activity is modulated by binding these regulatory domains. To explore the possibility of targeting PDE4 activation as a potential approach to prevent cardiac hypertrophy, we developed a specific peptide activator of PDE4 long isoforms. We hypothesized that a short fragment derived from UCR1, termed UCR1C, would bind UCR2 and modulate the activity of PDE4 long isoforms (Fig. 2C).
Figure 2. Development of an activator of PDE4 long isoforms.


(A) Schematic presentation of human PDE4 isoforms. As a note, rat long isoform PDE4A5 is the homolog of human PDE4A1, and rat super-short isoform PDE4A1 is the homolog of human PDE4A4. (B) UCR1C and UCR2 alignment in human PDE4 long isoforms. Arrow indicates the critical residues responsible for UCR1-UCR2 interaction. As a note, UCR1C and UCR2 are 100% conserved among different long isoforms from the same genes. Thus, representative long isoforms for each PDE4 genes (PDE4D3, PDE4B3, PDE4A1 and PDE4C2) were used for alignment. (C) Activation mode of PDE4 long isoforms: UCR1 binds UCR2 via electrostatic interactions between residues Arg98, and Arg101 of UCR1C and residues Glu146, Glu147, Asp149 of UCR2N in human PDE4D3. UCRs form a regulatory module, where UCR2 itself exerts an autoinhibitory effect on PDE activity. Phosphorylation as well as interaction of UCRs with lipids and proteins [PA/PS, XAP2, DISC1 and SH3 domains (Lyn and Fyn)] modulates PDE4 activity. Thus, we hypothesize that UCR1C (amino acids 81-136, in gray) binds and modulates PDE4 long isoforms. (D) Interaction of UCR1 and UCR1C with PDE4D3. HEK293T cells were co-transfected with VSV-PDE4D3 and FLAG-UCR1 expression constructs. Forty-eight hours later, cell lysates were harvested for immunoprecipitation (IP) with FLAG antibody. Ponceau S staining shows the successful IP (middle panels). UCR1C binding to PDE4D3 was determined by measuring the amount of VSV-PDE4D3 associated with the immunoprecipitants by Western blot. Both UCR1 (amino acids 17-136) and UCR1C bind PDE4D3. Double mutation of R98A and R101A (UCR1C R/A) abolished interaction. (E) UCR1C, but not UCR1C R/A, enhances activity of PDE4D3. Lysates from 2C were used for in vitro cAMP-PDE assay. Western blot analysis was performed to ensure equal expression of PDE4D3 and expressed of UCR1 mutants and control. The specific activity of PDE4D3 was calculated by normalizing the cAMP-PDE activity to PDE4D3 levels. (F) UCR1C activates long, but not short, PDE4A isoforms. HEK293T cells were co-transfected with GFP-tagged rat PDE4A isoforms and FLAG-UCR1C constructs. The long isoform GFP-PDE4A5 (homolog of human long isoform PDE4A1) and super short-isoform GFP-PDE4A1 (homolog of human super-short isoform PDE4A4) were immunoprecipitated then subjected to in vitro PDE assays and Western analysis. Specific activity was calculated from the cAMP-PDE assay normalized to GFP-PDE4A levels. Expression of UCR1C and its vector control were also shown for similar expression. InStat was used to perform for statistical analysis. A p-value < 0.05 was significant (*) while a p-value > 0.05 was not significant (n.s.). (G–H) UCR1C activates long PDE4B and PDE4C isoforms. HEK293T cells were co-transfected with FLAG-UCR1C constructs and V5-tagged human PDE4B3 (G) or VSV-tagged human PDE4C2 isoforms (H) respectively. The transfected lysates were used for in vitro PDE assays and Western analysis. Specific activity was calculated from the cAMP-PDE assay normalized to PDE4B3 or PDE4C2 respectively. Data represent the mean± s.e.m from 2 independent experiments.
In humans, there are four PDE4 genes (A, B, C, and D) responsible for expression of over 20 isoforms due to splicing (Fig. 2A). UCR1 and UCR2 are 100% conserved among the long isoforms derived from the same gene. Sequence alignment of the different PDE4 isoforms reveals that UCR2 is highly conserved among the 4 genes products, with arrows indicating conserved negatively charged amino acids thought to be important for UCR1 interaction (Fig. 2B). The cardiomyopathy phenotype of PDE4D-deficient mice prompted us to use PDE4D as a template to design fragments that might alter PDE4D function by interfering with UCR1/UCR2 interaction. We identified one such fragment, UCR1C, which spans amino acids 81-136 of human PDE4D3 and includes two positively charged Arg residues that are highly conserved between different PDE4 isoforms and critical for interaction with UCR2 (Figs. 2B, C, indicated by arrow) [51]. UCR1C bound PDE4D3 by co-immunoprecipitation (Fig 2D), and increased its activity (Fig 2E). Mutation of Arg98 and Arg101 of UCR1 to Ala abolished the interaction of UCR1 with UCR2 [51]. Similarly, these mutations in UCR1C (UCR1C R/A) abolished binding to PDE4D3 (Fig 2D) and enhancement of PDE activity (Fig 2E). Similar results were observed with additional PDE4 long isoforms PDE4B3 and PDE4C2 in which UCR1C stimulated PDE4B3 and PDE4C2 activity, and Arg/Ala mutations reduced these effects (Fig 2G, H). UCR1C also bound and activated rat PDE4A5, a long isoform, but not rat PDE4A1, a super-short isoform that lacks both UCR1 and UCR2 (Fig 2F). This effect of UCR1C on PDE4A5 activation but not PDE4A1 was not due to variance in UCR1C expression (Fig 2F). We also tested the effects of UCR1C on endogenous PDE4 activity. UCR1C activated PDE4 long isoform (Fig 3A) and PDE4B (Fig 3B). This effect was abolished by the Arg to Ala mutations in UCR1C (Figs 3A, B). Together, these results suggest that UCR1C acts as an activator of PDE4 long isoforms.
Figure 3. UCR1C activates endogenous PDE4 long isoforms.
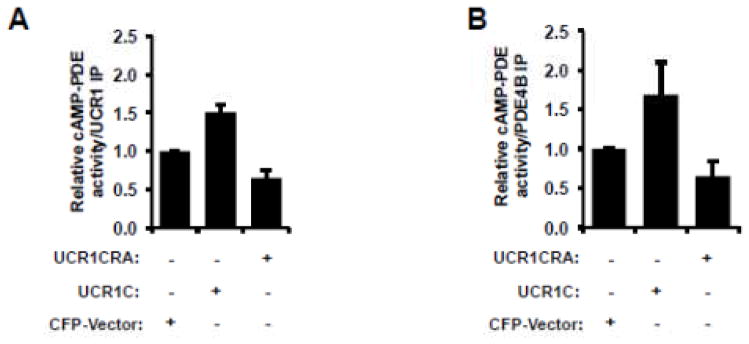
HEK293T cells were transfected with cerulean-vector, cerulean-UCR1C, or cerulean-UCR1C R/A. Forty-eight hours later, cell lysates were harvested, and 1.5 mg total protein was applied for immunoprecipitation using protein A-agarose beads, and anti-PDE4B pan-antibody or anti-UCR1 antibody. Relative PDE activities of endogenous PDE4 long isoforms (anti-UCR1 IP) are shown in (A), and endogenous PDE4B (anti-PDE4B IP) in (B). The data were shown as mean± s.e.m from two independent experiments.
3.3. UCR1C prevents ISO-induced cardiomyocyte hypertrophy
The importance of PDE4 activation on cardiomyocyte hypertrophy was further investigated by testing the effect of UCR1C expression on ISO-induced cardiomyocyte hypertrophy. Chronic ISO treatment increased cell size of NRVMs by 2 fold, and this effect was inhibited by expression of UCR1C (Figs 4A, B). This result suggests a critical role for activation of PDE4 long isoforms in preventing cardiomyocyte hypertrophy. As a note, UCR1C itself did not alter the cell size of cardiomyocytes in the absence of ISO.
Figure 4. UCR1C prevents ISO-induced cardiomyocyte hypertrophy.

UCR1C inhibits ISO-induced cardiomyocyte hypertrophy. NRVMs were transfected with Cerulean-UCR1C or Cerulean control vector then treated with ISO for 48 hours to induce hypertrophy. Cells were fixed, permeabilized, and stained with α-actinin (Red) for cell imaging. Representative images are shown in 4A. Cardiomyocytes with both α-actinin staining and Cerulean expression were selected for cell size measurements. The quantified results are shown in 4B. Data are expressed as mean ± s.e.m. The number of cells counted is indicated. Differences in quantitative variables were examined by one-way analysis of variance (ANOVA). A p-value < 0.001 was considered extremely significant (***) and < 0.05 was significant (*).
3.4. PDE4 activation by UCR1C does not result in global PKA inhibition
Both PDE4 inhibition and PDE4D ablation prolong ISO-induced cytoplasmic PKA activation in cardiomyocytes, suggesting the critical role of PDE4 in modulating PKA activity [52]. Thus, we postulated that activation of PDE4 long isoforms by UCR1C would decrease cAMP levels and lower PKA activity. To address this possibility, we utilized a novel FRET reporter of PKA activity AKAR2.2 (Fig 5A). An adenylyl cyclase activator FSK increased the FRET signal of AKAR2.2, which was reduced upon PKA inhibition with H89 (Figs 5B, C). Surprisingly, UCR1C did not alter FSK-induced global PKA activation as measured by AKAR2.2 (Figs 5B, C). In support of this result, Western blot analysis using a phosphospecific PKA substrate antibody showed no difference in FSK-induced phosphorylation of PKA substrates between control and UCR1C expressing cells (data not shown), confirming that UCR1C does not affect global PKA activity. Moreover, UCR1C did not modulate the overall phosphorylation of PDE4D3, consistent with the finding that UCR1C did not globally inhibit PKA (Fig 6). These observations raised the possibility that UCR1C-mediated activation of PDE4 leads to localized inhibition of PKA activity.
Figure 5. UCR1C does not alter global PKA activity.

(A) Diagram of A kinase activity reporter (AKAR2.2), adapted from [64]. In AKAR2.2, PKA phosphorylation of the P sites results in binding to FHA, which induces the proximity of CFP and citrine to increase the FRET ratio. AKAR2.2 was used to report the overall PKA activity inside cells. HEK293T cells were co-transfected with AKAR2.2 and RFP-tagged UCR1C or RFP control plasmid. Twenty-four hours later, cells were plated on slides and imaged after another 24 hours. (B) Representative pseudo-color images of AKAR2.2 FRET ratio change in response to adenylyl cyclase activator Forskolin (20 μM) stimulation and subsequent PKA inhibition using H89 (20 μM). (C) Time-course of average AKAR FRET ratio. Cells with similar RFP expression intensity were selected for analysis. Data are expressed as mean ± s.e.m.. Black squares represent cells expressing RFP vector (n=12 cells), open diamonds represent cells expressing RFP-UCR1C (n=12 cells).
Figure 6. UCR1C does not modulate PKA-mediated PDE4D3 phosphorylation.

HEK293T cells were co-transfected GFP-PDE4D3 with RFP-vector, RFP-UCR1C, or RFP-UCR1C R/A. 48 hours later, cells were treated with FSK or DMSO control, and cells lysates were harvested for PDE4D3 immunoprecipitation using protein A agarose beads and anti-GFP antibody. Western blot was then performed to investigate the phosphorylation level of PDE4D3. Image J quantification of two independent experiments on phosphor-PDE4D3/total were shown as mean± s.e.m.. Successful expression of UCR1C, UCR1C R/A mutants and vector in lysate and IP of PDE4D3 were also shown in the representative blot.
3.5. UCR1C activates AKAP-Lbc associated PDE4 and inhibits AKAP-Lbc phosphorylation
Being a PKA binding protein, we tested the interaction of AKAP-Lbc with PDE. Multiple proteomic screens using tandem MS to identify proteins associated with GST-tagged AKAP-Lbc fragments identified PDE as an AKAP-Lbc associated protein [37]. Analysis of immunoprecipitated AKAP-Lbc confirmed the presence of cAMP hydrolyzing activity, which was abolished by the general PDE inhibitor IBMX and the specific PDE4 inhibitor rolipram but not by PDE3-specific inhibitor cilostamide (Fig 7A). Furthermore, immunoprecipitation of endogenous AKAP-Lbc from mouse heart revealed the presence of associated PDE4D proteins, including long isoforms of PDE4D3, 5, 8, 9 (Figs 7B–F). These results confirmed that AKAP-Lbc bound PDE4, specifically PDE4 long isoforms. We next tested the binding preference of AKAP-Lbc for PDE isoforms lacking the UCRs by examining the interaction of AKAP-Lbc with PDE4B1 long isoforms versus PDE4B2 short isoform. In HEK293T cells, AKAP-Lbc co-immunoprecipitated endogenous PDE4B1 long isoform but not PDE4B2 short isoform (Fig 7G).
Figure 7. AKAP-Lbc interacts with PDE4 long isoforms.

(A) AKAP-Lbc immune complex contained cAMP-PDE4 activity. AKAP-Lbc was immunoprecipitated from mouse heart extract (3 mg) and used to perform in vitro cAMP-PDE assay. Specific PDE inhibitors were used to identify PDE activity in the AKAP-Lbc complex. Prior to PDE assay, AKAP-Lbc immunoprecipitates were treated with a general PDE inhibitor IBMX, a specific PDE3 inhibitor cilostamide, or a specific PDE4 inhibitor rolipram. mAKAP immunoprecipitates were used as a positive control and IgG immunoprecipitates were used as a negative control. cAMP-PDE activities were displayed mean ± s.e.m. of three independent assays performed in triplicate. (B–F) Co-immunoprecipitation (co-IP) of endogenous PDE4D with AKAP-Lbc purified from mouse heart lysate. Endogenous AKAP-Lbc was immunoprecipitated from mouse heart lysate (2 mg). Control IgG IPs were performed using an equal amount of heart lysate, demonstrating the specific interaction of PDE4D with AKAP-Lbc. Western blots show identification of PDE4D (using a pan antibody that detects all family members, (B)), PDE4D3 (C), PDE4D5 (D), PDE4D8 (E) and PDE4D9 (F) that associate with AKAP-Lbc. (G) Endogenous PDE4B1 long isoform, but not short form PDE4B2, binds AKAP-Lbc. HEK293T cells were transfected with V5-AKAP-Lbc, 48 hrs later, cell lysates were prepared, and AKAP-Lbc was immunoprecipitated using V5-agarose beads. PDE4B1 and PDE4B2 were detected by immunoblotting using antibodies specific for either PDE4B1 or PDE4B2.
Next we investigated whether UCR1C specifically modulates AKAP-Lbc-associated PDE activity. Consistent with the observation that UCR1 mediates the binding of PDE4 to AKAP-Lbc [53], UCR1C overexpression reduced PDE4D3 binding to AKAP-Lbc (Figs 8A, B). Nevertheless, the specific activity of PDE4D3 associated with AKAP-Lbc was enhanced by 3 fold in the presence of UCR1C (Fig 8C). Not surprisingly, UCR1C increased the total PDE activity associated with AKAP-Lbc (Fig 8D). This increased PDE activity in the AKAP-Lbc complex was reflected in a decrease in PKA-induced AKAP-Lbc phosphorylation (Figs 8E, F). The inhibitory effect of UCR1C on PKA activity in the AKAP-Lbc complex was not due to its modulation of PKA-RII binding to AKAP-Lbc as UCR1C did not affect AKAP-Lbc/PKA-RII interaction either in the resting state or following PKA stimulation (Fig 9). Together, these results suggest that UCR1C activates AKAP-Lbc-associated PDE4, thereby, inhibiting PKA activation and subsequent AKAP-Lbc phosphorylation by PKA.
Figure 8. UCR1C augments PDE4 activity and inhibits PKA in the AKAP-Lbc complex.

(A–D) HEK293T cells were co-transfected with GFP-AKAP-Lbc, VSV-PDE4D3 and RFP-UCR1C or RFP control vector. Forty-eight hours later, cells were treated with FSK (20 μM) or DMSO (vehicle control) for 20 minutes prior to AKAP-Lbc immunoprecipitation using GFP antibody. Samples were split in half for cAMP-PDE assays and Western blot analysis for quantification of protein levels. Representative Western blots are shown in 8A. Quantification of PDE4D3 binding to AKAP-Lbc by Image J is shown in 8B. Specific activity was calculated by normalizing PDE activities to levels of PDE4D3, and the result is shown in 8C. Total activity in AKAP-Lbc complex was calculated by dividing PDE activity to levels of AKAP-Lbc in IPs, and the result is shown in 8D. PDE activity is displayed mean ± s.e.m. from three independent experiments performed in triplicate. (E–F) UCR1C inhibits PKA-mediated AKAP-Lbc phosphorylation. HEK293T cells were co-transfected with V5-AKAP-Lbc and Cerulean-UCR1C or control vector. Forty-eight hours later, cells were treated with FSK, and then cell lysates were prepared and used for immunoprecipitation of AKAP-Lbc usingV5-agarose beads. PKA-mediated phosphorylation of AKAP-Lbc was measured by reactivity with an antibody to phospho-PKA substrates. Representative Western blot data are shown in 8E and quantification of results using Image J is shown in 8F as mean ± s.e.m.. InStat were performed for statistical analysis. A p-value < 0.05 was significant (*), a p-value < 0.01 was highly significant (**), while a p-value > 0.05 was not significant (n.s.).
Figure 9. UCR1C does not modulate AKAP-Lbc and PKA-RII interaction.

HEK293T cells were transfected with cerulean-vector, cerulean-UCR1C, or cerulean-UCR1C R/A. After forty-eight hours, cells were treated with FSK or DMSO control, lysates were harvested, and equivalent amounts of protein were used for AKAP-Lbc immunoprecipitation using Protein A agarose beads and anti-AKAP-Lbc antibody (VO96). Western blot was then performed to determine binding of PKA-RII to AKAP-Lbc. Image J was used for quantification data from two independent experiments. Relative PKA-RII/AKAP-Lbc IP is shown as mean±s.e.m. Representative Western blot shows equivalent expression of UCR1C, UCR1C R/A mutants and vector in lysate, and equal immunoprecipitation of AKAP-Lbc.
3.6. UCR1C specifically decreases nuclear PKA activity
PKA activation by cAMP in the perinuclear region results in translocation of the PKA catalytic subunits into the nucleus and phosphorylation of nuclear substrates, including CREB [54]. Because AKAP-Lbc is also found in the perinuclear region [39], we investigated whether AKAP-Lbc modulated nuclear PKA activity. Silencing AKAP-Lbc reduced FSK-induced CREB phosphorylation at Ser 133 (Fig 10A) while AKAP-Lbc overexpression enhanced FSK-induced CREB phosphorylation (Fig 10B) suggesting that AKAP-Lbc regulates nuclear PKA activation.
Figure 10. AKAP-Lbc regulates PKA-mediated CREB phosphorylation.

(A) AKAP-Lbc siRNA reduces PKA-mediated CREB phosphorylation. HEK293T cells were transfected with AKAP-Lbc siRNA or negative control siRNA. Seventy-two hours later, cells were treated with FSK, and cell lysates were harvested for Western blot. Representative Western blots, Image J quantification of AKAP-Lbc expression levels, and CREB phosphorylation level are shown. (B) AKAP-Lbc overexpression enhances PKA-mediated CREB phosphorylation. HEK293T cells were transfected with V5-RFP-AKAP-Lbc or control TagRFP. Forty-four hours later, cells were treated with FSK for 30 min, and cell lysates were harvested for Western blot. Representative blots, Image J quantification results of AKAP-Lbc expression level, and CREB phosphorylation level are shown. A p-value <0.05 was significant (*), and a p-value <0.001 was extremely significant (***).
Given the ability of UCR1C to block PKA activity in the AKAP-Lbc complex, and the ability of AKAP-Lbc to modulate nuclear PKA activity, we next tested whether UCR1C inhibited nuclear PKA activity using a modified AKAR2.2 FRET reporter targeted to the nucleus (nls-AKAR). FSK treatment led to a relatively slow increase in FRET, which was reduced by UCR1C expression (Figs 11A, B). Consistent with this result, UCR1C, but not the UCR1C R/A mutant, inhibited FSK-induced CREB phosphorylation (Figs 11C, D). Finally, UCR1C expression in NRVMs reduced CREB phosphorylation following ISO stimulation (Fig 12). These results suggest that UCR1C specifically inhibits nuclear PKA activity. Combined with the recent report that nuclear overexpression of PKA-C induces cardiomyocyte hypertrophy [55], these findings suggest that nuclear PKA activity is critical for cardiomyocyte hypertrophy.
Figure 11. UCR1C decreases nuclear PKA activity.

AKAR was amended with a nuclear localization signal (nls-AKAR) to specifically report nuclear PKA activity in situ. (A) Representative pseudo-color images of nls-AKAR FRET change in response to FSK stimulation. (B) Time course of average nls-AKAR FRET ratio in response to FSK stimulation. Black squares represent cells expressing RFP vector control (n=31 cells), open diamonds represent cells expressing RFP-UCR1C (n=37 cells). UCR1C but not UCR1C R/A inhibits PKA-mediated CREB phosphorylation. (C) HEK293T cells expressing Cerulean-UCR1, Cerulean-UCR1C R/A, or Cerulean vector control were stimulated with FSK for 40 min, prior to SDS-PAGE and assessment of CREB phosphorylation at PKA substrate site Ser133. (D) Quantification of CREB phosphorylation level using Image J is shown as mean ± s.e.m. from 3 independent experiments. InStat were performed for statistical analysis. A p-value < 0.05 was significant (*), a p-value < 0.001 was extremely significant (***) while a p-value > 0.05 was not significant.
Figure 12. UCR1C expression in NRVMs decreases ISO-induced CREB phosphorylation.

(A) Representative Image of ISO-induced CREB phosphorylation in NRVMs. NRVMs expressing Cerulean-UCR1C or Cerulean vector control were treated with ISO (10 μM for 30 min). NRVMs were then fixed and immunostained for the cardiomyocyte marker α-actinin (red) and phospho-CREB-Ser133 (green). (B) Quantification of CREB phosphorylation. Cerulean-positive and α-actinin–positive NRVMs were selected to determine phospho-CREB levels. Immunofluorescent signals were quantified using Metamorph Software. Results represent the average pCREB signal from the number of cells indicated in each condition shown as mean± s.e.m. A p-value < 0.001 were extremely significant (***), and a p-value < 0.05 were significant (*).
4. Discussion
To better understand the importance of compartmentalized cAMP signaling in heart functions, we investigated the role of localized PDE4 activation in reducing the hypertrophic response. We have identified a peptide sequence derived from the UCR1 region of PDE4D that inhibits cardiomyocyte hypertrophy by activating PDE4 long isoforms. Specifically, UCR1C activates PDE4 long isoforms associated with AKAP-Lbc, inhibits nuclear PKA activity, and reduces hypertrophy in response to β-AR stimulation.
There are discrepant reports in the literature regarding the specific PDE isoforms and activities present in hypertrophied heart [56–58]. However, as hearts progress toward failure, expression and activity of PDE4 long isoforms decrease [56]. Changes in PDE4 levels from compensated hypertrophy to decompensated heart failure suggest the possible involvement of PDE4 in hypertrophy and transition to heart failure. Consistent with this possibility, PDE4D deficient mice developed dilated hypertrophy, which predisposes mice to heart failure and arrhythmia [18]. Upon myocardial infarction, these PDE4D-deficient mice progressed to heart failure, partly due to promotion of PKA-mediated hyper-phosphorylation of ryanodine receptor 2 and phospholamban and a reduction of calcium release [47, 59]. In this study, we provide several lines of evidence supporting a role for PDE4 regulation of cardiomyocyte hypertrophy. First, PDE4 inhibition induces cardiomyocyte hypertrophy, mimicking chronic β-AR stimulation with ISO. This result suggests that basal PDE4 activity prevents cellular hypertrophy. Secondly, specific activation of PDE4 by UCR1C alone does not affect cardiomyocyte size, but rather attenuates the hypertrophic response to chronic ISO stimulation, indicating that PDE4 activity is critical in the regulation of humoral stimulation-induced cardiac hypertrophy. Thus, we propose that PDE4 activation may be efficacious in preventing hypertrophy.
Given the wide range of processes that are mediated by cAMP signaling and the fact that PDEs are the sole cellular enzymes capable of hydrolyzing cAMP, the development of reagents that modulate specific subcellular PDE activity (either inhibition or activation) is an important goal. Here, we demonstrate that PDE4 is activated by the PDE4D-derived peptide UCR1C. This increase in activity is likely due to binding of UCR1C to UCR2 in PDE4 long isoform as mutation of Arg98 and Arg101 to Ala disrupts the interaction of UCR1 with PDE4 long isoforms and abrogates the effect of UCR1C on PDE activity. Several lipids (e.g. PA/PS) and proteins (e.g. XIAP, DISC, antibody K116) are known to bind the UCRs of PDE4 and modulate PDE activity. However, the mechanism by which UCR1C binding to PDE4 results in activation is not clear. Partial endo-proteinase digestion (Lys-C) of PDE4D3 identified UCR2 as an autoinhibitory region that binds the catalytic subunit [21]. It has therefore been speculated that XAP2 or DISC binding of UCR2 stabilizes the closed conformation, blocking access of cAMP to the active site, thereby decreasing PDE enzymatic activity [60]. PKA-mediated phosphorylation of Ser54 in UCR1 of PDE4D3 also disrupts binding of UCR2 to the catalytic subunit, thereby releasing the auto-inhibitory effects of UCR2 on PDE activity [21]. Thus, we speculate that UCR1C activates PDE4 long isoforms by binding UCR2, stabilizing the open conformation. However, the possibility that UCR1C may also compete with protein binding to UCR2 (e.g. XAP2, DISC) and abrogate their inhibitory effects cannot be ruled out. While UCR1C activation of PDE4 does not appear to be through PKA-mediated phosphorylation of PDE4 (Fig 6), it is also possible that UCR1C activates PDE4 through other protein kinases (e.g. AMPK), especially considering MK2 can modulate PKA-mediated PDE4 activation [61]. Further structural studies of PDE4 binding to UCR1C will be required to determine the conformational change induced by UCR1C and provide clear insight into the molecular details of this activation mechanism.
Consistent with the observation that mice deficient in PKA-Cβ are resistant to hypertrophic stimuli [11], specific PKA inhibition abolishes ISO-induced cardiomyocyte hypertrophy, demonstrating the main contribution of PKA to ISO-induced cardiomyocyte hypertrophy. UCR1C inhibition of hypertrophic PKA signaling was investigated here. Although cAMP-mediated Epac activation induces hypertrophy [62], we did not study the effect of UCR1C on Epac activation. However, UCR1C may also impact Epac-activation and subsequent Epac-mediated hypertrophic signaling.
Being a general activator of PDE4 long isoforms, we anticipated that UCR1C would inhibit overall PKA activity. Unexpectedly, UCR1C did not inhibit global PKA activity, but rather, specifically inhibited nuclear PKA. This result is consistent with the observations that PDE4 modulates nuclear PKA activity. PDE4 inhibition by rolipram or PDE4D gene ablation enhances nuclear PKA activity [52]. Conversely, PDE4D3 overexpression inhibits nuclear PKA activity [63]. Importantly, nuclear PKA plays a central role in the hypertrophic response as nuclear PKA catalytic subunit overexpression in cardiomyocytes induces hypertrophy [55]. This result is consistent with our observation that UCR1C reduces nuclear PKA activity, which may explain the ability of UCR1C to inhibit ISO-induced hypertrophy.
Nuclear inhibition of cAMP-PKA signaling by UCR1C may be due to its compartmentalized specific activation of PDE4. Binding of PKA regulatory subunit (PKA-R) by cAMP leads to the dissociation of catalytically active PKA-C from the holoenzyme. PKA-C then translocates to the nucleus, resulting in increased nuclear PKA activity [54]. The perinuclear localization of AKAP-Lbc suggests its possible role in modulating nuclear PKA activity. Indeed, we found that silencing of AKAP-Lbc inhibited PKA-mediated phosphorylation of nuclear transcription factor CREB. Conversely, increasing AKAP-Lbc enhanced PKA-mediated CREB phosphorylation. These observations confirm the critical role of AKAP-Lbc in modulating nuclear PKA activity. As UCR1C activates PDE4 long isoforms associated with AKAP-Lbc and inhibits PKA-mediated AKAP-Lbc phosphorylation, we speculate that UCR1C specifically activates PDE4 in the AKAP-Lbc complex, inhibiting PKA activation at peri-nuclear sites and reducing nuclear PKA activity. However, other possibilities cannot be excluded and will be studied in the future. For example, UCR1C may activate PDE4 associated with other AKAPs such as mAKAP or may directly activate nuclear PDE4 and inhibit nuclear PKA. Regardless, our data are consistent with recent data and mechanistic modeling by Zhang and colleagues demonstrating the spatiotemporal regulation of nuclear PKA activity by AKAPs and PDE4 [63].
In conclusion, the pleiotropic effects of cAMP may be specifically modulated by directly targeting distinct PDE complexes. UCR1C represents a novel tool to study activation of PDE4 on localized PKA signaling. In this paper, we have examined the role of UCR1C in regulating hypertrophic PKA signaling. Our findings demonstrate that UCR1C specifically activates PDE4 in the AKAP-Lbc complex. However, our results do not rule out the possibility that UCR1C activates PDE4 associated with other complexes (i.e., β-AR) or in other distinct locations (nucleus, mitochondrial, Golgi, sarcomere and etc.). Nevertheless, this study demonstrates the utility of using UCR1C as a tool for studying spatial regulation of PDE4 on localized cAMP signaling.
Highlights.
We demonstrate that PDE4 but not PDE3 actively prevents cardiomyocyte hypertrophy.
We identify a novel peptide sequence, UCR1C, that activates PDE4 long isoforms.
We show that PDE4 activation by UCR1C prevents isoproterenol-induced cardiomyocyte hypertrophy.
We define the mechanism by which UCR1C modulates hypertrophic cAMP-PKA signaling.
We propose PDE4 activation as a potential therapeutic strategy in treating heart failure.
Acknowledgments
Funding
B.T.B. was supported by an American Heart Association Predoctoral Fellowship (14PRE18830077). A.V.K. received support from an NIH award (R21CA159179). J.P.O. received support from a Veteran’s Administration MERIT Award (1I01BX002095) and an NIH award (RO1CA116708). G.C.K. received support from an American Heart Association Scientist Development grant (11SDG5230003), the Center for Clinical and Translational Science at UIC through grant UL1TR000050 from the NIH National Center for Advancing Translational Sciences and start-up funds from the University of Illinois at Chicago.
The authors wish to thank Dr. Kimberly Dodge-Kafka for the VSV-PDE4D3 expression construct, and Dr. Jin Zhang for the AKAR2.2 FRET reporter. Also, we wish to thank the members of our laboratories for their helpful input during the course of these experiments.
Abbreviations
- PDEs
Phosphodiesterases
- AKAP
A-Kinase Anchoring Protein
- cAMP
3′, 5′-cyclic monophosphate
- Epac
exchange protein directly activated by cAMP
- PKA
protein kinase A
- PKC
protein kinase C
- PKD
protein kinase D
- UCR
Upstream conserved region
- β-AR
β-adrenergic receptor
- CREB
cAMP response element-binding protein
- NFAT
nuclear factor of activated T cells
- MEF2
myocyte enhancer factor 2
- PKA-Cβ
PKA catalytic subunit Cβ
- ACs
adenylyl cyclases
- NRVMs
neonatal rat ventricular myocytes
- KSR-1
kinase suppressor of Ras-1
- PA
phosphatidic acid
- PS
phosphatidylserine
- XAP2
X-associated protein 2
- DISC1
Deleted in Schizophrenia 1
- ISO
isoproterenol
- FSK
Forskolin
- IBMX
3-isobutyl-1-methylxanthine
- DMSO
dimethyl sulfoxide
- FRET
Fluorescence resonance energy transfer
Footnotes
Conflict of interests
None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hill JA, Olson EN. The New England journal of medicine. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Selvetella G, Hirsch E, Notte A, Tarone G, Lembo G. Cardiovascular research. 2004;63:373–380. doi: 10.1016/j.cardiores.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Frey N, Katus HA, Olson EN, Hill JA. Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 4.Frey N, Olson EN. Annual review of physiology. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 5.Heineke J, Molkentin JD. Nature reviews Molecular cell biology. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 6.Mudd JO, Kass DA. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 7.Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R, Chen Y, Yang Q, Zhang XD, Fan GC, Li H. Hypertension. 2013;61:1193–1202. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itaya T, Hashimoto H, Uematsu T, Nakashima M. British journal of pharmacology. 1990;99:572–576. doi: 10.1111/j.1476-5381.1990.tb12970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laurent AC, Breckler M, Berthouze M, Lezoualc’h F. Biochemical Society transactions. 2012;40:51–57. doi: 10.1042/BST20110642. [DOI] [PubMed] [Google Scholar]
- 10.Houslay MD. Trends in biochemical sciences. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 11.Enns LC, Bible KL, Emond MJ, Ladiges WC. BMC research notes. 2010;3:307. doi: 10.1186/1756-0500-3-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaccolo M, Pozzan T. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 13.Zaccolo M. British journal of pharmacology. 2009;158:50–60. doi: 10.1111/j.1476-5381.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapiloff MS, Chandrasekhar KD. Journal of cardiovascular pharmacology. 2011;58:337–338. doi: 10.1097/FJC.0B013E31822D5C08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guellich A, Mehel H, Fischmeister R. Pflugers Archiv : European journal of physiology. 2014;466:1163–1175. doi: 10.1007/s00424-014-1515-1. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto T, Kobayashi T, Kamata K. Journal of smooth muscle research = Nihon Heikatsukin Gakkai kikanshi. 2003;39:67–86. doi: 10.1540/jsmr.39.67. [DOI] [PubMed] [Google Scholar]
- 17.Field LA. 2013 [Google Scholar]
- 18.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Basic research in cardiology. 2011;106:249–262. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnart SE, Marks AR. Expert opinion on therapeutic targets. 2006;10:677–688. doi: 10.1517/14728222.10.5.677. [DOI] [PubMed] [Google Scholar]
- 21.Lim J, Pahlke G, Conti M. J Biol Chem. 1999;274:19677–19685. doi: 10.1074/jbc.274.28.19677. [DOI] [PubMed] [Google Scholar]
- 22.Sheppard CL, Lee LC, Hill EV, Henderson DJ, Anthony DF, Houslay DM, Yalla KC, Cairns LS, Dunlop AJ, Baillie GS, Huston E, Houslay MD. Cellular signalling. 2014;26:1958–1974. doi: 10.1016/j.cellsig.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 23.MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, Millen J, Beard MB, van Heeke G, Houslay MD. British journal of pharmacology. 2002;136:421–433. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baillie GS, MacKenzie SJ, McPhee I, Houslay MD. British journal of pharmacology. 2000;131:811–819. doi: 10.1038/sj.bjp.0703636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. The EMBO journal. 1999;18:893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Omori K, Kotera J. Circulation research. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 27.Grange M, Sette C, Cuomo M, Conti M, Lagarde M, Prigent AF, Nemoz G. J Biol Chem. 2000;275:33379–33387. doi: 10.1074/jbc.M006329200. [DOI] [PubMed] [Google Scholar]
- 28.Diviani D, Dodge-Kafka KL, Li J, Kapiloff MS. American journal of physiology Heart and circulatory physiology. 2011;301:H1742–1753. doi: 10.1152/ajpheart.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carnegie GK, Burmeister BT. Journal of cardiovascular pharmacology. 2011;58:451–458. doi: 10.1097/FJC.0b013e31821c0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carnegie GK, Means CK, Scott JD. IUBMB life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colledge M, Scott JD. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- 32.Wong W, Scott JD. Nature reviews Molecular cell biology. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 33.Beene DL, Scott JD. Current opinion in cell biology. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bristow MR. Int J Cardiol. 1984;5:648–652. doi: 10.1016/0167-5273(84)90179-7. [DOI] [PubMed] [Google Scholar]
- 35.Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Circulation research. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 36.Bisognano JD, Weinberger HD, Bohlmeyer TJ, Pende A, Raynolds MV, Sastravaha A, Roden R, Asano K, Blaxall BC, Wu SC, Communal C, Singh K, Colucci W, Bristow MR, Port DJ. Journal of molecular and cellular cardiology. 2000;32:817–830. doi: 10.1006/jmcc.2000.1123. [DOI] [PubMed] [Google Scholar]
- 37.Burmeister BT, Taglieri DM, Wang L, Carnegie GK. J Biol Chem. 2012;287:40535–40546. doi: 10.1074/jbc.M112.385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards HV, Scott JD, Baillie GS. The Biochemical journal. 2012;446:437–443. doi: 10.1042/BJ20120570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. Molecular cell. 2008;32:169–179. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diviani D, Abuin L, Cotecchia S, Pansier L. EMBO J. 2004;23:2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O’Donnell P, Taylor P, Taylor L, Zougman A, Woodgett JR, Langeberg LK, Scott JD, Pawson T. Current biology : CB. 2004;14:1436–1450. doi: 10.1016/j.cub.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 42.Carnegie GK, Smith FD, McConnachie G, Langeberg LK, Scott JD. Mol Cell. 2004;15:889–899. doi: 10.1016/j.molcel.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 43.Smith FD, Langeberg LK, Cellurale C, Pawson T, Morrison DK, Davis RJ, Scott JD. Nat Cell Biol. 2010;12:1242–1249. doi: 10.1038/ncb2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diviani D, Soderling J, Scott JD. J Biol Chem. 2001;276:44247–44257. doi: 10.1074/jbc.M106629200. [DOI] [PubMed] [Google Scholar]
- 45.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. J Cell Sci. 2005;118:5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 46.Beavo JA, Hardman JG, Sutherland EW. J Biol Chem. 1970;245:5649–5655. [PubMed] [Google Scholar]
- 47.Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones PP, Tian X, Wilson LS, Ahmad F, Chen SR, Movsesian MA, Manganiello V, Maurice DH, Conti M, Backx PH. Circulation research. 2011;109:1024–1030. doi: 10.1161/CIRCRESAHA.111.250464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nemoz G, Sette C, Conti M. Molecular pharmacology. 1997;51:242–249. doi: 10.1124/mol.51.2.242. [DOI] [PubMed] [Google Scholar]
- 49.Bolger GB, Peden AH, Steele MR, MacKenzie C, McEwan DG, Wallace DA, Huston E, Baillie GS, Houslay MD. J Biol Chem. 2003;278:33351–33363. doi: 10.1074/jbc.M303269200. [DOI] [PubMed] [Google Scholar]
- 50.Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- 51.Beard MB, Olsen AE, Jones RE, Erdogan S, Houslay MD, Bolger GB. J Biol Chem. 2000;275:10349–10358. doi: 10.1074/jbc.275.14.10349. [DOI] [PubMed] [Google Scholar]
- 52.Haj Slimane Z, Bedioune I, Lechene P, Varin A, Lefebvre F, Mateo P, Domergue-Dupont V, Dewenter M, Richter W, Conti M, El-Armouche A, Zhang J, Fischmeister R, Vandecasteele G. Cardiovascular research. 2014;102:97–106. doi: 10.1093/cvr/cvu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, Carnegie GK. Journal of visualized experiments : JoVE. 2013 doi: 10.3791/50529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meinkoth JL, Alberts AS, Went W, Fantozzi D, Taylor SS, Hagiwara M, Montminy M, Feramisco JR. Molecular and cellular biochemistry. 1993;127–128:179–186. doi: 10.1007/BF01076769. [DOI] [PubMed] [Google Scholar]
- 55.Yang JH, Polanowska-Grabowska RK, Smith JS, Shields CWt, Saucerman JJ. Journal of molecular and cellular cardiology. 2014;66:83–93. doi: 10.1016/j.yjmcc.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi K, Osanai T, Nakano T, Wakui M, Okumura K. Heart and vessels. 2002;16:249–256. doi: 10.1007/s003800200032. [DOI] [PubMed] [Google Scholar]
- 57.Mokni W, Keravis T, Etienne-Selloum N, Walter A, Kane MO, Schini-Kerth VB, Lugnier C. PloS one. 2010;5:e14227. doi: 10.1371/journal.pone.0014227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abi-Gerges A, Richter W, Lefebvre F, Mateo P, Varin A, Heymes C, Samuel JL, Lugnier C, Conti M, Fischmeister R, Vandecasteele G. Circulation research. 2009;105:784–792. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 60.Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, Thorsteinsdottir M, Hrafnsdottir S, Hagen T, Kiselyov AS, Stewart LJ, Gurney ME. Nature biotechnology. 2010;28:63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- 61.MacKenzie KF, Wallace DA, Hill EV, Anthony DF, Henderson DJ, Houslay DM, Arthur JS, Baillie GS, Houslay MD. The Biochemical journal. 2011;435:755–769. doi: 10.1042/BJ20101184. [DOI] [PubMed] [Google Scholar]
- 62.Metrich M, Morel E, Berthouze M, Pereira L, Charron P, Gomez AM, Lezoualc’h F. Pharmacological reports : PR. 2009;61:146–153. doi: 10.1016/s1734-1140(09)70017-9. [DOI] [PubMed] [Google Scholar]
- 63.Sample V, DiPilato LM, Yang JH, Ni Q, Saucerman JJ, Zhang J. Nature chemical biology. 2012;8:375–382. doi: 10.1038/nchembio.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]