

Abstract
The optical redox ratio as a measure of cellular metabolism is determined by an altered ratio between endogenous fluorophores NADH and flavin adenine dinucleotide (FAD). Although reported for other cancer sites, differences in optical redox ratio between cancerous and normal urothelial cells have not previously been reported. Here, we report a method for the detection of cellular metabolic states using flow cytometry based on autofluorescence, and a statistically significant increase in the redox ratio of bladder cancer cells compared to healthy controls. Urinary bladder cancer and normal healthy urothelial cell lines were cultured and redox overview was assessed using flow cytometry. Further localisation of fluorescence in the same cells was carried out using confocal microscopy. Multiple experiments show correlation between cell type and redox ratio, clearly differentiating between healthy cells and cancer cells. Based on our preliminary results, therefore, we believe that this data contributes to current understanding of bladder tissue fluorescence and can inform the design of endoscopic probes. This approach also has significant potential as a diagnostic tool for discrimination of cancer cells among shed urothelial cells in voided urine, and could lay the groundwork for an automated system for population screening for bladder cancer.
OCIS codes: (170.1530) Cell analysis, (110.0110) Imaging systems
1. Introduction
Bladder cancer (BCa) is amongst the 10 most common cancers worldwide and is a particular issue in ageing populations [1]. It can be categorised into muscle invasive (MIBC) and non-muscle invasive (NMIBC) disease, with the latter accounting for approximately 75% of the cases. The majority of NMIBC tumours carry a good prognosis; however, over two-thirds of them recur, with a small proportion progressing to MIBC [2]. Consequently, patients with the diagnosis of recurrent NMIBC require lifelong surveillance. Follow-up surveillance of NMIBC is conducted using cystoscopy and urinary cytology at regular intervals. The current approach creates a huge clinical workload and incurs a large financial cost for health care organisations. In the UK, BCa is estimated to be among the top five most expensive cancers to monitor and treat per patient [3]. Furthermore, the lack of sensitivity of gold standard detection techniques for low-grade cancers and Carcinoma in Situ (CIS) [4] imply that otherwise treatable and manageable bladder tumours have the opportunity to progress to more aggressive stages. It has been demonstrated that the sensitivity of WLC for bladder tumours can be as low as 62%, with particular limitations in the detection of CIS [5]. Furthermore, although tissue biopsy remains the most reliable method for tumour diagnosis, the necessity for repeat random biopsy is a source of discomfort and lay-up time for patients. Therefore, it is imperative that gold standard detection techniques which have a higher sensitivity for early-stage cancers are developed to prevent recurrence and progression of tumours, prevent patient morbidity, and reduce the costs of healthcare for patients. Photosensitising agents such as HAL, 5-ALA, and Hypericin have vastly improved sensitivity for bladder tumours, but at the expense of decreased specificity and increased cost [6, 7]. Therefore, it is imperative that gold standard detection techniques are developed which can balance sensitivity with specificity. Improvements to urinary cytology may be able to offer this opportunity. A number of non-invasive urinary biomarkers have been suggested, with a few approved by the Food and Drug Administration (FDA), USA [8]. Common biomarkers include NMP22 assay, BTA Stat, and Urovysion assays. However, none of these assays have made any significant impact on the clinical management of BCa. Moreover, NMP22 and BTA Stat assays suffer from low specificity for bladder cancer [9, 10], while UroVysion requires detailed specialist analysis, which is time consuming and can present inconclusive results in some cases. When compared to conventional voided urine cytology, these techniques show very little selective benefit.
Autofluorescence spectroscopy is an emerging technique based on the exploitation of endogenous fluorophores. A wide range of endogenous fluorophores exist within cells and tissues. These fluorophores can shed light on a number of important processes within the body, including metabolism. There exists a growing body of work on the application of autofluorescence spectroscopy to the diagnosis of a number of diseases and ailments, including many forms of cancer (bladder included) [11, 12]. Two of the most diagnostically important endogenous fluorophores are reduced Nicotinamide dinucleotide (NADH) and Flavin adenine dinucleotide (FAD), which can be used to monitor dramatic metabolic changes in cells and tissues. As tumours progress, cancer cells are often found to undergo a metabolic switch from favouring energy production through oxidative phosphorylation to energy production through aerobic glycolysis (known as the Warburg Effect). This switch has significant consequences for cancer cell physiology. Due to the fundamental makeup of the electron transport chain (ETC), with NADH as the electron donor responsible for generating the proton gradient, disrupting oxidative phosphorylation leads to an accumulation of NADH within cells [13]. It is possible to take advantage of the inherent fluorescent properties of NADH and its related metabolic co-factor FAD, which acts as an electron acceptor in the ETC, to give a measure of ETC shutdown in cells. Comparing the fluorescence intensities of NADH and FAD within cell populations enables the calculation of the Optical Redox Ratio (ORR) by dividing NADH fluorescence intensity by FAD fluorescence intensity. There already exists some evidence of the ORR being a useful tool for the differentiation of healthy and cancer breast cell lines, using fluorescence microscopy. In general, cancer cells are seen to have higher redox ratios than healthy cells owing to the increased cellular NADH resulting from glycolytic switching and the shutdown of the ETC.
In this paper, we present a comparison of the ORR of healthy and cancerous bladder cell lines using flow cytometry. Conventionally, flow cytometry is used for studying cells which have been tagged with strongly fluorescing conjugated antibodies against cell surface markers etc [14]. The fact that autofluorescence signals are removed from standard flow cytometry results suggests that they are of significant strength to warrant independent study [15]. There exists a small body of work suggesting the scientific merit of flow cytometry based on autofluorescence; however, this is a relatively unexplored field [16]. The sequential excitation of NADH and FAD in cell populations, along with appropriate data processing and gating, enables the 2D visualisation of their metabolic status in a so-called ‘redox overview’. This enables the comparison of visual information as well as numerical data, thereby allowing the calculation of average population ORRs and setting metabolic thresholds between cell populations. Our primary objective was to develop a flow cytometry setup for the detection of cellular autofluorescence and an analysis strategy to allow us to rapidly calculate the optical redox ratios of large cell populations. Based on reported literature, we hypothesise that BCa cells possess a higher optical redox ratio than healthy cells, and that we will be able to detect this difference using the technique employed in this study.
2. Materials and methods
2.1. Fluorescence spectroscopy of biomarkers
NADH and FAD were purchased as powders (Sigma-Aldrich) and prepared to 10µM solutions in sterile PBS to be studied using a ‘LAKK-M’ laser spectroscopy device (SPE- LAZMA, Moscow, Russia). A 5ml solution was added to a matte black cuvette (SPE- LAZMA, Moscow, Russia). Solutions of NADH and FAD were excited sequentially with UV (365nm) and Blue (450nm) sources to optimally excite the fluorophores. Data was visualised using custom-built software (SPE-LAZMA).
2.2 Cell culture
HUC bladder urothelial cell line (Caltag Medsystems, Whiteleaf Business Centre, Buckingham)) and 5637 BCa cell line (ATCC) were grown to confluence in 75cm2 culture flasks (Corning) with Dulbecco’s Modified Eagle Medium (Sigma-Aldrich) including 1% Penicillin-Streptomycin and 10% Fetal Bovine Serum. Cells were incubated in 5% CO1 incubator at 37°C.
Cells were prepared for study by trypsinising according to standard protocol. Cell suspension was passed through a 30µm filter into a FACS tube. Cells were suspended in PBS (1% FBS) at approximately 4 x 106/ml in all cases.
All cell lines used, and the working environment, were certified Mycoplasma free.
2.3 Flow cytometry
A BD Fortessa flow cytometer (Becton Dickinson) was used. System was cleaned and optimised prior to use, according to standard protocol. Both healthy and cancerous cells were passed through the system at “low” speed setting to set voltages and template population gates. Cell data was acquired at high speed from 10,000 events. In-built blue laser (488nm) was used at a power setting of 162V to analyse forward scatter and signal width of events. All populations of cells were plotted logarithmically with respect to forward scatter and signal width. All samples were gated identically on forward scatter and signal width to remove dead and doublet cells. Gating was based on co-ordinates
NADH and FAD were sequentially excited using UV laser (360nm) and blue laser (488nm), respectively. Excitation sources were kept as close to excitation maxima as possible for the molecules of interest (NADH Ex Max = 350nm; FAD Ex Max = 450nm). UV laser was run at a power setting of 211V, blue laser was run at a power setting of 397V. The NADH-specific signal was processed using a 450/50 bandpass filter (NADH Em Max = 450nm), and the FAD-specific signal was processed using a 530/30 bandpass filter (FAD Em Max 530nm). FCS data files containing all data were saved and transferred for analysis. Compensation was not required as all results were derived as ratios.
Cell viability was determined following flow cytometry on both cell types in suspension using trypan blue and confirmed to be over 95% for all samples.
FCS data files were analysed using FCS Express Flow Cytometry Package. Bivariate dot plots were generated on a logarithmic scale based on forward scatter and signal width of each event. An inclusive elliptical gate was drawn between co-ordinates 3.8K, 68K and 113K, 154K. The percentage of cells gated in for each sample were as follows: HUC sample 1 – 85.9%; HUC sample 2 – 68.3%; HUC sample 3 – 72%; 5637 sample 1 – 89.3%; 5637 sample 2 – 91.5%; 5637 sample 3 – 71.8%.
2.4 Confocal microscopy
BCa cells were grown in Petri dishes with a diameter of 3cm with coverslip thickness bottoms #1.5 (WillCoWells) in DMEM (10% FBS, 1% Pen-Strep), until 75% confluent. DMEM was removed from the cells and replaced with 1ml PBS (1% FBS) to minimise background fluorescence.
MitoTracker Red (Molecular Probes) was prepared to a 1mM stock solution in DMSO (Life Technologies) and diluted to 100nM solution in PBS (1% FBS). 1ml solution was added to each Petri dish. Cells were re-incubated for 45 minutes at 37°C, staining solution was removed and 1ml of pre-warmed PBS (1% FBS) was added.
A Carl Zeiss 710 confocal microscope was used. Cell sample was scanned in x, y, and z axes using a 40x objective lens to maximise resolution. NADH was excited at 350nm and FAD was excited at 488nm. Fluorescence was captured using a CCD camera with 450/20 and 530/20 bandpass filters to isolate NADH and FAD fluorescence, respectively. MitoTracker Red was excited at 579nm and detected at 599nm. A total of 50 cells were studied in this manner. The three excitation wavelengths were used sequentially and data stored for each frame.
Images of cells captured using CCD were processed using ImageJ (imageJ.nih.gov). Images from all fluorophores were merged to overlap mitochondrial fluorescence and autofluorescence.
2.5 Data analysis
Individual data on NADH and FAD data was exported from FCS Express, following gating, to Origin Pro 8 statistics software, to calculate means, medians, standard deviations, and standard errors. The average redox ratio was calculated for each population by dividing average NADH fluorescence across 400-500nm by average FAD fluorescence across 500-560nm to yield a real number between 0 and 10. Redox ratios of whole populations of cells were analysed for significant differences using student’s t test.
3. Results
3.1 NADH and FAD have specific excitation and emission profiles
NADH was found to have a fluorescence maximum at 490nm (Fig. 1(a) ), while FAD has its maximum at 550nm (Fig. 1(a) and 1(b)). NADH displays no fluorescence upon 450nm excitation (Fig. 1(b)); however, there is a significant degree of FAD fluorescence when excited at 365nm (Fig. 1(a)), which overlaps considerably with that of NADH.
Fig. 1.
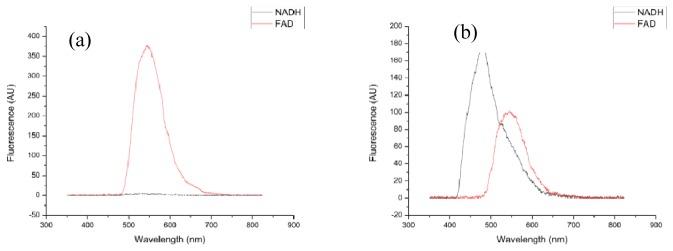
Spectra of NADH (black line) and FAD (red line) fluorescence under (a) excitation at 365nm and (b) excitation at 450nm.
3.2 Excitation and emission parameters specify mitochondrial fluorophores
Confocal microscopy was used to visualise mitochondria, the location of cellular respiration, and energy production through the ETC and, thus, the source of the vast majority of NADH and FAD. Super-imposing cellular autofluorescence signals in Fig. 2 from NADH (Green image) and FAD (Purple image) over MitoTracker red signal (Red image) enables direct analysis of the localisation of autofluorescence. Figure 2 clearly shows that autofluorescence excited and recorded at the parameters used is arising from the mitochondria.
Fig. 2.
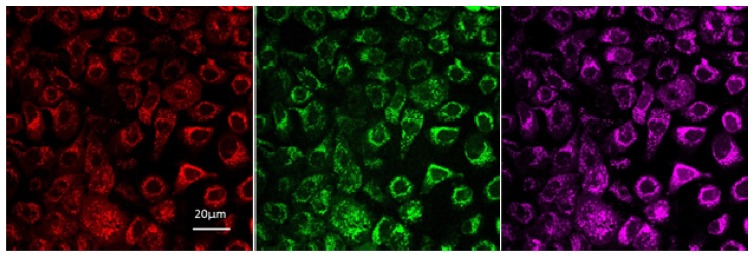
Confocal microscopy image of fluorescence arising in BCa cells from Mitotracker Red (red), NADH (green) and FAD (purple).
3.3 Cells in a population can be discriminated and plotted based on their metabolic activity
Bivariate dot plot was created with increasing NADH fluorescence intensity on the x axis and increasing FAD fluorescence intensity on the y axis. This “redox overview” gives an indication of how a population of cells is distributed metabolically. The spread of redox ratios of different populations of cells can be directly compared in this manner. Figure 3 displays the redox overviews of healthy (HUC) cells (3a), and BCa cells (3b). Healthy cells display linear distribution of redox ratios, suggesting a dependent relationship between NADH and FAD. Cancerous cells display a relationship trending towards increased NADH level.
Fig. 3.

Redox overviews of cell populations in triplicate (x = NADH, y = FAD): (a) Healthy cells; (b) Cancer cells.
3.4 Healthy bladder and bladder cancerous cells display significantly different redox ratios
Figure 4 displays the average redox ratios and standard error of the mean of healthy and cancerous cell populations (healthy cells = 0.457+/−0.022, cancerous cells = 0.588+/−0.018). The p value was calculated using student’s t test and was found to be <0.05, thereby indicating that these results are statistically significant and that we can reject the null hypothesis that cancerous cells and healthy cells have the same redox ratios.
Fig. 4.
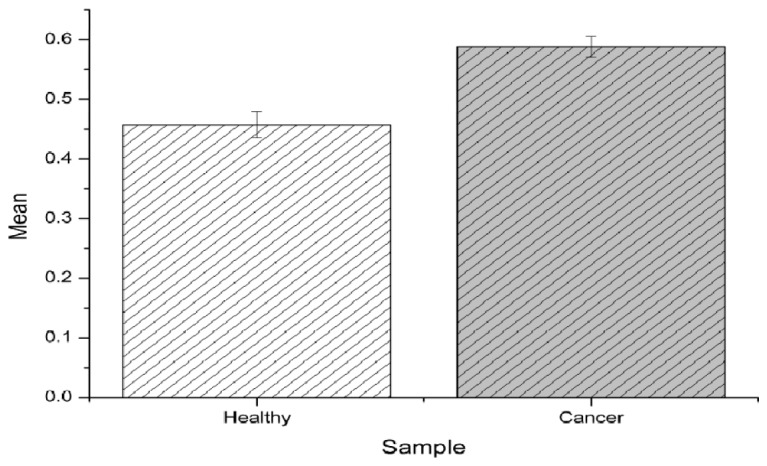
Mean values of redox ratios for “HUC” healthy control cells and “5637” BCa cells.
4. Discussion
We hypothesised that BCa cells would possess an increased optical redox ratio compared to healthy cells owing to their selective accumulation of NADH. Furthermore, we aimed to translate current methods for the calculation of optical redox ratio into an automated flow cytometry setup for rapid throughput and automatic calculation of redox ratios. We demonstrated that BCa cells possess a significantly increased redox ratio compared to healthy cells, in keeping with research on other cancers, and proved that the autofluorescence flow cytometric method is sensitive to metabolic differences among cell populations. This finding improves our understanding of the metabolic processes occurring at the cellular level in BCa. The applicability of flow cytometry to the study of cellular redox ratios suggests its worth as a diagnostic tool for the screening of BCa.
New techniques for the detection of BCa have been sought for a number of years after it became apparent that gold standard techniques were not providing adequate sensitivity for early stage disease. PDT rectified this issue to an extent, resulting in higher rates of cancer detection, but at the expense of increased cost and reduced specificity [7]. Many avenues of contemporary research are focused on endogenous fluorescence (diagnosis without the need for photosensitisers), which can yield a vast amount of information on tissue structure, inflammation, and metabolic state [17, 18].
In recent years, fluorescence spectroscopy induced by laser excitation has been used in the study of a wide range of diseases [19]. With respect to BCa, several biopsy studies have explored the endogenous fluorescence patterns of healthy and cancerous epithelia, with a view to improve the sensitivity and specificity of early detection. These studies have drawn a number of conclusions regarding discrepancies in the chemical constitution of healthy and cancerous tissue and proposed some useful algorithms for determining the presence of cancer [20]. In many types of cancer, the optical redox ratio has received much attention as a diagnostic algorithm to discriminate between healthy and cancerous cells. Glycolytic switches, which often accompany cellular transformation to malignancy, occur early in the process of tumour development. Increases in NADH can even be seen in pre-cancerous tissue [11, 21]. This suggests that NADH levels can be a powerful diagnostic marker in early stage tumours. To account for variations in size and granularity of cells, NADH is rationed against the proton acceptor FAD to measure levels of electron transport chain shutdown in cells, enabling redox ratios to be used for direct comparisons among cell types. Much of the ground- breaking work occurring in autofluorescence spectroscopy at present is concerned with studying the optical redox ratio of cell lines [22–25]. To our knowledge, the work presented in this paper is the first demonstration of an increased optical redox ratio in BCa cell lines compared to healthy control. Based on these findings, we conclude that transitional cell carcinoma of the bladder is similar to many other epithelial cancers and is associated at an early stage with a glycolytic switch, shutdown of the electron transport chain, and accumulation of NADH within cells. Our work contributes to the growing evidence of glycolytic switch and increased optical redox ratio being common to a wide range of cancers.
As tissue is a complex medium and fluorophores are known to interact with one another, diagnostic information from individual fluorophores can often be lost or misinterpreted [26]. Additionally, physiological changes such as tissue thickening or hardening may affect fluorescence measurements irrespective of fluorophore. In their study, Zheng et al. discuss the reduction in NADH and FAD fluorescence in malignant tissue as a possible result of tissue thickening [27]. Finally, the highly scattering nature of blood to many wavelengths of light often distorts photonic information in freshly resected tissue. Therefore, when studying specific fluorophores to draw conclusions, it is imperative to avoid non-specific tissue features and delineate between overlapping and interacting fluorophores. Cellular models are particularly useful for metabolic autofluorescence studies of NADH and FAD due to the lack of spectral contamination from other fluorophores or the scattering properties of blood.
A potential clinical application of these findings is in the screening of voided urine. Population-wide VUC presents the opportunity to detect up to 100% of BCa cases at an early stage, but is time consuming and not directly translatable to large populations owing to the cost [28]. Tests such as NMP22 are being considered for population screening, while other techniques are constantly being devised [29]. In many urinary cytology methodologies, flow cytometry has been suggested as a useful adjunct for the rapid throughput of large populations of cells. Dual modality flow cytometry allows the measurement of antigen expression and nuclear chromatin volumes [30]. This indicates that cell yield in voided urine (<100 cells/ml) is sufficient to run high throughput analysis on. Detection of the metabolic status of cells shed in urine could be used in conjunction with nuclear and antigenic information from cells to improve the sensitivity and specificity of cancer detection. In theory, the flow cytometric method is capable of detecting even 1 cell/ml, however the diagnostic yield is limited by dead/dying cells and doublet cells. Patient studies will allow us to determine the minimum cell number required for detection. Although originally proposed in the 1980’s [16], variation in cell size and granularity may have been an obstacle to autofluorescence flow cytometry in the past. The advent and validation of the optical redox ratio provides a method by which to account for cellular variations in size and granularity. We know from cell viability analysis that in each case, 90–95% cells studied from each experiment survived analysis. There remains a small caveat in that external environments will vary considerably from the native bladder interior, but prompt processing and analysis should preserve the cellular metabolic profile.
One of the greatest advantages of this methodology for autofluorescence analysis of cells is in the high throughput of cells, thereby enabling the generation of redox overviews and the direct comparison of different populations of cells. We demonstrated obvious differences in the redox overviews of cancerous and healthy cells, thereby suggesting that diagnostic thresholding based on this data is possible. Using the redox overview and based on the linear distribution of healthy cell populations, this technique could therefore present exquisite sensitivity for the detection of cells with aberrant metabolism (those falling below the healthy distribution line due to an increase in NADH) – theoretically, we could detect as low as 1 aberrant cell in 10,000. As stated earlier, more work is required, particularly with patient samples, to define the healthy cell redox parameters and optimize diagnostic thresholding. Healthy cells generally display higher NADH and FAD levels than cancerous cells, but this is a function of the increased size and granularity of the healthy cell line rather than an inherent difference between phenotypes. This disparity serves to underline the importance of the redox ratio; this is because if measurements were made solely on NADH levels, healthy cells would appear to contain much higher levels, thereby contradicting the actual result.
Autofluorescence flow cytometry for the detection of BCa presents a number of attractive qualities. Firstly, it is non-invasive so can be done in an out-patient clinic or GP surgery without the need for costly and time-consuming surgery. It is a rapid technique which allows the throughput of vast numbers of cells, providing an indication of full cell populations. The implementation of autofluorescence flow cytometry as a diagnostic tool largely relies on three things: the presence of sufficient, easily detectable live cells in urine, an affordable system for clinical integration, and sufficient laser power to generate autofluorescence. The first steps in validating this system for clinical application will be a study of patient-specific voided urine. Building on our current proof of concept work, the results from these studies will answer questions of specificity and sensitivity and also enable the calculation of accurate diagnostic thresholds. It would also be of worth to determine the ability of this technique to detect different stages of BCa. This is not something we have explored currently, but in theory the progressive accumulation of NADH in increasingly malignant cells could serve to even further skew the optical redox ratio as bladder tumours progress. The accuracy of autofluorescence flow cytometry for the detection of different stages of bladder cancer could be further enhanced by including additional endogenous fluorophores for analysis, such as tryptophan, tyrosine and lipofuscein. Validation of the diagnostic worth of this system will be followed by efforts to produce compact and cost-effective systems. Conventional bench-top flow cytometers are indispensable for multi-parametric (up to 16 colour) investigations of antigen expression; however, this makes the devices large, bulky, and expensive to run and maintain. Recently, one colour conventional flow cytometry has been reported using a lab-on-chip and camera from a conventional smart phone [31]. The applicability of autofluorescence flow cytometry on a small scale is likely to be aided significantly by the development of compact UV and blue diodes of sufficient optical power to excite the fluorophores, such as those included in contemporary multifunctional laser diagnostic systems, including the LAKK-M (SPE- LAZMA). Applying flow cytometric methodologies to compact diagnostic systems may enable the development of portable and affordable devices for rapid two-colour screening of cellular autofluorescence, sufficient for the calculation of metabolic overviews and clinical application of this technology.
5. Conclusion
We optimised a flow cytometry setup for the study of the mitochondrial fluorophores NADH and FAD to allow the calculation of optical redox ratios from large populations of cells. Statistically significant differences in the redox ratios of healthy and cancerous cells were observed, along with notably different population redox overviews. We hope these findings can contribute to the greater understanding of bladder cancer etiology and, with some modifications to existing instrumentation, provide the basis for a screening tool for voided urine cytology.
Acknowledgments
This work was funded by the EU FP7 project ABLADE (Advanced Bladder Cancer Diagnosis and Therapy), with further contribution from NHS and EPSRC. We acknowledge the assistance of Sam Swift and Alan Prescott, Microscopy core facility, University of Dundee, and Rosemary Clark and Arlene Whigham in the Flow Cytometry core facility, University of Dundee.
References and links
- 1.Shariat S. F., Milowsky M., Droller M. J., “Bladder cancer in the elderly,” Urol. Oncol. 27(6), 653–667 (2009). 10.1016/j.urolonc.2009.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Heijden A. G., Witjes J. A., “Recurrence, Progression, and follow-up in non-muscle-invasive bladder cancer,” Eur. Urol. Suppl. 8(7), 556–562 (2009). 10.1016/j.eursup.2009.06.010 [DOI] [Google Scholar]
- 3.Sievert K. D., Amend B., Nagele U., Schilling D., Bedke J., Horstmann M., Hennenlotter J., Kruck S., Stenzl A., “Economic aspects of bladder cancer: what are the benefits and costs?” World J. Urol. 27(3), 295–300 (2009). 10.1007/s00345-009-0395-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphrey P. A., “Urothelial carcinoma in situ of the bladder,” J. Urol. 187(3), 1057–1058 (2012). 10.1016/j.juro.2011.12.020 [DOI] [PubMed] [Google Scholar]
- 5.Jocham D., Stepp H., Waidelich R., “Photodynamic diagnosis in urology: state-of-the-art,” Eur. Urol. 53(6), 1138–1150 (2008). 10.1016/j.eururo.2007.11.048 [DOI] [PubMed] [Google Scholar]
- 6.Jichlinski P., Lovisa B., Erling C., Aymon D., van den Berg H., Wagnieres G., “Fluorescence cystoscopy. Perspective in clinical practice and research,” Urologe A 47(8), 975–977 (2008). 10.1007/s00120-008-1778-2 [DOI] [PubMed] [Google Scholar]
- 7.Mowatt G., N’Dow J., Vale L., Nabi G., Boachie C., Cook J. A., Fraser C., Griffiths T. R., Aberdeen Technology Assessment Review (TAR) Group , “Photodynamic diagnosis of bladder cancer compared with white light cystoscopy: Systematic review and meta-analysis,” Int. J. Technol. Assess. Health Care 27(1), 3–10 (2011). 10.1017/S0266462310001364 [DOI] [PubMed] [Google Scholar]
- 8.Parker J., Spiess P. E., “Current and emerging bladder cancer urinary biomarkers,” ScientificWorldJournal 11, 1103–1112 (2011). 10.1100/tsw.2011.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oge O., Atsü N., Sahin A., Ozen H., “Comparison of BTA stat and NMP22 tests in the detection of bladder cancer,” Scand. J. Urol. Nephrol. 34(6), 349–351 (2000). 10.1080/003655900455404 [DOI] [PubMed] [Google Scholar]
- 10.Hosseini J., Golshan A. R., Mazloomfard M. M., Mehrsai A. R., Zargar M. A., Ayati M., Shakeri S., Jasemi M., Kabiri M., “Detection of recurrent bladder cancer: NMP22 test or urine cytology?” Urol. J. 9(1), 367–372 (2012). [PubMed] [Google Scholar]
- 11.Georgakoudi I., Jacobson B. C., Müller M. G., Sheets E. E., Badizadegan K., Carr-Locke D. L., Crum C. P., Boone C. W., Dasari R. R., Van Dam J., Feld M. S., “NAD(P)H and collagen as in vivo quantitative fluorescent biomarkers of epithelial precancerous changes,” Cancer Res. 62(3), 682–687 (2002). [PubMed] [Google Scholar]
- 12.De Veld D. C., Witjes M. J., Sterenborg H. J., Roodenburg J. L., “The status of in vivo autofluorescence spectroscopy and imaging for oral oncology,” Oral Oncol. 41(2), 117–131 (2005). 10.1016/j.oraloncology.2004.07.007 [DOI] [PubMed] [Google Scholar]
- 13.Pelicano H., Xu R. H., Du M., Feng L., Sasaki R., Carew J. S., Hu Y., Ramdas L., Hu L., Keating M. J., Zhang W., Plunkett W., Huang P., “Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism,” J. Cell Biol. 175(6), 913–923 (2006). 10.1083/jcb.200512100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barlogie B., Raber M. N., Schumann J., Johnson T. S., Drewinko B., Swartzendruber D. E., Göhde W., Andreeff M., Freireich E. J., “Flow cytometry in clinical cancer research,” Cancer Res. 43(9), 3982–3997 (1983). [PubMed] [Google Scholar]
- 15.Mosiman V. L., Patterson B. K., Canterero L., Goolsby C. L., “Reducing cellular autofluorescence in flow cytometry: an in situ method,” Cytometry 30(3), 151–156 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Thorell B., “Flow-cytometric monitoring of intracellular flavins simultaneously with NAD(P)H levels,” Cytometry 4(1), 61–65 (1983). 10.1002/cyto.990040109 [DOI] [PubMed] [Google Scholar]
- 17.Aihara H., Tajiri H., Suzuki T., “Application of autofluorescence endoscopy for colorectal cancer screening: rationale and an update,” Gastroenterol. Res. Pract. 2012, 971383 (2012). 10.1155/2012/971383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavlova I., Williams M., El-Naggar A., Richards-Kortum R., Gillenwater A., “Understanding the biological basis of autofluorescence imaging for oral cancer detection: high-resolution fluorescence microscopy in viable tissue,” Clin. Cancer Res. 14(8), 2396–2404 (2008). 10.1158/1078-0432.CCR-07-1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramanujam N., “Fluorescence spectroscopy of neoplastic and non-neoplastic tissues,” Neoplasia 2(1-2), 89–117 (2000). 10.1038/sj.neo.7900077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koenig F., McGovern F. J., Althausen A. F., Deutsch T. F., Schomacker K. T., “Laser induced autofluorescence diagnosis of bladder cancer,” J. Urol. 156(5), 1597–1601 (1996). 10.1016/S0022-5347(01)65456-9 [DOI] [PubMed] [Google Scholar]
- 21.Skala M. C., Riching K. M., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., White J. G., Ramanujam N., “In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia,” Proc. Natl. Acad. Sci. U.S.A. 104(49), 19494–19499 (2007). 10.1073/pnas.0708425104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ostrander J. H., McMahon C. M., Lem S., Millon S. R., Brown J. Q., Seewaldt V. L., Ramanujam N., “Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status,” Cancer Res. 70(11), 4759–4766 (2010). 10.1158/0008-5472.CAN-09-2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varone A., Xylas J., Quinn K. P., Pouli D., Sridharan G., McLaughlin-Drubin M. E., Alonzo C., Lee K., Münger K., Georgakoudi I., “Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues,” Cancer Res. 74(11), 3067–3075 (2014). 10.1158/0008-5472.CAN-13-2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh A., Cook R. S., Rexer B., Arteaga C. L., Skala M. C., “Optical imaging of metabolism in HER2 overexpressing breast cancer cells,” Biomed. Opt. Express 3(1), 75–85 (2012). 10.1364/BOE.3.000075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mujat C., Greiner C., Baldwin A., Levitt J. M., Tian F., Stucenski L. A., Hunter M., Kim Y. L., Backman V., Feld M., Münger K., Georgakoudi I., “Endogenous optical biomarkers of normal and human papillomavirus immortalized epithelial cells,” Int. J. Cancer 122(2), 363–371 (2008). 10.1002/ijc.23120 [DOI] [PubMed] [Google Scholar]
- 26.Jyothikumar V., Sun Y., Periasamy A., “Investigation of tryptophan-NADH interactions in live human cells using three-photon fluorescence lifetime imaging and Förster resonance energy transfer microscopy,” J. Biomed. Opt. 18(6), 060501 (2013). 10.1117/1.JBO.18.6.060501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng W., Lau W., Cheng C., Soo K. C., Olivo M., “Optimal excitation-emission wavelengths for autofluorescence diagnosis of bladder tumors,” Int. J. Cancer 104(4), 477–481 (2003). 10.1002/ijc.10959 [DOI] [PubMed] [Google Scholar]
- 28.Larré S., Catto J. W., Cookson M. S., Messing E. M., Shariat S. F., Soloway M. S., Svatek R. S., Lotan Y., Zlotta A. R., Grossman H. B., “Screening for bladder cancer: rationale, limitations, whom to target, and perspectives,” Eur. Urol. 63(6), 1049–1058 (2013). 10.1016/j.eururo.2012.12.062 [DOI] [PubMed] [Google Scholar]
- 29.Pattari S. K., Dey P., “Urine: beyond cytology for detection of malignancy,” Diagn. Cytopathol. 27(3), 139–142 (2002). 10.1002/dc.10135 [DOI] [PubMed] [Google Scholar]
- 30.Palmeira C. A., Oliveira P. A., Seixas F., Pires M. A., Lopes C., Santos L., “DNA image cytometry in bladder cancer: state of the art,” Anticancer Res. 28(1B), 443–450 (2008). [PubMed] [Google Scholar]
- 31.Zhu H., Mavandadi S., Coskun A. F., Yaglidere O., Ozcan A., “Optofluidic fluorescent imaging cytometry on a cell phone,” Anal. Chem. 83(17), 6641–6647 (2011). 10.1021/ac201587a [DOI] [PMC free article] [PubMed] [Google Scholar]