

Abstract
During the last 20 years, it has been well established that a finely tuned, continuous crosstalk between neurons and astrocytes not only critically modulates physiological brain functions but also underlies many neurological diseases. In particular, this novel way of interpreting brain activity is markedly influencing our current knowledge of epilepsy, prompting a re-evaluation of old findings and guiding novel experimentation. Here, we review recent studies that have unraveled novel and unique contributions of astrocytes to the generation and spread of convulsive and nonconvulsive seizures and epileptiform activity. The emerging scenario advocates an overall framework in which a dynamic and reciprocal interplay among astrocytic and neuronal ensembles is fundamental for a fuller understanding of epilepsy. In turn, this offers novel astrocytic targets for the development of those really novel chemical entities for the control of convulsive and nonconvulsive seizures that have been acknowledged as a key priority in the management of epilepsy.
Keywords: glutamate, GABA, connexin, potassium, temporal lobe epilepsy, absence epilepsy
Astrocyte-Neuron Reciprocal Signaling
Being unable to generate action potentials, astrocytes were for a long time considered nonexcitable cells. However, in studies from the 1990s, a Ca2+-based form of excitability underlying long-distance communication between astrocytes in cultures was revealed by the observation that Ca2+ increases evoked in individual astrocytes by the neurotransmitter glutamate could propagate to neighboring astrocytes as an intercellular Ca2+ wave (Charles and others 1991; Cornell-Bell and others 1990; Finkbeiner 1992). This pioneering finding challenged the common belief that astrocytes merely act in the brain as neuron-supportive elements and paved the way for a new era in astrocyte research. Studies in brain slice preparations then reported that astrocytes respond to the synaptic release of glutamate (Pasti and others 1997; Porter and McCarthy 1996), GABA (Kang and others 1998), noradrenaline (Duffy and MacVicar 1995; Kulik and others 1999), and acetylcholine (Araque and others 2002; Navarrete and others 2012; Shelton and McCarthy 2000; Takata and others 2011) with complex, often repetitive cytosolic Ca2+ elevations, which primarily depend on the activation of G protein–coupled metabotropic receptors (Verkhratsky and Kettenmann 1996; Verkhratsky and others 1998) linked to phospholipase C, production of inositol 1,4,5-trisphosphate (InsP3), activation of InsP3 receptors, and finally, release of Ca2+ from intracellular Ca2+ compartments (Berridge 1993; Haydon and Carmignoto 2006; Pozzan and others 1994; Shigetomi and others 2011). Brain slice results were then validated in in vivo experiments that used imaging of fluorescence Ca2+ signals by two-photon laser scanning microscopy in anesthetized as well as awake, freely moving mice. By using this new in vivo approach, Hirase and colleagues (2004) could reveal neuronal activity–evoked Ca2+ elevations in the large majority of cortical astrocytes. Correlated Ca2+ increases between adjacent astrocytes were also detected, suggesting that in the neuron-astrocyte network, in vivo neurons can signal simultaneously to multiple astrocytes. Subsequent studies confirmed that astrocytes from different brain regions, including the olfactory bulb (Petzold and others 2008), cerebellum, and neocortex (Schummers and others 2008; Wang and others 2006), can respond to sensory stimulation with complex Ca2+ elevations. The amplitude, frequency, kinetics, and also spatial extent of astrocyte Ca2+ elevations were revealed to be strictly dependent on the pattern and strength of neuronal activity (Pasti and others 1997; Todd and others 2010), suggesting an astrocyte encoding of neuronal information (Bezzi and others 1998; Kang and others 1998; Pasti and others 1997).
These studies proved beyond a doubt that neurons “excite” astrocytes through the same signals that transfer information to postsynaptic neurons. What is most relevant for the topic of this article is that this “neuronal excitation” promotes in astrocytes either a feedback or a feedforward signal or both that can take the form of a Ca2+-dependent release of diverse neuroactive molecules, the so-called gliotransmitters, which contribute to the control of neuronal network excitability (Fig. 1). Among these are classic chemical transmitters, such as glutamate (Bezzi and others 1998; Parpura and others 1994), D-serine (Henneberger and others 2010; Zhuang and others 2010), ATP (Zhang and others 2003), (Bowser and Khakh 2004) and GABA (Kozlov and others 2006; Le Meur and others 2012), but also vasoactive agents, such as prostaglandins, and inflammatory signals, such as interleukin-1β (IL-β), high-mobility group box 1 (HMGB1), and tumor necrosis factor α (Bezzi and others 2001; Maroso and others 2010; Vezzani and others 2008). The regulated, activity-dependent release of different gliotransmitters has been shown 1) to modulate short- and long-term changes in synaptic transmission (Brockhaus and Deitmer 2002; Henneberger and others 2010; Jourdain and others 2007; Panatier and others 2006; Pascual and others 2005; Pasti and others 1997; Perea and Araque 2007; Serrano and others 2006; Shigetomi and others 2011; Zhang and others 2003), 2) to control cerebral blood flow (Haydon and Carmignoto 2006; Petzold and others 2008; Zonta and others 2003), 3) to favor neuronal synchrony in the neuronal network (Angulo and others 2004; Fellin and others 2004), and 4) to contribute to brain inflammatory and immune responses (Aronica and others 2012; Hamby and Sofroniew 2010) (Fig. 1). While our effort in the evaluation of the richness in gliotransmitter modulatory actions in the brain leads to a new understanding of the astrocyte’s role in brain function, we are also beginning to realize how deep and wide the impact of gliotransmission might be on the pathogenesis of different brain disorders, including epilepsy (Fig. 2).
Figure 1.

Regulated release of gliotransmitters that can affect epileptiform activities. (1) Classic neurotransmitters activate G protein–coupled receptors (GPCRs) and cytosolic Ca2+ elevations in astrocytes. (2) Transient receptor potentials, such as TRPA1, mediate Ca2+ influx and contribute to cytosolic Ca2+ regulation. The gliotransmitters that contribute to the regulation of neural network excitability, such as glutamate, D-serine, GABA, and ATP, are released via both Ca2+-dependent (3) and Ca2+-independent (4), nonmutually exclusive mechanisms. (5) Other signaling molecules, including inflammatory mediators, such as prostaglandins (PGs) and diverse cytokines, are also released, but through undefined mechanisms. By targeting microglia, neurons, and also astrocytes, these molecules can enhance seizure generation.
Figure 2.
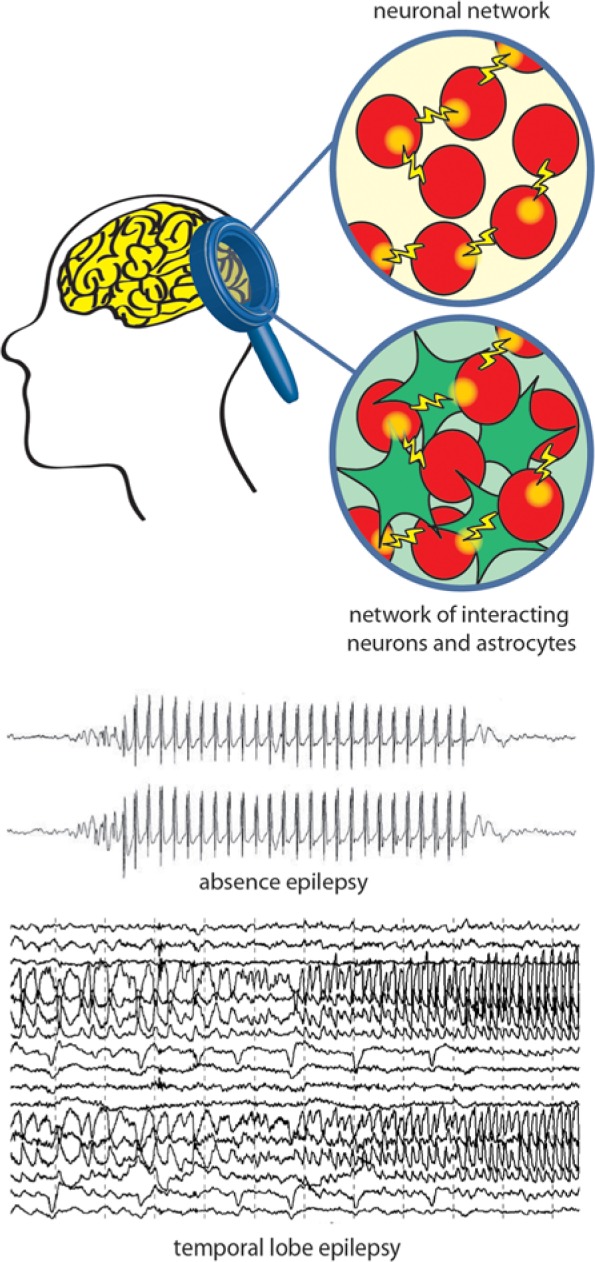
A schematic drawing of the abnormal electrical activity during epileptic seizures. Absence and temporal lobe seizures (see electroencephalogram traces) originate from malfunctions in neurons (classic view, top circle) or in the finely tuned crosstalk between astrocytic (green) and neuronal (red) networks (novel view, bottom circle).
Astrocytes and Epilepsy
Epilepsy is a neurological disease that is characterized by the periodic occurrence of spontaneous seizures, which can be convulsive or nonconvulsive in nature depending on the type of epilepsy (Noebels and others 2012). Among the most common and severe forms of convulsive epilepsies is temporal lobe epilepsy (TLE). The clinical manifestation of this disorder is represented by recurrent seizures that arise as an intense, synchronous discharge in a relatively high number of neurons from a restricted region of the medial (mTLE) or lateral temporal lobe, that is, the epileptogenic focus, and eventually generalize to both temporal lobes and extratemporal structures through a progressive recruitment of other neuronal populations (Avoli and others 2002; Cammarota and others 2013; Pinto and others 2005; Traub and Wong 1982; Trevelyan and others 2006). As we mentioned above, through the release of different gliotransmitters, astrocytes contribute to the regulation of both neuronal excitability and synaptic transmission and favor neuronal synchrony (Fellin and others 2004). The reactive gliosis revealed from neurosurgical specimens of patients presenting with mTLE may have an impact on this astrocyte signaling, leading to an increase in local network excitability and synchronous discharges that predispose neurons to generate focal epileptiform activities in TLE. In support of this view, various membrane channels, receptors, and transporters in astrocytic membranes are altered in the epileptic brain. Excitingly, recent evidence, reviewed below, suggests that in the course of the pathogenesis of mTLE, these glial changes alter homeostatic network functions, temporally precede alterations in neurons, and thus seem to be causative for the disorder.
At the other end of the clinical spectrum of epilepsies are the nonconvulsive absence seizures, which consist of sudden and brief periods of impairment of consciousness that are always accompanied by generalized and synchronous spike and wave discharges on electroencephalograms (Avoli 2012; Blumenfeld 2005; Crunelli and Leresche 2002; Panayiotopoulos 1997). Absence seizures are present, either alone or most commonly in association with convulsive seizures, in many idiopathic generalized epilepsies and are known to be generated by paroxysmal activity within cortical and thalamic networks with little or no involvement of other brain regions (Bai and others 2010; Crunelli and Leresche 2002; Vergnes and Marescaux 1992; Williams 1953). Differently from convulsive seizures, clinical evidence indicates that drugs that increase GABAergic function aggravate and induce absence seizures in patients with absence seizures and in healthy individuals, respectively (Ettinger and others 1999; Panayiotopoulos 2001; Perucca and others 1998). Thus, an increased GABAergic function may be an underlying cause of these seizures: indeed, it has been demonstrated that the enhancement of tonic GABAA receptor function in the thalamus of genetic absence epilepsy models is due to a malfunction of the astrocytic GABA transporter GAT-1 (Cope and others 2009).
In this review, we summarize the current evidence of astrocyte dysfunctions in epilepsy and discuss potential underlying mechanisms (see also Sifert and others 2006, 2010). Specifically, we discuss how reciprocal signaling between neurons and astrocytes at the level of local brain circuits may contribute to the generation of epileptogenic foci in mTLE models. We also clearly demonstrate a critical role of astrocytes in the disturbance of K+ and transmitter homeostasis, as well as GABA transporter function and cytokine regulation, and the impact that these abnormalities have on the generation of both convulsive and nonconvulsive seizures. Thus, although research on astrocytes in epilepsy is still in its infancy, these findings might eventually lead to a classification of mTLE and absence seizures as disorders of astrocyte-neuron signaling rather than neuronal diseases. Importantly, because currently available anticonvulsant drugs and therapies are insufficient to control mTLE and absence seizures in about one third of patients, ongoing research on astrocytic abnormalities in epilepsy is providing promising new astrocytic targets, which may lead to more specific antiepileptogenic therapeutic strategies.
K Channels, Gap Junctions, and Aquaporins in mTLE
K+ Uptake and K+ Spatial Buffering
Neuronal activity elicits transient increases in the extracellular potassium concentration ([K+]o), which under pathological conditions like epilepsy can reach up to 12 mM (Heinemann and Lux 1977). Even moderate rises in [K+]o increase neuronal excitability and synaptic transmission (Walz 2000), underscoring the necessity of tight control of K+ homeostasis for normal brain function. This task is mainly accomplished by astrocytes, which display very negative resting potentials due to a high resting permeability for K+. Responsible for this property are inwardly rectifying K+ channels of the Kir4.1 subtype (Seifert and others 2009) (Fig. 3). Astrocytes control [K+]o by two mechanisms: K+ uptake and spatial buffering. Net uptake of K+ is mainly mediated by Na+/K+ pumps and Na+/K+/Cl– co-transporters (Kofuji and Newman 2004). It is rather unlikely that this mechanism alone is sufficient for efficient clearance of excess [K+]o because intracellular K+ accumulation results in water influx and cell swelling. The spatial buffering model (Orkand and others 1966) describes another more effective mechanism for [K+]o clearance. It is based on the fact that astrocytes are electrically connected to each other via gap junction (GJ) channels to form a functional syncytium (Fig. 3). According to the spatial buffering model, excessive extracellular K+ is taken up by astrocytes at sites of high neuronal activity, redistributed through the astrocytic network, and released at regions of lower [K+]o. This uptake and release of K+ occur passively, driven by the respective electrochemical gradients. Kir4.1 channels are particularly well suited for this task because they possess a high open probability at rest and their conductance increases at high [K+]o.
Figure 3.

Epilepsy-associated alterations of functional properties in astrocytes. (1) Seizure activity leads to an increase in extracellular K+ concentration. Down-regulation of Kir channels was observed in astrocytes in human and experimental epilepsy. (2) Gap junctions (GJs) mediate the spatial redistribution of K+. Genetic ablation of GJs entails impaired K+ buffering and hyperactivity through neuronal depolarization-induced glutamate release and synchronization. (3) The dislocation of water channels contributes to impaired K+ buffering. (4) Astrocytes are primarily responsible for glutamate uptake. Reduction of the EAAT1 and EAAT2 proteins is observed in the human epileptic hippocampus. Elevated extracellular glutamate decreases the threshold for seizure induction. (5) Glutamate is converted into glutamine through glutamine synthetase (GS). In the human epileptic hippocampus, loss of GS resulted in elevated extracellular glutamate levels. In experimental epilepsy, down-regulation of GS was observed in the chronic phase. (6) The release of glutamate and ATP from astrocytes through hemichannels may promote hyperexcitability. Modified and reproduced with permission from Steinhäuser and Seifert (2012).
Kir4.1 Channels and K+ Buffering in Epilepsy
Increased [K+]o has been associated with the pathophysiology of epilepsy (Fisher and others 1976), and it is known that high [K+]o is sufficient to trigger epileptiform activity in vitro (Traynelis and Dingledine 1988). To assess the impact of Kir4.1 channels in K+ buffering, the effect of Ba2+-induced Kir channel block on stimulus-triggered rises in [K+]o or iontophoretically applied K+ was analyzed. In these experiments, Ba2+ enhanced [K+]o accumulation under control conditions, but not in sclerotic hippocampi, indicating the disturbance of Ba2+-sensitive K+ uptake in sclerosis (Kivi and others 2000). This hypothesis was confirmed with patch clamp analysis, demonstrating reduced Kir currents in the sclerotic CA1 region of specimens from patients with mTLE (Bordey and Sontheimer 1998; Hinterkeuser and others 2000). Western blot (Das and others 2012) and immunohistochemistry (Heuser and others 2012) also revealed a significant loss of the Kir4.1 protein in human hippocampal sclerosis (HS). These studies imply that impaired K+ clearance and increased seizure susceptibility in mTLE-HS result from the reduced expression of Kir4.1 channels. However, it remains unclear whether this reduction represents a cause, effect, or adaptive response in TLE. In favor of a causative role, David and colleagues (2009) showed in an albumin model of epilepsy that Kir4.1 down-regulation occurs before the onset of epileptic activity. However, another study performed in a kainate model found no changes in astrocytic Kir currents in post–status epilepticus (SE) (Takahashi and others 2010).
Further support for a crucial role of Kir4.1 in glial K+ buffering emerged from Kir4.1 knockout mice. Global deletion of Kir4.1 resulted in motor impairments and premature death (Neusch and others 2001). Mice with glia-specific deletion of Kir4.1 (cKir4.1–/– mice) displayed a similarly severe phenotype and demonstrated that loss of Kir4.1 causes epilepsy (Chever and others 2010; Haj-Yasein and others 2011).
Missense variations in KCNJ10, the gene encoding Kir4.1, have been linked to seizure susceptibility in man (Buono and others 2004). Loss-of-function mutations in KCNJ10 underlie an autosomal recessive disorder characterized by seizures, ataxia, sensorineural deafness, mental retardation, and tubulopathy (EAST/SeSAME syndrome) (Bockenhauer and others 2009; Scholl and others 2009). Patients suffering from this disorder display focal and generalized tonic-clonic seizures since childhood. Heuser and colleagues (2010) showed that a combination of three single nucleotide polymorphisms (SNPs) in the AQP4 gene (encoding a water channel), together with two SNPs in the KCNJ10 gene, was associated with mTLE. Association analysis in patients with a history of febrile seizures (mTLE-FS) revealed that a combination of SNPs in KCNJ10, AQP4, and the area between KCNJ10 and KCNJ9 was associated with mTLE-FS (Heuser and others 2012).
Potential Roles of GJs in Epilepsy
Astrocytes in the adult brain are connected to each other via GJ channels (Fig. 3) composed of connexin 43 (Cx43) and Cx30 (Nagy and Rash 2000), allowing the intercellular exchange of ions, second messengers, metabolites, and amino acids. The astroglial syncytium has important functions, including spatial buffering of K+ ions(see above), delivery of energetic metabolites to neurons (Giaume and others 1997), intercellular propagation of Ca2+ waves (Scemes and Giaume 2006), volume regulation (Scemes and Spray 1998), and adult neurogenesis (Kunze and others 2009).
The role of interastrocytic coupling in the development and progression of epilepsy is still controversial (Carlen 2012; Nemani and Binder 2005; Steinhäuser and others 2012). According to the spatial buffering concept (see above), the astroglial network is expected to possess antiepileptic function because a reduction of astrocytic coupling would result in the accumulation of extracellular K+, neuronal depolarization, and a lowered threshold for seizure generation. In line with this hypothesis are results from mice with coupling-deficient astrocytes. In these mice, clearance of K+ and glutamate was disturbed. They displayed spontaneous epileptiform events, a reduced threshold for the generation of epileptic activity, and increased synaptic transmission (Pannasch and others 2011; Wallraff and others 2006). Although these findings strongly support an anticonvulsive role of glial GJ networks, a potential seizure-promoting role emerged from the results by Rouach and colleagues (2008), who demonstrated that astroglial GJs mediate activity-dependent intercellular trafficking of metabolites from blood vessels to sites of high energy demand. The trafficking seemed to be essential for the maintenance of synaptic activity under pathological conditions such as epilepsy. The involvement of GJ channels in the intercellular spread of Ca2+ waves also would favor a proconvulsive role of the syncytium through neuronal synchronization and spread of ictal activity (Gomez-Gonzalo and others 2010). Thus, astroglial GJ networks might play a dual, proepileptic and antiepileptic, role in epilepsy. Future work will elucidate which of the mechanisms prevail under various circumstances.
Cx Expression and Coupling in Epileptic Tissue
Seizure-induced changes in Cx expression have been observed in a variety of animal models and human tissue. The results are conflicting and do not allow us to draw definitive conclusions. In experimental epilepsy, increased (Condorelli and others 2002; Gajda and others 2003; Mylvaganam and others 2010; Samoilova and others 2003; Szente and others 2002; Takahashi and others 2010), unchanged (Khurgel and Ivy 1996; Li and others 2001; Söhl and others 2000; Xu and others 2009), and decreased (David and others 2009; Elisevich and others 1997a, 1998; Xu and others 2009) Cx43 and/or Cx30 transcript and/or protein have been reported. This inconsistency might be explained by differences between animal models, seizure duration, and investigated brain areas. In human specimens, mostly up-regulation of the Cx43 transcript and/or protein has been described (Aronica and others 2001; Collignon and others 2006; Fonseca and others 2002; Naus and others 1991), although unchanged levels have also been reported (Elisevich and others 1997b). However, Cx expression does not necessarily reflect functional coupling because posttranslational modifications may alter unitary conductance, open probability, trafficking, or internalization. Hence, functional coupling analyses are indispensable. Increased astrocytic coupling has been reported by Takahashi and colleagues (2010) in a post-SE model and by Samoilova and colleagues (2003) in hippocampal slice cultures chronically exposed to bicuculline. In contrast, Xu and colleagues (2009) observed reduced coupling in the hippocampal CA1 region in a model of tuberous sclerosis complex. Human coupling studies have so far only been performed on astrocyte cultures derived from epileptic specimens (Lee and others 1995) in which enhanced coupling was found with fluorescence recovery after photobleaching.
Another approach to assess the role of GJ channels in epilepsy is the pharmacological inhibition of interastrocytic communication. Such experiments have been performed in a variety of in vivo and in vitro models of epilepsy (Bostanci and Bagirici 2006, 2007; Gajda and others 2003; Jahromi and others 2002; Kohling and others 2001; Medina-Ceja and others 2008; Perez-Velazquez and others 1994; Ross and others 2000; Samoilova and others 2003, 2008; Szente and others 2002; Voss and others 2009). Most of these studies reported aniticonvulsive effects of GJ blockade, although opposite effects were observed by Voss and colleagues (2009). In neocortical slices from patients with mTLE or focal cortical dysplasia, GJ inhibitors attenuated epileptiform activity (Gigout and others 2006). Major problems with GJ blockers are their significant side effects and poor Cx isoform specificity.
In conclusion, Cx expression studies and functional coupling analyses yield an inconsistent picture of the role of the astroglial network in the pathophysiology of epilepsy. Further work is needed to clarify this issue.
Role of Cx Hemichannels in Epilepsy
In addition to intercellular communication, functional membrane-spanning Cx hemichannels (HCs) have been demonstrated in astrocytes (Fig. 3). These channels are nonselective and permeable for large molecules, such as ATP, glutamate, glucose, and glutathione. Under normal conditions, these channels are closed, but the open probability increases upon depolarization, altered intracellular and extracellular Ca2+ concentration, metabolic inhibition, or proinflammatory cytokines (Orellana and others 2013; Theis and Giaume 2012). Activated HCs might promote neuronal hyperactivity and hypersynchronization through the excessive release of ATP and glutamate, which in turn increases excitability and Ca2+ wave propagation (Bedner and Steinhäuser 2013). Furthermore, GJ channels and HCs are oppositely regulated by proinflammatory cytokines (Meme and others 2006; Retamal and others 2007) and may play differential roles in epilepsy. In hippocampal slice cultures exposed to bicuculline, Yoon and colleagues (2010) showed that selective inhibition of HCs by low concentrations of mimetic peptides had protective effects, while blockade of both HCs and GJs by high doses of the peptide exacerbated lesions.
Role of Aquaporin-4 in Regulation of Excitability
Aquaporins mediate transmembrane water movements in response to osmotic gradients. Among the isoforms identified so far, aquaporin-4 (AQP4) is the predominant water channel in the brain, where it is mainly localized to astrocyte perivascular end-feet and perisynaptic processes (Papadopoulos and Verkman 2013) (Fig. 3). Moreover, AQP4 is implicated in the pathogenesis of epilepsy mainly due to its role in regulating extracellular fluid osmolarity and extracellular space (ECS) volume (Schwartzkroin and others 1998). Osmolarity-induced shrinkage of the ECS causes hyperexcitability (Chebabo and others 1995; Dudek and others 1990; Pan and Stringer 1996; Roper and others 1992), while increasing the ECS volume fraction attenuates epileptiform activity (Dudek and others 1990; Haglund and Hochman 2005; Pan and Stringer 1996; Traynelis and Dingledine 1989). In addition to ECS regulation, the spatial overlap of AQP4 with Kir4.1 in glial end-feet gave rise to the hypothesis that AQP4 may be involved in K+ homeostasis (Binder and others 2012).
Insight into the role of AQP4 in ECS regulation, K+ clearance, and excitability came from transgenic mice: AQP4-deficient (AQP4–/–) mice display mild ECS volume expansion as assessed by Fluorescence Recovery After Photobleaching (FRAP) and theTetraMethylAmmonium+ (TMA+) method (Binder and others 2004b; Yao and others 2008). In acute seizure models, AQP4–/– mice exhibited an elevated seizure threshold but prolonged seizure duration (Binder and others 2004a; Lee and others 2012). In addition, in vivo and in situ studies using K+-sensitive electrodes or a fluorescent K+ sensor revealed impaired stimulus-induced [K+]o clearance in AQP4-deficient mice (Binder and Steinhäuser 2006). Interestingly, K+ spatial buffering was enhanced in AQP4–/– mice probably due to improved GJ coupling (Strohschein and others 2011). The expanded ECS volume found in AQP4-deficient mice offers an explanation for the elevated seizure threshold of these mice, while the impaired K+ uptake might account for the prolonged duration and increased frequency of seizures. However, the mechanistic link between AQP4 expression and K+ homeostasis has not been resolved yet. The view of a functional interaction between AQP4 and Kir4.1 (Nagelhus and others 1999) is not supported by studies showing AQP4-independent Kir4.1 function (Ruiz-Ederra and others 2007; Strohschein and others 2011; Zhang and Verkman 2008). Rather, astrocytic K+ uptake during neuronal activity might trigger AQP4-dependent water uptake and a reduction of the ECS. In turn, ECS shrinkage might increase [K+]o and consequently further K+ uptake by astrocytes (Jin and others 2013; Papadopoulos and Verkman 2013).
AQP4 Expression and Regulation in Epileptic Tissue
Ultimately, AQP4 expression was investigated in hippocampi of patients with mTLE using reverse trascrition-PCR, immunohistochemistry, and gene chip analysis (Lee and others 2004). The authors found enhanced AQP4 levels in HS but reduced expression of the dystrophin gene, which encodes the protein that is involved in anchoring AQP4 in perivascular end-feet, and speculated that polarity in astrocytic AQP4 distribution got lost. This hypothesis was subsequently confirmed with immunogold electron microscopy and Western blot analysis (Eid and others 2005). This locally restricted reduction of AQP4 was accompanied by a loss of perivascular dystrophin, indicating that AQP4 mislocalization was caused by a disrupted dystrophin complex. Similar losses of perivascular AQP4 and dystrophin were found in tissue from patients with focal cortical dysplasia (Medici and others 2011). A further indication for the involvement of AQP4 in epilepsy and for the proposed interplay between AQP4 and Kir4.1 comes from genetic studies showing that several SNPs in the KCNJ10 and AQP4 genes are associated with mTLE (Heuser and others 2010) (see also below).
In a recent study, Alvestad and colleagues (2013) explored in a kainate model whether the loss of perivascular AQP4 found in human mTLE is involved in epileptogenesis or merely represents a consequence of the condition. They demonstrated that AQP4 mislocalization precedes the chronic phase of epilepsy, suggesting that astrocytic dysfunction is of pathophysiological relevance.
Glutamate: Transporters and Degradation
Extracellular Glutamate Levels in TLE
The rapid clearance of excessive glutamate from the ECS and the recycling of glutamate are important processes for brain function. To prevent excitotoxic accumulation in the ECS, glutamate is taken up by astrocytes via transporters, converted to glutamine by the enzyme glutamine synthetase (GS), and shuttled back to neurons for resynthesis of glutamate (Fig. 3). Dysfunction of glutamate metabolism seems to be critically involved in the pathophysiology of epilepsy (Coulter and Eid 2012). Accordingly, glutamate and its analogs cause seizures and neuronal loss in experimental epilepsy (Ben-Ari 1985; Fremeau and others 2002; Nadler and Cuthbertson 1980; Olney and others 1972). Increased extracellular glutamate levels were found in the hippocampi of patients with mTLE (Cavus and others 2005; During and Spencer 1993). Patients with HS displayed the highest interictal glutamate levels (Petroff and others 2003). However, the source of glutamate in HS is obscure because one of the hallmarks of this pathology is the loss of glutamatergic neurons in the hippocampal CA1 region.
Astrocytic Glutamate Uptake in Epilepsy
Uptake of glutamate from the ECS is driven by the electrochemical gradients. Five transporter isoforms have been identified, and two of them, GLAST (EAAT1) and GLT1 (EAAT2), are preferentially expressed in astrocytes (Fig. 3). The impact of astrocytic glutamate uptake became obvious from mice devoid of transporter proteins. GLT1 knockout mice displayed enhanced seizure susceptibility (Tanaka and others 1997). The pharmacological inhibition of GLT1 in the rat neocortex reduced the threshold for evoking epileptiform activity (Campbell and Hablitz 2004; Demarque and others 2004). In contrast, knockdown of GLT1 caused increased extracellular glutamate, but not seizures (Rothstein and others 1996). Mice deficient in GLAST showed no spontaneous seizures, but amygdala kindling or provoked seizures were of longer duration, were more severe, and occurred after a shorter latency (Watanabe and others 1999).
In patients with mTLE, data on the regulation of GLT1 and GLAST are inconsistent. Decreased GLT1 and GLAST levels were found by Mathern and colleagues (1999) and Proper and colleagues (2002), while Tessler and colleagues (1999) and Eid and colleagues (2004) reported no changes. In kindled rats, unchanged GLT1 and GLAST levels were described (Akbar and others 1997; Miller and others 1997; Simantov and others 1999), while decreased levels were found in pilocarpine (Lopes and others 2013) and albumin models (David and others 2009) as well as in tuberous sclerosis (Wong and others 2003). Finally, Guo and colleagues (2010) observed decreased GLAST but unaffected GLT1 expression in the hippocampus of spontaneously epileptic rats.
Together, these studies indicate that astrocytic glutamate uptake plays a crucial role in protecting neurons from hyperexcitability. However, how exactly this mechanism is disturbed in epilepsy is still under discussion.
Regulation of GS in Epilepsy
In the CNS, GS is predominantly expressed by astrocytes, where it converts glutamate and ammonia to glutamine (Fig. 3). Impaired GS activity plays a crucial role in the pathogenesis of mTLE (Eid and others 2013b). Thus, intrahippocampal infusion of a GS inhibitor caused recurrent seizures and neuropathological changes similar to human mTLE (Eid and others 2008; Perez and others 2012; Wang and others 2009). Moreover, decreased GS protein and enzyme activities, probably due to posttranscriptional modification (Eid and others 2013a), have been found in patients with mTLE-HS (Eid and others 2004; van der Hel and others 2005) (Fig. 4). Finally, mutations in the GS encoding gene, GLUL, are associated with reduced GS activity and seizures (Haberle and others 2005, 2006, 2011). One argument against a causative role for GS dysfunction in epileptogenesis, however, arises from the study by Hammer and colleagues (2008), who in a kainate model confirmed decreased GS proteins in the chronic phase but reported increased GS levels in the latent period (prior to seizure onset). The link between GS down-regulation and seizure development is based on the assumption that GS deficiency causes intracellular glutamate accumulation. This accumulation, in turn, slows down glutamate uptake, leading to increased glutamate levels in the ECS. Indeed, GS inhibition entails glutamate accumulation in hippocampal astrocytes (Perez and others 2012).
Figure 4.

Decreased glutamine synthetase (GS) immunoreactivity in the CA1 region of patients with medial temporal lobe epilepsy (mTLE). There is a dense and even distribution of GS-positive cells in the subiculum and CA1 region of autopsy (A) and non-mTLE hippocampi (D). High-power fields of the subiculum in autopsy (B) and non-mTLE (E) hippocampi show that staining is confined to astrocytes. High-power fields of the CA1 area in autopsy (C) and non-mTLE (F) hippocampi also show many GS-positive astrocytes. In the mTLE hippocampus (G), there are many GS-positive cells in the subiculum, but the CA1 area is severely deficient in GS staining. High-power view of the subiculum in G (H) confirms the presence of staining in astrocytes, which have fewer processes than GS-positive astrocytes in the corresponding areas of autopsy (B) and non-mTLE hippocampi (E). (I) High-power view of the CA1 area in G confirms the lack of GS staining in this region. Specificity controls with GS antiserum (J) and preimmune serum (K) on adjacent sections of the non-mTLE hippocampus shown in D to F reveal no staining in K. DG = dentate gyrus. Modified and reproduced with permission from Eid and others (2004).
Interruption of the glutamate-glutamine cycle through GS inhibition impairs inhibitory GABAergic transmission (Liang and others 2006) but has little effect on excitatory synaptic function (Kam and Nicoll 2007). The consequences of this relationship have been studied in a model of astrocytic gliosis with GS down-regulation, which produced a deficit in inhibitory, but not excitatory, synaptic transmission. Employing voltage-sensitive dye imaging, the authors showed that these inhibitory deficits entail network hyperexcitability, which could partially be reversed by exogenously supplied glutamine. These data emphasize the importance of proper GS function for inhibitory neurotransmission and the prevention of hyperexcitability (Ortinski and others 2010).
Astrocyte Ca2+ Signaling, Gliotransmitter Release, and Epileptiform Discharges in TLE
As a consequence of Ca2+ elevations, astrocytes can release gliotransmitters that differently modulate neuronal excitability and synaptic transmission. Some of these gliotransmitters, such as glutamate, D-serine, and ATP, have been proposed to be released through a Ca2+-dependent mechanism that involves vesicle exocytosis (Bezzi and others 2004; Martineau and others 2013; Parpura and Zorec 2010), lysosome exocytosis (Li and others 2008; Zhang and others 2007), or permeation through ion channel openings upon intracellular Ca2+ elevations (Takano and others 2005; Woo and others 2012) (Fig. 1). Gliotransmitter Ca2+-independent release mechanisms have been also identified and rely on the opening of large pore channels, such as anion channels and P2X7 receptors, or the reversal operation of glutamate transporters (Fellin and Carmignoto 2004; Parpura and others 2004). While we cannot exclude that Ca2+-dependent and Ca2+-independent gliotransmitter release mechanisms coexist and are operative under different conditions, from the presence of astrocyte Ca2+ elevations, we can infer that gliotransmitter release is operative in these cells. Below, we discuss recent studies that described an enhancement of Ca2+ signals in astrocytes and a synchronization of neuronal activities by gliotransmitter release during epileptiform activity.
Astrocyte Ca2+ Signals
If astrocytes contribute to the generation of seizures, it is expected that not only neurons but also astrocytes are hyperactive in epileptic brain circuits. Recent studies provided evidence that during epileptiform activities, Ca2+ signaling in astrocytes is indeed enhanced. For example, in rat hippocampal slices in which network excitability was enhanced by reducing external Mg2+ and by blocking inhibitory synapses with the GABAA receptor antagonist picrotoxin, both the number of astrocytes displaying Ca2+ oscillations and the frequency of Ca2+ peaks were significantly increased (Fellin and others 2006). The increased Ca2+ oscillation frequency correlated with an increased frequency of the slow inward currents (SICs) observed under these conditions in pyramidal neurons. In vivo studies also showed that epileptiform activities induced by various pharmacological treatments were associated with an increased frequency of Ca2+ oscillations in cortical astrocytes (Ding and others 2007; Tian and others 2005). Interestingly, in the same in vivo models of epilepsy, the increase in Ca2+ oscillation frequency was reduced by the systemic administration of anticonvulsant drugs, such as valproate, gabapentin, and phenytoin (Tian and others 2005). Consistent with an increased activity of astrocytes in the epileptic brain, the astrocytic expression of the metabotropic glutamate receptors (mGluRs), which mediates the Ca2+ response of astrocytes to synaptically released glutamate, is increased in animal models of TLE (Aronica and others 2000; Ulas and others 2000).
Altogether, these data indicate that a significant correlation exists between an increase in astrocyte Ca2+ oscillations and the emergence of epileptiform activities and suggest that astrocytes may contribute to the generation of epileptic discharges.
Gliotransmitter Release
An experimental observation that provided further support to the potential role of astrocytes and Ca2+-dependent gliotransmission in the generation of epileptiform activities was obtained in young rat hippocampal slices (Fellin and Carmignoto 2004). In these experiments, Ca2+ elevations evoked in astrocytes by various stimuli that activated G protein–linked receptors, including the synaptic release of glutamate induced by Schaffer collateral stimulation, were followed by the generation in pyramidal CA1 neurons by SICs (Fellin and others 2004). These events appear to originate from a Ca2+-dependent release of glutamate from astrocytes as they were insensitive to the block of action potential–dependent glutamate release from neurons by tetrodotoxin (TTX); could be induced by photolysis of a Ca2+-caged compound loaded into individual astrocytes; and were mediated by extrasynaptic, high-affinity N-methyl D-aspartate receptor 2B (NR2B)–containing N-methyl-D-aspartate receptors (NMDARs) (Fellin and Carmignoto 2004). Similar events have been subsequently observed in different brain regions, including the thalamus (Parri and others 2001), nucleus accumbens (D’Ascenzo and others 2007; Fellin and others 2007), olfactory bulb (Kozlov and others 2006), brain stem (Reyes-Haro and others 2010), spinal cord (Bardoni and others 2010), and neocortex (Ding and others 2007). In relation to a potential role of astrocytic glutamate in epileptiform activity, it is worth stressing here that SICs can occur with a high level of synchrony among two nearby pyramidal neurons (Angulo and others 2004; Fellin and Carmignoto 2004). Given that excessive neuronal synchronization in local networks is one of the hallmarks of epileptic disorders, SICs may be involved in the generation of epileptiform activity. In addition, the observation that SICs can depolarize the neuronal membrane to the action potential discharge threshold (Fellin and others 2006; Pirttimaki and others 2011) suggests that astrocytic glutamate, initially acting on one or two neurons, may enlarge its action to synaptically connected neurons and promote focal epileptiform discharges by recruiting large neuronal populations into synchronous bursts. Consistent with this hypothesis, confocal microscope Ca2+ imaging experiments, which allow us to evaluate neuronal network activities by monitoring simultaneously the Ca2+ signal dynamics from tens of neurons, revealed that astrocytic glutamate triggers simultaneous NMDA receptor–mediated Ca2+ increases in groups of CA1 neurons, an event that was termed a “domain response” (Fellin and others 2004). It is important to underline that the proconvulsant action of astrocytic glutamate may have more impact in regions that are rich with recurrent axon collaterals, such as the hippocampus and other limbic structures, where it may represent a nonneuronal mechanism that favors neuronal synchrony (Carmignoto and Fellin 2006).
The hypothesis that the activation-induced glutamate release from astrocytes is directly involved in the generation of epileptic events was specifically investigated in a number of in vitro and in vivo studies. A study by Tian and colleagues (2005), using different slice models of chemically induced epilepsy, proposed the paroxysmal depolarizing shifts (PDSs) that characterize interictal epileptiform events to correspond in large part to the depolarizing events generated by SICs and thus result from the release of glutamate from astrocytes, which were strongly activated under these experimental conditions. In favor of an astrocytic origin of PDSs, the authors showed that a large majority of these events were insensitive to action potential inhibition by the Na+ channel blocker TTX. These observations were, however, at variance with results obtained in a number of studies reporting a sensitivity to TTX of interictal discharges (Fellin and others 2006; Gomez-Gonzalo and others 2010; Perreault and Avoli 1991; Stasheff and others 1993). Also, differences in the sensitivity of PDSs and SICs to NMDA receptor blockers—that is, D-AP5 abolished SICs, but not PDSs—render unlikely that PDSs coincide with astrocyte-mediated SICs. It thus appears that PDSs and SICs share similar kinetics but are distinct events generated by two different cellular sources (D’Ambrosio 2006; Wetherington and others 2008). Subsequent studies clarified that astrocyte Ca2+ signals leading to glutamate release cooperate with neurons in the generation of an epileptogenic focus rather than represent a direct cause of the epileptic discharge. In a young rat cortical slice model of TLE, Gomez-Gonzalo and colleagues (2010) showed that local NMDA applications to entorhinal cortex layer V to VI neurons, in the presence of the proconvulsant 4-aminopyridine and low external Mg2+, can trigger focal seizure–like discharges. The authors observed that two subsequent, but not single, NMDA pulse stimulations evoked both a synchronous and intense firing discharge in a small group of neurons and early Ca2+ elevations in astrocytes. This local response was regularly followed by a seizure-like event that emerged at the focus with a few seconds’ delay and later propagated to distant brain regions. The early Ca2+ elevation in astrocytes represented a central event in NMDA-induced seizure generation. Indeed, after a drastic reduction of the Ca2+ increase by the Ca2+ chelator 1,2-bis(o-amino-phenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA), by patching individual astrocytes with a BAPTA-containing pipette that allows the spreading of BAPTA throughout the astrocyte syncytium, double NMDA stimulation, which normally triggered a seizure discharge, became unsuccessful. Importantly, weak NMDA stimulation, composed of a single NMDA pulse that failed to trigger both Ca2+ signals in astrocytes and seizures, became successful when it was coupled with a stimulus that triggered Ca2+ elevations in astrocytes, such as L- Threonyl- L- phenylalanyl- L- leucyl- L- leucyl- L-argininamide (TFLLR), that is, a peptide that activates thrombin protease-activated receptor 1. Of note, this receptor is highly, if not selectively, expressed in astrocytes of the entorhinal cortex, and it is known to mediate glutamate release in these cells (Fellin and others 2004). As a whole, these data suggest that astrocytic Ca2+ signals induced in astrocytes by neurons through NMDA applications evoke the release of gliotransmitters, such as glutamate and possibly D-serine, which enhanced NMDA receptor activation. This, in turn, leads to the recruitment of a critical mass of neurons that, in turn, generates a focal seizure discharge (Gomez-Gonzalo and others 2010).
The initiation of a seizure discharge at the epileptogenic focus may thus be due to the intense firing of a critical mass of neurons in a recruitment process that involves an excitatory loop between neurons and astrocytes. When astrocytes are consistently engaged by an episode of hyperactivity in a group of neurons, they signal back by a Ca2+-dependent gliotransmitter activity that increases the basic excitability of brain microcircuits. If this feedback signal operates on a brain network that is abnormally hyperactive as a consequence of trauma, high temperature, or genetic defects, it will contribute to drive neurons towards the seizure discharge threshold.
A recent study in Drosophila provides further support to this conclusion. The study by Melom and Littleton (2013) reported that in the Drosophila brain, a mutation of the glial-specific K+-dependent Na+/Ca2+ exchanger in brain glia leads to a series of defective features in these cells, including an increase in Ca2+ basal levels and a suppression of microdomain Ca2+ oscillations that are normally observed in controls. Most importantly, these zyd mutants exhibit an enhancement of seizure susceptibility to increased temperature. Given that these glial cells form in the Drosophila brain a honeycomb network, in a fashion similar to the mosaic distribution of astrocytes in the mammalian brain, and also that they encapsulate individual neuronal somata similarly to the typical anatomic features of astrocytic processes, these glial cells are in a privileged position to exchange with neurons’ functional signals. The authors indeed suggested that in zyd mutants, glia exhibiting high Ca2+ basal levels respond to a stressful environmental stimulus with an additional Ca2+ increase that, through glia-to-neuron signaling, initiates a seizure-inducing process. Consistent with the role of glia Ca2+ elevations in seizure generation, they induced a selective expression of the heat-activated TRPA1 channels in glia and observed a strong, immobilizing seizure within seconds of TRPA1 stimulation by a temperature shift to 30°C. Although it remains to be fully investigated how glia signal to neurons in this model, this study provides further support for a direct role of glia in seizure generation.
In addition to enhancing the excitability of single neurons and favoring neuronal synchronies, the release of glutamate from astrocytes contributes to the delayed neuronal death that characterizes chronic epileptic conditions (Ding and others 2007). During SE, astrocytes are highly activated, and the consequent glutamate release can trigger neuronal damage through an excessive activation of NR2B-containing NMDA receptors. The inhibition of astrocyte Ca2+ signals by the mGluR antagonist 2-Methyl-6-(phenylethynyl)pyridine (MPEP) as well as the block by ifenprodil of extrasynaptic 2B subunit–containing NMDA receptors, that is, the neuronal targets of astrocytic glutamate, reduce the delayed neuronal death that characterizes this pathological condition (Ding and others 2007). Consistent with the role of extrasynaptic NR2B-containing NMDA receptors in cell death during SE is the finding that activation of this receptor subtype results in mitochondrial dysfunction and stimulates both dephosphorylation of the cyclic adenosine monophosphate response element binding protein and neuronal death following hypoxic ischemic insults or head injuries (Hardingham and others 2002).
Role of Astrocytes in Absence Seizures
Astrocytic GABA Transporters
Classically, epilepsy is viewed as originating either from enhanced glutamatergic transmission, decreased GABAergic transmission, or both. Thus, the mutations in GABAA receptor genes of patients with absence seizures (Kananura and others 2002; Lachance-Touchette and others 2011; Maljevic and others 2006; Macdonald and others 2010; Wallace and others 2001) have always been interpreted as leading to a widespread loss of function in GABAAR-mediated synaptic transmission. However, in transgenic mice carrying one of this human GABAAR point mutations (i.e., R43Q in the γ subunit) (Wallace and others 2001), abnormalities in GABAergic transmission (i.e., decreased inhibitory postsynaptic current [IPSC] frequency) are present in cortical but not in thalamic reticular or thalamocortical neurons (Tan and others 2007). Indeed, many pieces of independent evidence indicate that in thalamocortical neurons of sensory thalamic nuclei of many absence epilepsy models, the GABAergic function is not decreased but is either increased or unchanged. In particular, 1) in Genetic Absence Epilepsy Rats from Strasbourg (GAERS), a well-established genetic absence model (Danober and others 1998), thalamocortical neurons show rhythmic bursts of GABAA inhibitory postsynaptic potentials (IPSPs) during absence seizures (Pinault and others 1998); 2) the thalamic injection of penicillin (Kostopoulos 2000) or bicuculline (Steriade and Contreras 1995) fails to elicit absence seizures; 3) in many mouse models, there is no change or an increased phasic GABAAR-mediated inhibition (i.e., IPSPs or IPSCs) in thalamocortical neurons compared to their respective nonepileptic control strains (Bessaih and others 2006; Caddick and others 1999; Cope and others 2009; Tan and others 2008); 4) GABA levels in the thalamus of GAERS are higher than in the nonepileptic control strain (Richards and others 1995); and 5) GABABR agonists induce absence seizures in naïve animals and aggravate them in different models of this nonconvulsive epilepsy (Aizawa and others 1997; Danober and others 1998; Snead 1992). Importantly, drugs that increase GABA levels, that is, vigabatrin and tiagabine, induce absence seizures in animals and humans as well as aggravate them in animal models of, and in patients suffering from, absence epilepsy (Danober and others 1998; Ettinger and others 1999; Hosford and Wang 1997; Panayiotopoulos 2001; Perucca and others 1998).
In line with these findings, tonic GABAA inhibition in thalamocortical neurons is increased in mouse and rat genetic and pharmacological models of absence epilepsy, including GAERS, stargazer, and lethargic and succinic semialdehyde dehydrogenase knockout animals as well as in the γ-hydroxybutyric acid (GHB) and 4,5,6,7-tetrahydroisoxazolo-[5,4-C]pyridine-3-ol (THIP) models (Cope and others 2009; Errington and others 2011b). Moreover, absence seizures in GAERS are blocked by the thalamic injection of a GABAAR δ subunit–specific antisense oligodeoxynucleotide and cannot be induced in GABAAR δ-subunit knockout mice (Cope and others 2009). Together, these data suggest a potential therapeutic role for inverse agonists at perisynaptic/extrasynaptic δ-GABAARs in absence epilepsy (Errington and others 2011a).
The increased tonic GABA-A current in thalamocortical neurons of genetic mouse and rat models does not result from an enhanced activity of perisynaptic/extrasynaptic δ subunit–containing GABAARs, but it is due to a loss of function of one of the GABA transporters, GAT-1 (Cope and others 2009) (Fig. 5), which in the thalamus of both humans and rodents is exclusively located in astrocytes (Borden 1996; De Biasi and others 1998; Pow and others 2005). This conclusion is based both on indirect evidence (i.e., measurements of the tonic GABAA current in GAERS and stargazer mice) (Cope and others 2009) and direct evidence showing that the GABA transporter current measured from patch-clamped astrocytes in GAERS thalamic slices is not affected by NO-711 (a selective blocker of GAT-1) but is abolished by SNAP5114, a selective blocker of GAT-3 (Pirttimaki and others 2012).
Figure 5.
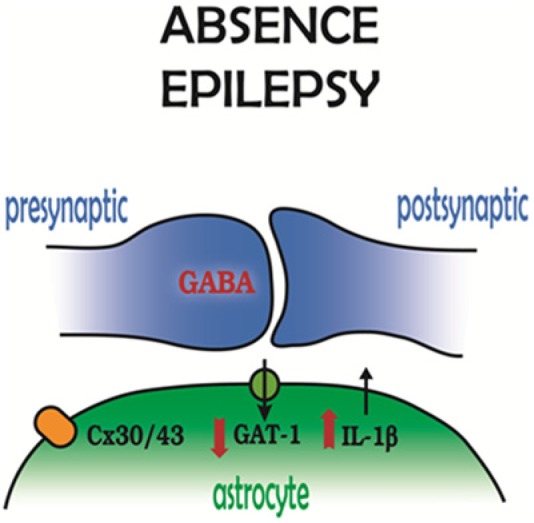
Astrocytic molecular players in absence epilepsy. GABAergic synapses in the sensory thalamus show a decreased function of the astrocytic GABA transporter GAT-1, while in the somatosensory cortex, interleukin-1β levels are increased. Astrocytic thalamic gap junctions, that is, connexin 30 and connexin 43, also contribute to absence seizure generation. Modified and reproduced with permission from Crunelli and Carmignoto (2013).
Interestingly, the thalamic expression of GAT-1 in GAERS and stargazer mice is similar to that of their respective nonepileptic control strains, and only a silent mutation is present in the GAT-1 gene of these two genetic models (Cope and others 2009). So, it may be possible that the inability of GAT-1 to regulate GABA levels is because this transporter remains as an immature intracellular protein or, alternatively, that the phosphorylation of GAT-1, and thus its function, is compromised. Moreover, one might expect GAT-3, the only other GABA transporter in the thalamus (Borden 1996; De Biasi and others 1998; Pow and others 2005), to compensate for the malfunctioning GAT-1, but this is not the case. It might also be that GAT-3 is located far away from the perisynaptic/extrasynaptic GABAARs that are responsible for the tonic GABAA current, although a recent investigation concluded that GAT-1 is “primarily localized near GABAergic synapses whereas GAT-3 is localized both near and far away from synapses” (Beenhakker and Huguenard 2010). Clearly, more direct studies investigating the relative position of these transporters with respect to synaptic and perisynaptic/extrasynaptic GABAARs are necessary to resolve this issue.
Astrocyte-Neuron Signaling in Absence Seizures
Slow outward currents (SOCs) are the characteristic signatures of GABAergic astrocyte-to-neuron signaling (Kozlov and others 2006). Possibly as a consequence of reduced GAT-1 activity in the thalamus, some properties (i.e., amplitude, rise time, and decay time) of SOCs are altered in GAERS thalamocortical neurons (Pirttimaki and others 2011, 2012). At present, one cannot ascribe a precise mechanistic significance for these changes in thalamic SOCs to the pathophysiological processes underlying absence seizures. Nevertheless, it is interesting that vigabatrin, which elicits and/or exacerbates absence seizures in animals and humans (see above), can increase the frequency of SOCs in thalamic neurons (Jimenez-Gonzalez and others 2011).
Importantly, no differences are observed in the properties of SICs, the characteristic signature of glutamatergic astrocyte-to-neuron signaling (Angulo and others 2004; Fellin and others 2004), between GAERS prior to seizure onset and age-matched nonepileptic rats (Pirttimaki and others 2012). Moreover, no change in astrocytic glutamate transporters has been reported at this age in the thalamus of this genetic model (Dutuit and others 2002).
It is now well established that, contrary to the classic view, absence seizures are not generalized from their very start, but there is an “initiation site” in a localized cortical region both in experimental models (Meeren and others 2002; Polack and others 2007) and in humans (Bai and others 2010; Holmes and others 2004; Moeller and others 2010; Westmijse and others 2009). Thus, it is important that future studies investigate whether similar abnormalities to those present in the activity of GAT-1 and SOCs are also present in the cortical “initiation site” of rat genetic models and humans suffering from this form of epilepsy. Indeed, in cortical neurons, one might expect to see alterations in SICs because glutamate uptake is reduced in this brain region of preseizure GAERS (Touret and others 2007) as a result of a decreased expression of the astrocytic glutamate transporters GLT1 and GLAST (Dutuit and others 2002).
Other Astrocytic Abnormalities in Absence Seizures
Another recent finding that stresses the potential role of astrocytic abnormalities in the expression of absence seizures is the selective induction of IL-1β in activated astrocytes within the cortical “initiation site” of absence seizures (i.e., the peri-oral region of the somatosensory cortex) (Fig. 5), but not in other cortical regions or in the thalamus of GAERS prior to seizure onset (Akin and others 2011). This enhanced cortical expression of IL-1β is not simply an epiphenomenon because the systemic injection of a specific blocker of IL-1β synthesis drastically decreases absence seizures in this model (Akin and others 2011) and the injection of an IL-1β inducer increases the number of absence seizures in WAG/Rij rats (Kovács and others 2006), another well-established model of absence epilepsy (Coenen and Van Luijtelaar 2003). Unfortunately, no data are available on the levels of IL-1β in humans before or after the onset of absence seizures.
Finally, it is important to note that the ability of the GJ blocker carbenoxolone to drastically reduce absence seizures in both rat (WAG/Rij) and mouse (lethargic) models when it is injected intrathalamically or systemically (Gareri and others 2005) (Fig. 5), respectively, has been mainly explained as resulting from a block of neuronal GJs. While strong evidence exists for neuronal GJs in the thalamus (Hughes and others 2004, 2011; Landisman and others 2002; Nagy and Rash 2000; Söhl and others 2005), both the reticular thalamic nucleus and the sensory thalamic nuclei also exhibit strong immunoreactivity for the astroglia-specific Cx30 and Cx43 (Nagy and Rash 2000) (Fig. 3). Thus, astrocytic GJs are also likely to play a role in the antiabsence effect of carbenoxolone and in the mechanisms underlying the expression of absence seizures (Fig. 5).
Conclusions
The findings reviewed here highlight novel key elements of astrocytic involvement in epilepsy, in particular, abnormalities in the astrocyte control of extracellular K+, Ca2+ signaling, and gliotransmitter release in mTLE and abnormalities in GAT-1, Cx30- and Cx43-based GJs, and the IL-1β pathway in absence seizures. This shift in research from neurons to astrocyte-neuron interactions (Crunelli and Carmignoto 2013; Steinhäuser and others 2012) has allowed us to unravel novel mechanisms underlying the generation of seizures (Baulac and Pitkanen 2009). These novel astrocyte-based targets should pave the way to a rational discovery program for the development of fourth-generation antiepileptogenic drugs with increased efficacy that many expert groups have identified as a key priority for the improved clinical management of convulsive and nonconvulsive epilepsy (Baulac and Pitkanen 2009; Galanopoulou and others 2012; Löscher and Schmidt 2011; Simonato and others 2012).
Acknowledgments
The authors thank Drs. I. Nauroth and M. Zonta for graphic support and Dr. C.E. Elger for providing some of the electroencephalogram traces in Figure 2.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received the following financial support for the research, authorship, and/or publication of this article: V.C. was supported by the Wellcome Trust (grant 91882) and EU (FP7-202167 NeuroGLIA); G.C. by the EU (FP7-202167 NeuroGLIA), Telethon Italy (GGP10138B, GGP12265), Italian Institute of Technology (IIT), and Cariparo Foundation; and C.S. by the DFG (STE 552/3) and EU (FP7-202167 NeuroGLIA, ESF EuroEPINOMICS).
References
- Aizawa M, Ito Y, Fukuda H. 1997. Pharmacological profiles of generalized absence seizures in lethargic, stargazer and gamma-hydroxybutyrate-treated model mice. Neurosci Res 29:17–25. [DOI] [PubMed] [Google Scholar]
- Akbar MT, Torp R, Danbolt NC, Levy LM, Meldrum BS, Ottersen OP. 1997. Expression of glial glutamate transporters GLT-1 and GLAST is unchanged in the hippocampus in fully kindled rats. Neuroscience 78:351–9. [DOI] [PubMed] [Google Scholar]
- Akin D, Ravizza T, Maroso M, Carcak N, Eryigit T, Vanzulli I, and others. 2011. IL-1b is induced in reactive astrocytes in the somatosensory cortex of rats with genetic absence epilepsy at the onset of spike-and-wave discharges, and contributes to their occurrence. Neurobiol Dis 44:259–69. [DOI] [PubMed] [Google Scholar]
- Alvestad S, Hammer J, Hoddevik EH, Skare Ø, Sonnewald U, Amiry-Moghaddam M, Ottersen OP. 2013. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res 105:30–41. [DOI] [PubMed] [Google Scholar]
- Angulo MC, Kozlov AS, Charpak S, Audinat E. 2004. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martín ED, Perea G, Arellano JI, Buño W. 2002. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci 22:2443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Jansen GH, Leenstra S, Yankaya B, Troost D. 2001. Expression of connexin 43 and connexin 32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol (Berl) 101:449–59. [DOI] [PubMed] [Google Scholar]
- Aronica E, Ravizza T, Zurolo E, Vezzani A. 2012. Astrocyte immune responses in epilepsy. Glia 60:1258–68. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. 2000. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci 12:2333–44. [DOI] [PubMed] [Google Scholar]
- Avoli M. 2012. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia 53:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, D’Antuono M, Louvel J, Kohling R, Biagini G, Pumain R, and others. 2002. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol 68:167–207. [DOI] [PubMed] [Google Scholar]
- Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, and others. 2010. Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci 30:5884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Ghirri A, Zonta M, Betelli C, Vitale G, Ruggieri V, and others. 2010. Glutamate-mediated astrocyte-to-neuron signalling in the rat dorsal horn. J Physiol 588:831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac M, Pitkanen A. 2009. Research priorities in epilepsy for the next decade. Epilepsia 50:571–83. [Google Scholar]
- Bedner P, Steinhäuser C. 2013. Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem Int 63:682–7. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. 2010. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci 30:15262–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. 1985. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14:375–403. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361:315–25. [DOI] [PubMed] [Google Scholar]
- Bessaih T, Bourgeais L, Badiu CI, Carter DA, Toth TI, Ruano D, and others. 2006. Nucleus-specific abnormalities of GABAergic synaptic transmission in a genetic model of absence seizures. J Neurophysiol 96:3074–81. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, and others. 1998. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391:281–5. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, and others. 2001. CXCR4-activated astrocyte glutamate release via TNF: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–10. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhäuser C, Pilati E, Volterra A. 2004. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci 7:613–20. [DOI] [PubMed] [Google Scholar]
- Binder DK, Nagelhus EA, Ottersen OP. 2012. Aquaporin-4 and epilepsy. Glia 60:1203–14. [DOI] [PubMed] [Google Scholar]
- Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. 2004a. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 15:259–62. [DOI] [PubMed] [Google Scholar]
- Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. 2004b. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci 24:8049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Steinhäuser C. 2006. Functional changes in astroglial cells in epilepsy. Glia 54:358–68. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H. 2005. Cellular and network mechanisms of spike-wave seizures. Epilepsia 46 Suppl 9:21–33. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, and others. 2009. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden LA. 1996. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int 29:335–56. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. 1998. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res 32:286–303. [DOI] [PubMed] [Google Scholar]
- Bostanci MO, Bagirici F. 2007. Anticonvulsive effects of carbenoxolone on penicillin-induced epileptiform activity: an in vivo study. Neuropharmacology 52:362–7. [DOI] [PubMed] [Google Scholar]
- Bostanci MO, Bagirici F. 2006. The effects of octanol on penicillin induced epileptiform activity in rats: an in vivo study. Epilepsy Res 71:188–94. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. 2004. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci 24:8606–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockhaus J, Deitmer JW. 2002. Long-lasting modulation of synaptic input to Purkinje neurons by Bergmann glia stimulation in rat brain slices. J Physiol 545:581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buono RJ, Lohoff FW, Sander T, Sperling MR, O’Connor MJ, Dlugos DJ, and others. 2004. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res 58:175–83. [DOI] [PubMed] [Google Scholar]
- Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA. 1999. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4lh) and tottering (Cacna1atg) mouse thalami. J Neurophysiol 81:2066–74. [DOI] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M, Carmignoto G. 2013. Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol 591:807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SL, Hablitz JJ. 2004. Glutamate transporters regulate excitability in local networks in rat neocortex. Neuroscience 127:625–35. [DOI] [PubMed] [Google Scholar]
- Carlen PL. 2012. Curious and contradictory roles of glial connexins and pannexins in epilepsy. Brain Res 1487:54–60. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Fellin T. 2006. Glutamate release from astrocytes as a non-synaptic mechanism for neuronal synchronization in the hippocampus. J Physiol Paris 99:98–102. [DOI] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, and others. 2005. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol 57:226–35. [DOI] [PubMed] [Google Scholar]
- Charles AC, Merrill JE, Dirksen ER, Sanderson MJ. 1991. Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6:983–92. [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG. 1995. Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695:203–16. [DOI] [PubMed] [Google Scholar]
- Chever O, Djukic B, McCarthy KD, Amzica F. 2010. Implication of kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional kir4.1 knock-out mice. J Neurosci 30:15769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenen AM, Van Luijtelaar EL. 2003. Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. Behav Genet 33:635–55. [DOI] [PubMed] [Google Scholar]
- Collignon F, Wetjen NM, Cohen-Gadol AA, Cascino GD, Parisi J, Meyer FB, and others. 2006. Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. J Neurosurg 105:77–87. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Mudo G, Trovato-Salinaro A, Mirone MB, Amato G, Belluardo N. 2002. Connexin-30 mRNA is up-regulated in astrocytes and expressed in apoptotic neuronal cells of rat brain following kainate-induced seizures. Mol Cell Neurosci 21:94–113. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orban G, Errington AC, Lorincz ML, and others. 2009. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15:1392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. 1990. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247:470–3. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. 2012. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60:1215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Carmignoto G. 2013. New vistas on astroglia in convulsive and non-convulsive epilepsy highlight novel astrocytic targets fro treatment. J Physiol 591:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. 2002. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 3:371–82. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R. 2006. Does glutamate released by astrocytes cause focal epilepsy? Epilepsy Curr 6:173–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. 1998. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol 55:27–57. [DOI] [PubMed] [Google Scholar]
- Das A, Wallace GC, Holmes C, McDowell ML, Smith JA, Marshall JD, and others. 2012. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220:237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, and others. 2007. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci U S A 104:1995–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. 2009. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci 29:10588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi S, Vitellaro-Zuccarello L, Brecha NC. 1998. Immunoreactivity for the GABA transporter-1 and GABA transporter-3 is restricted to astrocytes in the rat thalamus: a light and electron-microscopic immunolocalization. Neuroscience 83:815–28. [DOI] [PubMed] [Google Scholar]
- Demarque M, Villeneuve N, Manent JB, Becq H, Represa A, Ben Ari Y, Aniksztejn L. 2004. Glutamate transporters prevent the generation of seizures in the developing rat neocortex. J Neurosci 24:3289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, and others. 2007. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci 27:10674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Tasker JG. 1990. Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett 120:267–70. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. 1995. Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J Neurosci 15:5535–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Spencer DD. 1993. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341:1607–10. [DOI] [PubMed] [Google Scholar]
- Dutuit M, Touret M, Szymocha R, Nehlig A, Belin M-F, Didier-Blazès M. 2002. Decreased expression of glutamate transporters in genetic absence epilepsy rats before seizure occurrence. J Neurochem 80:1029–38. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, and others. 2008. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131:2061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjørnsen LP, Spencer DD, and others. 2005. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A 102:1193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Lee TS, Wang Y, Perez E, Drummond J, Lauritzen F, and others. 2013a. Gene expression of glutamate metabolizing enzymes in the hippocampal formation in human temporal lobe epilepsy. Epilepsia 54:228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, and others. 2004. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363:28–37. [DOI] [PubMed] [Google Scholar]
- Eid T, Tu N, Lee TS, Lai JC. 2013b. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int 63:670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisevich K, Rempel SA, Smith B, Allar N. 1997a. Connexin 43 mRNA expression in two experimental models of epilepsy. Mol Chem Neuropathol 32:75–88. [DOI] [PubMed] [Google Scholar]
- Elisevich K, Rempel SA, Smith B, Edvardsen K. 1997b. Hippocampal connexin 43 expression in human complex partial seizure disorder. Exp Neurol 145:154–64. [DOI] [PubMed] [Google Scholar]
- Elisevich K, Rempel SA, Smith B, Hirst K. 1998. Temporal profile of connexin 43 mRNA expression in a tetanus toxin-induced seizure disorder. Mol Chem Neuropathol 35:23–37. [DOI] [PubMed] [Google Scholar]
- Errington AC, Cope DW, Crunelli V. 2011a. Augmentation of tonic GABAA inhibition in absence epilepsy: therapeutic value of inverse agonists at extrasynaptic GABAA receptors. Adv Pharmacol Sci 2011:790590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington AC, Gibson KM, Crunelli V, Cope DW. 2011b. Aberrant GABAA receptor-mediated inhibition in cortico-thalamic networks of succinic semialdehyde dehydrogenase deficient mice. PLoS One 6:e19021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AB, Bernal OG, Andriola MR, Bagchi S, Flores P, Just C, and others. 1999. Two cases of nonconvulsive status epilepticus in association with tiagabine therapy. Epilepsia 40:1159–62. [DOI] [PubMed] [Google Scholar]
- Fellin T, Carmignoto G. 2004. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol 559:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, D’Ascenzo M, Haydon PG. 2007. Astrocytes control neuronal excitability in the nucleus accumbens. ScientificWorldJournal 7:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. 2006. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci 26:9312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. 2004. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–43. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. 1992. Calcium waves in astrocytes-filling in the gaps. Neuron 8:1101–8. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Pedley TA, Moody WJ, Jr, Prince DA. The role of extracellular potassium in hippocampal epilepsy. Arch Neurol. 1976. February;33(2):76-83. [DOI] [PubMed] [Google Scholar]
- Fonseca CG, Green CR, Nicholson LF. 2002. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res 929:105–16. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr., Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, and others. 2002. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A 99:14488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajda Z, Gyengesi E, Hermesz E, Ali KS, Szente M. 2003. Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia 44:1596–600. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS, Buckmaster PS, Staley KJ, Moshé SL, Perucca E, Engel J, Jr., and others. 2012. Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia 53:571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato-Salinaro A, and others. 2005. Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice). Neuropharmacology 49:551–63. [DOI] [PubMed] [Google Scholar]
- Giaume C, Tabernero A, Medina JM. 1997. Metabolic trafficking through astrocytic gap junctions. Glia 21:114–23. [PubMed] [Google Scholar]
- Gigout S, Louvel J, Kawasaki H, D’Antuono M, Armand V, Kurcewicz I, and others. 2006. Effects of gap junction blockers on human neocortical synchronization. Neurobiol Dis 22:496–508. [DOI] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, and others. 2010. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol 8:e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Sun F, Yu JL, Wang QH, Tu DY, Mao XY, and others. 2010. Abnormal expressions of glutamate transporters and metabotropic glutamate receptor 1 in the spontaneously epileptic rat hippocampus. Brain Res Bull 81:510–6. [DOI] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, and others. 2005. Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353:1926–33. [DOI] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Toutain A, Rutsch F, Benoist JF, Gelot A, and others. 2006. Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis 29:352–8. [DOI] [PubMed] [Google Scholar]
- Haberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T. 2011. Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab 103:89–91. [DOI] [PubMed] [Google Scholar]
- Haglund MM, Hochman DW. 2005. Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol 94:907–18. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, and others. 2011. Evidence that compromised K(+) spatial buffering contributes to the epileptogenic effect of mutations in the human kir4.1 gene (KCNJ10). Glia 59:1635–42. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Sofroniew MV. 2010. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7:494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer J, Alvestad S, Osen KK, Skare O, Sonnewald U, Ottersen OP. 2008. Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia 56:856–68. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. 2002. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–14. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. 2006. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–31. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD. 1977. Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res 120:231–49. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. 2010. Long-term potentiation depends on release of D-serine from astrocytes. Nature 463:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Tauboll E, and others. 2012. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol 71:814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser K, Nagelhus EA, Tauboll E, Indahl U, Berg PR, Lien S, and others. 2010. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res 88:55–64. [DOI] [PubMed] [Google Scholar]
- Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, and others. 2000. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci 12:2087–96. [DOI] [PubMed] [Google Scholar]
- Hirase H, Qian L, Barthó P, Buzsáki G. 2004. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol 2:E96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MD, Brown M, Tucker DM. 2004. Are “generalized” seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence. Epilepsia 45:1568–79. [DOI] [PubMed] [Google Scholar]
- Hosford DA, Wang Y. 1997. Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia 38:408–14. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lorincz M, Cope DW, Blethyn KL, Kekesi KA, Parri HR, and others. 2004. Synchronized oscillations at alpha and theta frequencies in the lateral geniculate nucleus. Neuron 42:253–68. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lorincz ML, Blethyn K, Kekesi KA, Juhasz G, Turmaine M, and others. 2011. Thalamic gap junctions control local neuronal synchrony and influence macroscopic oscillation amplitude during EEG alpha rhythms. Front Psychol 2:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahromi SS, Wentlandt K, Piran S, Carlen PL. 2002. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol 88:1893–902. [DOI] [PubMed] [Google Scholar]
- Jimenez-Gonzalez C, Pirttimaki T, Cope DW, Parri HR. 2011. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABA(A) receptors. Eur J Neurosci 33:1471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin BJ, Zhang H, Binder DK, Verkman AS. 2013. Aquaporin-4-dependent K(+) and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol 141:119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, and others. 2007. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10:331–9. [DOI] [PubMed] [Google Scholar]
- Kam K, Nicoll R. 2007. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kananura C, Haug K, Sander T, Runge U, Gu W, Hallmann K, and others. 2002. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol 59:1137–41. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. 1998. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1:683–92. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Ivy GO. 1996. Astrocytes in kindling: relevance to epileptogenesis. Epilepsy Res 26:163–75. [DOI] [PubMed] [Google Scholar]
- Kivi A, Lehmann TN, Kovacs R, Eilers A, Jauch R, Meencke HJ, and others. 2000. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci 12:2039–48. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. 2004. Potassium buffering in the central nervous system. Neuroscience 129:1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Gladwell SJ, Bracci E, Vreugdenhil M, Jefferys JG. 2001. Prolonged epileptiform bursting induced by 0-Mg(2+) in rat hippocampal slices depends on gap junctional coupling. Neuroscience 105:579–87. [DOI] [PubMed] [Google Scholar]
- Kostopoulos GK. 2000. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin Neurophysiol 111 Suppl 2:S27–38. [DOI] [PubMed] [Google Scholar]
- Kovács Z, Kekesi KA, Szilagyi N, Abraham I, Szekacs D, Kiraly N, and others. 2006. Facilitation of spike-wave discharge activity by lipopolysaccharides in Wistar Albino Glaxo/Rijswijk rats. Neuroscience 140:731–42. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. 2006. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A 103:10058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Haentzsch A, Luckermann M, Reichelt W, Ballanyi K. 1999. Neuron-glia signaling via alpha(1) adrenoceptor-mediated Ca2+ release in Bergmann glial cells in situ. J Neurosci 19:8401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Hüttmann K, Bedner P, and others. 2009. Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A 106:11336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance-Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, and others. 2011. Novel a1 and g2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci 34:237–49. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL, Connors BW. 2002. Electrical synapses in the thalamic reticular nucleus. J Neurosci 22:1002–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK. 2012. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol 235:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Magge S, Spencer DD, Sontheimer H, Cornell-Bell AH. 1995. Human epileptic astrocytes exhibit increased gap junction coupling. Glia 15:195–202. [DOI] [PubMed] [Google Scholar]
- Lee TS, Eid T, Mane S, Kim JH, Spencer DD, Ottersen OP, De Lanerolle NC. 2004. Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol (Berl) 108:493–502. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Mendizabal-Zubiaga J, Grandes P, Audinat E. 2012. GABA release by hippocampal astrocytes. Front Comput Neurosci 6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ropert N, Koulakoff A, Giaume C, Oheim M. 2008. Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J Neurosci 28:7648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shen H, Naus CC, Zhang L, Carlen PL. 2001. Upregulation of gap junction connexin 32 with epileptiform activity in the isolated mouse hippocampus. Neuroscience 105:589–98. [DOI] [PubMed] [Google Scholar]
- Liang SL, Carlson GC, Coulter DA. 2006. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci 26:8537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MW, Soares FM, de Mello N, Nunes JC, Cajado AG, de Brito D, and others. 2013. Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res 226:153–63. [DOI] [PubMed] [Google Scholar]
- Löscher W, Schmidt D. 2011. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52:657–78. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ. 2010. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol 588:1861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, and others. 2006. A mutation in the GABAA receptor a1-subunit is associated with absence epilepsy. Ann Neurol 59:983–7. [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, and others. 2010. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 16:413–9. [DOI] [PubMed] [Google Scholar]
- Martineau M, Shi T, Puyal J, Knolhoff AM, Dulong J, Gasnier B, and others. 2013. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. J Neurosci 33:3413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, and others. 1999. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52:453–72. [DOI] [PubMed] [Google Scholar]
- Medici V, Frassoni C, Tassi L, Spreafico R, Garbelli R. 2011. Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res 1367:330–9. [DOI] [PubMed] [Google Scholar]
- Medina-Ceja L, Cordero-Romero A, Morales-Villagran A. 2008. Antiepileptic effect of carbenoxolone on seizures induced by 4-aminopyridine: a study in the rat hippocampus and entorhinal cortex. Brain Res 1187:74–81. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. 2002. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci 22:1480–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melom JE, Littleton JT. 2013. Mutation of a NCKX eliminates glial microdomain calcium oscillations and enhances seizure susceptibility. J Neurosci 33:1169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C. 2006. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. FASEB J 20:494–6. [DOI] [PubMed] [Google Scholar]
- Miller HP, Levey AI, Rothstein JD, Tzingounis AV, Conn PJ. 1997. Alterations in glutamate transporter protein levels in kindling-induced epilepsy. J Neurochem 68:1564–70. [DOI] [PubMed] [Google Scholar]
- Moeller F, LeVan P, Muhle H, Stephani U, Dubeau F, Siniatchkin M, Gotman J. 2010. Absence seizures: individual patterns revealed by EEG-fMRI. Epilepsia 51:2000–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylvaganam S, Zhang L, Wu C, Zhang ZJ, Samoilova M, Eubanks J, and others. 2010. Hippocampal seizures alter the expression of the pannexin and connexin transcriptome. J Neurochem 112:92–102. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Cuthbertson GJ. 1980. Kainic acid neurotoxicity toward hippocampal formation: dependence on specific excitatory pathways. Brain Res 195:47–56. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, and others. 1999. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26:47–54. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. 2000. Connexins and gap junctios of astrocytes and oligodendrocytes in the CNS. Brain Res Rev 32:29–44. [DOI] [PubMed] [Google Scholar]
- Naus CCG, Bechberger JF, Paul DL. 1991. Gap junction gene expression in human seizure disorder. Exp Neurol 111:198–203. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Perea G, de Sevilla DF, Gomez-Gonzalo M, Nunez A, Martin ED, Araque A. 2012. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol 10:e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Binder DK. 2005. Emerging role of gap junctions in epilepsy. Histol Histopathol 20:253–9. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. 2001. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 21:5429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV. 2012. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Oxford: Oxford University Press. [PubMed] [Google Scholar]
- Olney JW, Sharpe LG, Feigin RD. 1972. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol 31:464–88. [DOI] [PubMed] [Google Scholar]
- Orellana JA, Martinez AD, Retamal MA. 2013. Gap junction channels and hemichannels in the CNS: regulation by signaling molecules. Neuropharmacology 75:567–82. [DOI] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966. July;29(4):788-806 [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, and others. 2010. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13:584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan E, Stringer JL. 1996. Influence of osmolality on seizure amplitude and propagation in the rat dentate gyrus. Neurosci Lett 207:9–12. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet J-P, Toquet B, Pollegioni L, Poulain DA, Oliet SHR. 2006. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125:775–84. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP. 1997. Absences epilepsies. In: Engel JJ, Pedley TA. (eds). Epilepy: A Comprehensive Textbook. Philadelphia: Lippincott-Raven; p. 2327–46. [Google Scholar]
- Panayiotopoulos CP. 2001. Treatment of typical absence seizures and related epileptic syndromes. Paediatr Drugs 3:379–403. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, and others. 2011. Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A 108:8467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. 2013. Aquaporin water channels in the nervous system. Nat Rev Neurosci 14:265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. 1994. Glutamate-mediated astrocyte-neuron signalling. Nature 369:744–7. [DOI] [PubMed] [Google Scholar]
- Parpura V, Scemes E, Spray DC. 2004. Mechanisms of glutamate release from astrocytes: gap junction “hemichannels,” purinergic receptors and exocytotic release. Neurochem Int 45:259–64. [DOI] [PubMed] [Google Scholar]
- Parpura V, Zorec R. 2010. Gliotransmission: exocytotic release from astrocytes. Brain Res Rev 63:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. 2001. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci 4:803–12. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, and others. 2005. Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–6. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. 1997. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci 17:7817–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. 2007. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317:1083–6. [DOI] [PubMed] [Google Scholar]
- Perez EL, Lauritzen F, Wang Y, Lee TS, Kang D, Zaveri HP, and others. 2012. Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis 47:331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Velazquez JL, Valiante TA, Carlen PL. 1994. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J Neurosci 14:4308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault P, Avoli M. 1991. Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J Neurophysiol 65:771–85. [DOI] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. 1998. Antiepileptic drugs as a cause of worsening seizures. Epilepsia 39:5–17. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Errante LD, Kim JH, Spencer DD. 2003. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology 60:1646–51. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Albeanu DF, Sato TF, Murthy VN. 2008. Coupling of neural activity to blood flow in olfactory glomeruli is mediated by astrocytic pathways. Neuron 58:897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. 1998. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol 509(Pt 2):449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto DJ, Patrick SL, Huang WC, Connors BW. 2005. Initiation, propagation, and termination of epileptiform activity in rodent neocortex in vitro involve distinct mechanisms. J Neurosci 25:8131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Hall SD, Parri HR. 2011. Sustained neuronal activity generated by glial plasticity. J Neurosci 31:7637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttimaki TM, Parri HR, Crunelli V. 2012. Astrocytic GAT-1 dysfunction in experimental absence seizures. J Physiol 591:823–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. 2007. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci 27:6590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. 1996. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16:5073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV, Sullivan RK, Williams SM, Scott HL, Dodd PR, Finkelstein D. 2005. Differential expression of the GABA transporters GAT-1 and GAT-3 in brains of rats, cats, monkeys and humans. Cell Tissue Res 320:379–92. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. 1994. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 74:595–636. [DOI] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, and others. 2002. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125:32–43. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, Giaume C. 2007. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 27:13781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Haro D, Muller J, Boresch M, Pivneva T, Benedetti B, Scheller A, and others. 2010. Neuron-astrocyte interactions in the medial nucleus of the trapezoid body. J Gen Physiol 135:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Lemos T, Whitton PS, Bowery NG. 1995. Extracellular GABA in the ventrolateral thalamus of rats exhibiting spontaneous absence epilepsy: a microdialysis study. J Neurochem 65:1674–80. [DOI] [PubMed] [Google Scholar]
- Roper SN, Obenaus A, Dudek FE. 1992. Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann Neurol 31:81–5. [DOI] [PubMed] [Google Scholar]
- Ross FM, Gwyn P, Spanswick D, Davies SN. 2000. Carbenoxolone depresses spontaneous epileptiform activity in the CA1 region of rat hippocampal slices. Neuroscience 100:789–96. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, and others. 1996. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–86. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. 2008. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322:1551–5. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. 2007. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 K+ channels in retinal Muller cells. J Biol Chem 282:21866–72. [DOI] [PubMed] [Google Scholar]
- Samoilova M, Li J, Pelletier MR, Wentlandt K, Adamchik Y, Naus CC, Carlen PL. 2003. Epileptiform activity in hippocampal slice cultures exposed chronically to bicuculline: increased gap junctional function and expression. J Neurochem 86:687–99. [DOI] [PubMed] [Google Scholar]
- Samoilova M, Wentlandt K, Adamchik Y, Velumian AA, Carlen PL. 2008. Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Exp Neurol 210:762–75. [DOI] [PubMed] [Google Scholar]
- Scemes E, Giaume C. 2006. Astrocyte calcium waves: what they are and what they do. Glia 54:716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Spray DC. 1998. Increased intercellular communication in mouse astrocytes exposed to hyposmotic shocks. Glia 24:74–84. [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, and others. 2009. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A 106:5842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. 2008. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320:1638–43. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW. 1998. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 32:275–85. [DOI] [PubMed] [Google Scholar]
- Seifert G, Carmignoto G, Steinhäuser C. 2010. Astrocyte dysfunction in epilepsy. Brain Res Rev 63:212–21. [DOI] [PubMed] [Google Scholar]
- Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C. 2009. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 29:7474–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhäuser C. 2006. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7:194–206. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. 2006. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci 26:5370–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton MK, McCarthy KD. 2000. Hippocampal astrocytes exhibit Ca2+-elevating muscarinic cholinergic and histaminergic receptors in situ. J Neurochem 74:555–63. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. 2011. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci 15:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simantov R, Crispino M, Hoe W, Broutman G, Tocco G, Rothstein JD, Baudry M. 1999. Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity. Mol Brain Res 65:112–23. [DOI] [PubMed] [Google Scholar]
- Simonato M, Löscher W, Cole AJ, Dudek E, Engel J, Jr., Kaminski RM, and others. 2012. Finding a better drug for epilepsy: preclinical screening strategies and experimental trial design. Epilepsia 53:1860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead OC., 3rd 1992. Evidence for GABAB-mediated mechanisms in experimental generalized absence seizures. Eur J Pharmacol 213:343–9. [DOI] [PubMed] [Google Scholar]
- Söhl G, Güldenagel M, Beck H, Teubner B, Traub O, Gutierrez R, and others. 2000. Expression of connexin genes in hippocampus of kainate-treated and kindled rats under conditions of experimental epilepsy. Mol Brain Res 83:44–51. [DOI] [PubMed] [Google Scholar]
- Söhl G, Maxeiner S, Willecke K. 2005. Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6:191–200. [DOI] [PubMed] [Google Scholar]
- Stasheff SF, Hines M, Wilson WA. 1993. Axon terminal hyperexcitability associated with epileptogenesis in vitro, I: origin of ectopic spikes. J Neurophysiol 70:961–75. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Seifert G. 2012. Astrocyte dysfunction in epilepsy. In: Noebels J, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV. (eds). Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Oxford: Oxford University Press; p. 606–17. [Google Scholar]
- Steinhäuser C, Seifert G, Bedner P. 2012. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60:1192–202. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. 1995. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci 15:623–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohschein S, Hüttmann K, Gabriel S, Binder DK, Heinemann U, Steinhäuser C. 2011. Impact of aquaporin-4 channels on K(+) buffering and gap junction coupling in the hippocampus. Glia 59:973–80. [DOI] [PubMed] [Google Scholar]
- Szente M, Gajda Z, Said AK, Hermesz E. 2002. Involvement of electrical coupling in the in vivo ictal epileptiform activity induced by 4-aminopyridine in the neocortex. Neuroscience 115:1067–78. [DOI] [PubMed] [Google Scholar]
- Takahashi DK, Vargas JR, Wilcox KS. 2010. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol Dis 40:573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, and others. 2005. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A 102:16466–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K, Hirase H. 2011. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J Neurosci 31:18155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Chiu C, Jones MV, Petrou S. 2008. Increased thalamic inhibition in the absence seizure prone DBA/2J mouse. Epilepsia 49:921–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, and others. 2007. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci U S A 104:17536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, and others. 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–702. [DOI] [PubMed] [Google Scholar]
- Tessler S, Danbolt NC, Faull RLM, Storm-Mathisen J, Emson PC. 1999. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 88:1083–91. [DOI] [PubMed] [Google Scholar]
- Theis M, Giaume C. 2012. Connexin-based intercellular communication and astrocyte heterogeneity. Brain Res 1487:88–98. [DOI] [PubMed] [Google Scholar]
- Tian G-F, Azmi H, Takano T, Xu Q, Peng W, Lin J, and others. 2005. An astrocytic basis of epilepsy. Nat Med 11:973–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KJ, Darabid H, Robitaille R. 2010. Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30:11870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touret M, Parrot S, Denoroy L, Belin M-F, Didier-Blazès M. 2007. Glutamatergic alterations in the cortex of genetic absence epilepsy rats. BCM Neurosci 8:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK. 1982. Cellular mechanism of neuronal synchronization in epilepsy. Science 216:745–7. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. 1988. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol 59:259–76. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. 1989. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol 61:927–38. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. 2006. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci 26:12447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulas J, Satou T, Ivins KJ, Kesslak JP, Cotman CW, Balazs R. 2000. Expression of metabotropic glutamate receptor 5 is increased in astrocytes after kainate-induced epileptic seizures. Glia 30:352–61. [PubMed] [Google Scholar]
- van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, De Graan PN. 2005. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64:326–33. [DOI] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C. 1992. Cortical and thalamic lesions in rats with genetic absence epilepsy. J Neural Transm Suppl 35:71–83. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H. 1996. Calcium signalling in glial cells. Trends Neurosci 19:346–52. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. 1998. Glial calcium: homeostasis and signaling function. Physiol Rev 78:99–141. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Balosso S, Ravizza T. 2008. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun 22:797–803. [DOI] [PubMed] [Google Scholar]
- Voss LJ, Jacobson G, Sleigh JW, Steyn-Ross A, Steyn-Ross M. 2009. Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia 50:1971–8. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, and others. 2001. Mutant GABAA receptor g2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 28:49–52. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. 2006. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26:5438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz W. 2000. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36:291–300. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, and others. 2006. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci 9:816–23. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Lee TS, Eid T. 2009. The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol 220:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. 1999. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res 845:92–6. [DOI] [PubMed] [Google Scholar]
- Westmijse I, Ossenblok P, Gunning B, van Luijtelaar G. 2009. Onset and propagation of spike and slow wave discharges in human absence epilepsy: a MEG study. Epilepsia 50:2538–48. [DOI] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. 2008. Astrocytes in the epileptic brain. Neuron 58:168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. 1953. A study of thalamic and cortical rhythms in petit mal. Brain 76:50–69. [DOI] [PubMed] [Google Scholar]
- Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, and others. 2003. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol 54:251–6. [DOI] [PubMed] [Google Scholar]
- Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, and others. 2012. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell 151:25–40. [DOI] [PubMed] [Google Scholar]
- Xu L, Zeng LH, Wong M. 2009. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis 34:291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Hrabetova S, Nicholson C, Manley GT. 2008. Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J Neurosci 28:5460–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JJ, Green CR, O’Carroll SJ, Nicholson LF. 2010. Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res 92:153–62. [DOI] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. 2008. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci 37:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-M, Wang H-K, Ye C-Q, Ge W, Chen Y, Jiang Z-L, and others. 2003. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40:971–82. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chen G, Zhou W, Song A, Xu T, Luo Q, and others. 2007. Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9:945–53. [DOI] [PubMed] [Google Scholar]
- Zhuang Z, Yang B, Theus MH, Sick JT, Bethea JR, Sick TJ, Liebl DJ. 2010. EphrinBs regulate D-serine synthesis and release in astrocytes. J Neurosci 30:16015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann K-A, Pozzan T, Carmignoto G. 2003. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. [DOI] [PubMed] [Google Scholar]