

ABSTRACT
FLT3, a type III receptor tyrosine kinase, expresses on most acute leukemia cells as well as normal hematopoietic stem/progenitor cells. Mutation in the FLT3 gene is the most frequent genetic alteration in acute myeloid leukemia (AML) and is well known as an important driver mutation for the development of myeloid malignancies. FLT3 mutation is a strong poor prognostic factor for the long-term survival in AML patients, while neither high-dose chemotherapy nor allogeneic hematopoietic stem cell transplantation can overcome a poor prognosis. Development of an FLT3 inhibitor is, therefore, much awaited. To date, several potent FLT3 inhibitors have been developed and some of them were evaluated for efficacy in clinical trials, although no FLT3 inhibitor has been yet approved. Moreover, several problems for clinical use, such as adverse effects, blood concentration and resistance have been apparent. Recently developed AC220 is a highly selective and sensitive FLT3 inhibitor. In Phase I and II trials, AC220 so far showed the best efficacy of AML cells harboring FLT3 mutation among clinically evaluated FLT3 inhibitors, while severe bone marrow suppression and QTc prolongation should be resolved for the clinical use. In this review, I summarize the characteristics of FLT3 inhibitors in clinical development and discuss important issues to be resolved for clinical use.
Key Words: FLT3, inhibitors, leukemia, molecular target, resistance
INTRODUCTION
Acute myeloid leukemia (AML) is a phenotypically and genetically heterogeneous disease.1,2) Although recent advances in chemotherapy and an application of allogeneic hematopoietic stem cell transplantation have improved the prognosis of patients with AML, more than 50% of them remain uncured. Furthermore, half of them cannot undergo intensive chemotherapy, which could provide them with complete remission and/or cure, because of the complications or their poor general condition. In contrast, prognosis of the patients with acute promyelocytic leukemia (APL) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph-ALL), has been dramatically improved by the application of molecular-targeting agents, such as all-trans retinoic acid and imatinib, indicating that development of other targeting agents is necessary for improving the prognosis of AML patients. Many genetic alterations, which are closely associated with the development and/or progression of AML, have been identified, and some of them are expected to be therapeutic targets.3,4) The FLT3 mutation is the most frequently identified genetic alteration in AML, and induces the constitutive activation of FLT3 kinase. Therefore, FLT3 inhibitor serves as a promising molecular target in the treatment of leukemia. To date, several FLT3 kinase inhibitors were developed and their efficacy and safety were evaluated in phase I/II studies, but none of them were approved for clinical use. In this review, I would like to summarize the recent advances of FLT3 inhibitors and problems to be resolved for clinical use.
FLT3 MUTATION
FLT3 (FMS-like tyrosine kinase 3) belongs to a type III receptor tyrosine kinase together with KIT, FMS and PDGF-receptor, and consists of five immunoglobulin-like domains in the extracellular region, a juxtamembrane (JM) domain, a tyrosine kinase (TK) domain separated by a kinase insert (KI) domain and a C-terminal domain in the intracellular region.5-7) FLT3 expresses on the surface of normal hematopoietic stem/progenitor cells. FLT3 ligand (FL) is expressed by bone marrow stroma cells, and FL-FLT3 interaction plays an important role in the survival, proliferation and differentiation of normal hematopoietic stem/progenitor cells.8-13) In addition, FLT3 expresses in most AML and ALL cells, and FL-stimulation enhances proliferation and reduces apoptosis of leukemia cells.14) In 1996, an internal tandem duplication mutation in the JM domain-coding sequence of the FLT3 gene (FLT3-ITD) was first identified in AML cells.15) Subsequently, a missense point mutation at the D835 residue and point mutations, deletions and insertions in the codons surrounding D835 within a TK domain of FLT3 (FLT3-KDM) were identified (Figure 1).16-22) FLT3 mutations are identified in about 30% of the adult patients with AML, and are highly associated with leukocytosis and poor prognosis.23-28) The WHO and European LeukemiaNet, therefore, recommend that FLT3 mutations should be analyzed at the diagnosis of AML.2,29)
Fig. 1.

FLT3 mutations.
There are two types of FLT3 mutations, FLT3-ITD and FLT3-KDM. FLT3-ITD occurs between exons 14 and 15. When leukemia cells have FLT3-ITD, PCR products give a wild-type band and a larger ITD band (A). D835 and I836 codons are encoded by the nucleotide GATATC which forms the Eco RV restriction site. The amplified products of wild type FLT3 are digested to two bands by the Eco RV. When amplified products contain D835-mutations (FLT3-KDM), undigested bands are observed (B).
BIOLOGICAL EFFECTS OF FLT3 MUTATIONS
The binding of FL to the extracellular domain of FLT3 leads to the dimerization and transphosphorylation of the A-loop, resulting in the activation of FLT3, followed by induction of multiple intracellular signaling pathways, such as MAPK- and AKT-signals, leading to cell proliferation and activation. In contrast, mutant FLT3 is ligand-independently dimeralized and activated. Of note is that mutant FLT3 activates STAT5 in addition to MAPK- and AKT-signals, indicating that the phosphorylation level of STAT5 is a surrogate marker of the mutant FLT3 activation.30-34) The constitutively active mutant FLT3 kinase induces autonomous proliferation to cytokine-dependent cell lines, such as Ba/F3 and 32D cells. Furthermore, when mutant FLT3-transfected hematopoietic stem cells are transplanted, the mice develop an oligoclonal myeloproliferative disorder (MPD), but not AML, suggesting that mutant FLT3 is sufficient to induce a MPD, and thus additional mutations that impair hematopoietic differentiation and/or proliferation might be necessary for the development of monoclonal AML.35)
FIRST-GENERATION FLT3 INHIBITORS
Since FLT3 mutation is the most frequent genetic alteration and an independent poor prognostic factor in AML, mutant FLT3 serves as an important molecular target in the treatment of AML. At first, tyrosine kinase inhibitors (TKIs), which have a potency to inhibit the FLT3 kinase, were subjected to clinical trials (Figure 2).
Fig. 2.
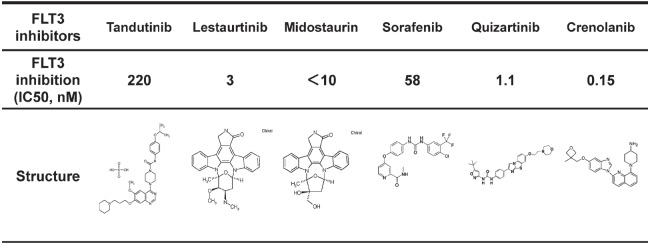
Structure and inhibitory activity of FLT3 inhibitors.
Tandutinib, Lestaurtinib and Midostaurin are first-generation FLT3 inhibitors. The second-generation FLT3 inhibitors, Sorafenib and Quizartinib, show clinical efficacy, while several problems regarding adverse effects and resistant mutations should be resolved before clinical use. Crenolanib is a recently developed FLT3 inhibitor, which is potent against both FLT3-ITD and FLT3-KDM.
Tandutinib (Millenium) is a derivative of quinazoline and inhibits the kinase activity of FLT3-ITD, but not FLT3-KDM.36) Although this compound also has potency against PDGFRB and KIT, the inhibitory spectrum is relatively selective to FLT3.37) In a Phase 1 study of 40 patients with relapsed or refractory AML, three patients had 40 to 50% reduction in the number of bone marrow (BM) blasts. In this study, no adverse effects were observed, while the peak plasma concentration of this compound did not reach to the biologically effective level. In a Phase 2 study of 25 patients with relapsed or refractory AML harboring FLT3-ITD, decrease of peripheral and BM leukemia cells was observed in seven of the 15 patients, while clinical response could not be evaluated in eight patients because of rapid disease progression or the toxicity, such as ptosis and QTc prolongation.38) Therefore, further clinical development was discontinued. Although this compound has a high selectivity against FLT3, a lower inhibitory effect (IC50 is 220 nM) might cause clinically unimpressive results.
Sunitinib (Pfizer) is a derivative of indolinone and has been approved for renal cell carcinoma, gastrointestinal stromal tumor (GIST) and neuroendocrine tumor (NET) in Japan. This compound has a unique inhibiting property to tyrosine kinases and inhibits KIT, PDGFR and KDR kinases more sensitively than FLT3 kinase. In a Phase 1 study of 15 patients with advanced AML, the tentative reduction of peripheral blast cells was observed in seven patients.39) Since two patients died of cardiotoxicity and the plasma concentration was brought to the biologically effective level, a further clinical development for AML has been discontinued.
Midostaurin (Novartis) is a benzoylstaurosporine and initially developed as a KDR inhibitor.40) This compound inhibits the kinase activity both of FLT3-ITD and FLT3-KDM. In a Phase 2 study of 61 patients including 11 FLT3-ITD and 4 FLT3-KDM patients, half of the enrolled patients showed over a 50% reduction in the number of bone marrow blasts.41) Although one AML patient with FLT3-ITD achieved complete remission, the duration was very short. Pharmacokinetic properties showed that the plasma concentration of this compound could not be maintained at the biologically effective level, because it consists of an indolocarbazole molecule, which is known to be highly associated with acid-α-glycoprotein (AGP) in human plasma. Notably, two patients died of pulmonary edema, which was considered a drug-induced toxicity. Since it was concluded that a monotherapy of midostaurin did not have sufficient clinical activity in AML patients with FLT3 mutations, a combination with chemotherapeutic regimens has been conducted. A randomized phase 3 study (RATIFY study) is now conducting for evaluating the superiority of midostaurin in addition to the conventional induction and consolidation therapies.
Lestaurtinib (Cephalon) was derived from indolocarbazole and structurally very similar as midosutaurin. This compound has a high potency against FLT3 kinases (IC50 is 3 nM).42,43) In a Phase 2a study of 12 AML patients with FLT3 mutations, the reduction of peripheral blast cells under 5% or the loss of BM blasts was observed in 4 patients, while the complete remission (CR) was not achieved in any patients.44) Since this compound is also a derivative of indolocarbazole, the plasma concentration is lower than expected. The clinical efficacy of this compound alone seemed to be limited, so a Phase 2 study with a combination of lestaurtinib and conventional chemotherapeutic agents was conducted.45) In this study, 224 patients with the first relapsed AML harboring FLT3 mutations were randomly assigned to chemotherapy (MEC or High-dose AraC) alone or a combination of chemotherapy and lestaurtinib. The CR/CRp rate in the combination group (16%) was not statistically better than that in the chemotherapy group (21%). Based on these results, further clinical development of lestaurtinib has been discontinued.
SECOND-GENERATION FLT3 INHIBITORS
The early phase studies of the first-generation FLT3 inhibitors for AML were unimpressive. However, these early studies have disclosed several problems (e.g.; maintaining the effective plasma concentration and serious adverse events) to be resolved before clinical use. Since the first-generation FLT3 inhibitors were not originally screened for the sensitivity and selectivity against the activated FLT3 kinase, the discovery and clinical development of novel FLT3 inhibitors in the second generation is required (Figure 2).
We developed a novel FLT3 inhibitor, KW-2449, which has a potent and unique kinase inhibition profile against FLT3 (IC50 is 6 nM), ABL, T315I-mutant ABL (ABL-T315I) tyrosine kinases as well as Aurora kinase.46) In a Phase 1 study of 37 patients including 11 FLT3 mutations, 10 of the 31 (32%) evaluable patients showed more than 50% reduction of leukemia cells in peripheral blood and BM. Although the maximum tolerated dose (MTD) was not established by the Phase 1 study, the compound was more rapidly metabolized than expected and the dephosphorylations of FLT3 and STAT5 were observed only for 8 hours. We, therefore, conducted the next phase 1/2 study, in which the administration schedule was changed from BID to TID or QID. However, we discontinued the study because of the gastrointestinal toxicity.
Sorafenib (Bayer) is a multikinase inhibitor, which has potency against RAF-1, VEGFR, PDGFR, KIT and FLT3, and has been approved for hepatocellular carcinoma and renal cell cancer.47) The IC50 value against FLT3 is 58 nM. Although clinical efficacy of sorafenib monotherapy was limited to the transient blast reduction in two Phase 1 studies, a combination with chemotherapy revealed a good CR rate in the relapsed and/or refractory AML patients with FLT3 mutations.48) However, since a combination of chemotherapy and sorafenib did not show better overall and event-free survivals than a chemotherapy alone in elderly AML patients, further studies are required to evaluate the efficacy and safety of the combination therapy.49)
Quizartinib (Ambit) has been screened for the affinity against FLT3 using the KinomeScan technique and has a high selectivity and sensitivity to FLT3. The IC50 value against FLT3 is less than 1 nM.50) Although the IC50 value against dephosphorylation of FLT3-ITD is 1.1 nM, this compound does not have a potency against FLT3-KDM. The preclinical study showed the high bioavailabilty and AUC.51) In a Phase 1 study, the QTc prolongation was the dose-limiting toxicity, while one CR and 4 incomplete CR (CRi) were observed in 18 AML patients harboring FLT3-ITD. In a Phase 2 study, 40 of the 79 (50.6%) patients with relapsed and/or refractory AML harboring FLT3-ITD achieved CRp or CRi. However, no CR was observed due to the BM suppression. In addition, the QTc prolongation was also observed even at the recommended dose, which was determined by the Phase 1 study. These results indicated the strong potency of quizartinib against AML cells harboring FLT3-ITD, while it is necessary to determine the optimal dose and/or schedule in the clinical use.
RESISTANT MECHANISM OF FLT3 INHIBITORS
Although no FLT3 inhibitors are approved for clinical use, several resistant mechanisms of FLT3 inhibitors have been disclosed through the early clinical studies. These resistant mechanisms are classified into primary and secondary resistances (Table 1).52,53) The primary resistance includes a different potency against types of FLT3 mutations, other activating signals and the lower potency against leukemia stem cells. In particular, different potencies between FLT3-ITD and FLT3-KDM have been apparent in many FLT3 inhibitors. KDMs trigger the activation loop into an active conformation affecting the binding affinity of FLT3 inhibitors.
Table 1.
Primary and secondary resistant mechanism of FLT3 inhibitor.
| Primary resistance | Secondary resistance |
|---|---|
| Low potency against FLT3-KDM | Acquiring resistant mutations |
| Other activating signals | FL stimulation |
| Low potency against leukemia stem cells | Overexpression of FLT3 |
The secondary resistance includes FL-dependent resistance, an acquisition of resistant mutations and overexpression of FLT3. It has been reported that the growth-inhibitory effect of FLT3 inhibitors is reduced by the addition of FL in vitro (Table 2).54) In a Phase 2 study of lestaurtinib, the plasma concentration of FL was increased during the treatment of lestaurtinib, and the high FL concentration reduced the clinical efficacy. The most important resistance mechanism is the acquisition of resistant mutations. The resistant mutation was not clearly understood in Phase 1/2 studies of the first-generation FLT3 inhibitors because of their low efficacies. However, several resistant mutations have been identified in the patients who were treated with quizartinib.55) It has been reported that D835Y, D835V, D835F or F691L mutations were additionally acquired at relapse in eight patients with FLT3-ITD who achieved CR by the quizartinib monotherapy. Since these mutated residues structurally exist around the ATP-binding pocket and the F691L mutation corresponds to the gate-keeper mutation, T315I, in the BCR-ABL gene, the binding of quizartinib to mutant FLT3 is blocked (Table 3 and Figure 3). These results raise further questions. The first-generation FLT3 inhibitors, except for tandutinib, are multikinase inhibitors, while their clinical efficacies were unimpressive and several adverse effects were apparent. Therefore, more selective and sensitive FLT3 inhibitors are thought to be better for clinical use. A selective and sensitive FLT3 inhibitor, quizartinib, shows the best clinical efficacy in clinically developed FLT3 inhibitors, but easily induces resistant mutation. It remains unclear whether FLT3-selective inhibitor is clinically better than multikinase inhibitor. However, development of a novel-type FLT3 inhibitor, which is selective and sensitive against both FLT3-ITD and FLT3-KDM, is much awaited. At present, ponatinib and crenolanib, which have potency against both FLT3-ITD and FLT3-KDM, are under clinical evaluation (Table 3).56,57)
Table 2.
Comparison of GI50 values of FLT3 inhibitors between FL absent and present conditions.
| FLT3 Inhibitors | GI50 value against MOLM14 (nM) | Inhibitory ratio | ||
|---|---|---|---|---|
| FL | 0 ng/mL | 10 ng/mL | ||
| Lestaurtinib | 3.3 | 6.8 | 2.1 | |
| Midostaurin | 7.2 | 25 | 3.5 | |
| Sorafenib | 3.3 | 12 | 3.6 | |
| Quizartinib | 0.38 | 1.3 | 3.4 | |
Addition of FL reduces the GI50 values of FLT3 inhibitors against human AML cell lines harboring FLT3-ITD, MOLM14.
Table 3.
GI50 values of FLT3 inhibitors against mutation types.
| Mutation type | GI50 (nM) [relative ratio to ITD] | |||
|---|---|---|---|---|
| Quizartinib | Sorafenib | Ponatinib | Crenolanib | |
| FLT3/ITD | 0.13 [1] | 1.3 [1] | 3.0 [1] | 1.3 [1] |
| FLT3/ITD+F691L | 102 [785] | 1189 [915] | 52 [17] | 67.8 [52.2] |
| FLT3/ITD+F691I | 122 [938] | 648 [498] | 4.2 [1.4] | – |
| FLT3/ITD+D835V | 120 [923] | 2209 [1699] | 349 [116] | – |
| FLT3/ITD+D835Y | 28 [215] | 675 [519] | 284 [95] | 8.7 [6.7] |
| FLT3/ITD+D835F | 166 [1277] | 2374 [1826] | 414 [138] | – |
| FLT3/ITD+D835H | 5.5 [42] | 164 [126] | 211 [70] | – |
| FLT3/ITD+D839G | 1.9 [15] | 112 [86] | 54 [18] | – |
| FLT3/ITD+Y842C | 33 [254] | 469 [361] | 229 [76] | – |
| FLT3/ITD+Y842H | 18 [138] | 260 [200] | 80 [27] | – |
Several resistant mutations have been acquired after the treatment of quizartinib, and most of them are resistant to sorafenib. However, ponatinib and crenolanib have inhibitory activities against those resistant mutations.
Fig. 3.
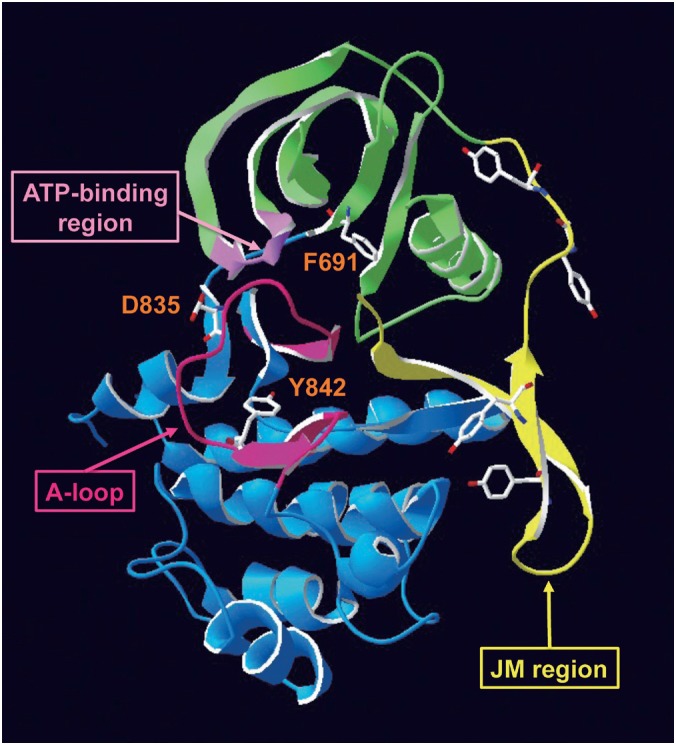
Structure of FLT3 and resistant mutations.
In the inactive form of wild-type FLT3, the JM domain blocks activation of the kinase and may inhibit self-dimerization. Mutations of D835 and Y842 residues in the A-loop induce a conformational change blocking the binding of FLT3 inhibitors to the ATP-binding-pocket. Since the F691 residue is a gatekeeper of the ATP-binding-pocket, its mutation also blocks the binding of FLT3 inhibitors.
CONCLUSION
Since FLT3 mutation is a poor prognostic factor for the long-term survival in AML patients, FLT3 is a promising therapeutic target. It has been proved by the quizartinib study that selective and continuous FLT3 inhibition could provide patients with CR. Development of FLT3 inhibitors will make for a more efficacious therapeutic strategy for leukemia therapy.
ACKNOWLEDGEMENTS
I would like to thank Ms. Yukie Konishi, Ms. Manami Kira and Ms. Emi Kohno for secretarial assistance.
CONFLICT OF INTEREST
H. Kiyoi receives research funding from Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Dainippon Sumitomo Pharma, Zenyaku Kogyo and FUJIFILM Corporation.
REFERENCES
- 1).Estey E, Dohner H. Acute myeloid leukaemia. Lancet, 2006; 368: 1894–1907. [DOI] [PubMed]
- 2).Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD; European LeukemiaNet. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood, 2010; 115: 453–474. [DOI] [PubMed]
- 3).Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med, 2013; 368: 2059–2074. [DOI] [PMC free article] [PubMed]
- 4).Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, Usui N, Kanamori H, Kiguchi T, Imai K, Uike N, Kimura F, Kitamura K, Nakaseko C, Onizuka M, Takeshita A, Ishida F, Suzushima H, Kato Y, Miwa H, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Naoe T. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia, 2014 Aug; 28(8): 1586–1595 [DOI] [PubMed]
- 5).Rosnet O, Marchetto S, deLapeyriere O, Birnbaum D. Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene, 1991; 6: 1641–1650. [PubMed]
- 6).Rosnet O, Schiff C, Pébusque MJ, Marchetto S, Tonnelle C, Toiron Y, Birg F, Birnbaum D. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood, 1993; 82: 1110–1119. [PubMed]
- 7).Lyman SD, James L, Zappone J, Sleath PR, Beckmann MP, Bird T. Characterization of the protein encoded by the flt3 (flk2) receptor-like tyrosine kinase gene. Oncogene, 1993; 8: 815–822. [PubMed]
- 8).Dehmel U, Zaborski M, Meierhoff G, Rosnet O, Birnbaum D, Ludwig WD, Quentmeier H, Drexler HG. Effects of FLT3 ligand on human leukemia cells. I. Proliferative response of myeloid leukemia cells. Leukemia, 1996; 10: 261–270. [PubMed]
- 9).Dehmel U, Quentmeier H, Drexler HG. Effects of FLT3 ligand on human leukemia cells. II. Agonistic and antagonistic effects of other cytokines. Leukemia, 1996; 10: 271–278. [PubMed]
- 10).DaSilva N, Hu ZB, Ma W, Rosnet O, Birnbaum D, Drexler HG. Expression of the FLT3 gene in human leukemia-lymphoma cell lines. Leukemia, 1994; 8: 885–888. [PubMed]
- 11).Drexler HG. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia, 1996; 10: 588–599. [PubMed]
- 12).Drexler HG, Meyer C, Quentmeier H. Effects of FLT3 ligand on proliferation and survival of myeloid leukemia cells. Leuk Lymphoma, 1999; 33: 83–91. [DOI] [PubMed]
- 13).Rosnet O, Bühring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D, Arnoulet C, Chabannon C, Kanz L, Hannum C, Birnbaum D. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia, 1996; 10: 238–248. [PubMed]
- 14).Zheng R, Levis M, Piloto O, Brown P, Baldwin BR, Gorin NC, Beran M, Zhu Z, Ludwig D, Hicklin D, Witte L, Li Y, Small D. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood, 2004; 103: 267–274. [DOI] [PubMed]
- 15).Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, Sonoda Y, Fujimoto T, Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia, 1996; 10: 1911–1918. [PubMed]
- 16).Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R, Naoe T. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood, 2001; 97: 2434–2439. [DOI] [PubMed]
- 17).Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol, 2001; 113: 983–988. [DOI] [PubMed]
- 18).Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, Radich JP. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood, 2001; 97: 3589–3595. [DOI] [PubMed]
- 19).Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, Ehninger G, Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood, 2002; 99: 4326–4335. [DOI] [PubMed]
- 20).Spiekermann K, Bagrintseva K, Schoch C, Haferlach T, Hiddemann W, Schnittger S. A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood, 2002; 100: 3423–3425. [DOI] [PubMed]
- 21).Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E, Ehninger G, Schiller G, Klimek V, Nimer SD, Gratwohl A, Choudhary CR, Mueller-Tidow C, Serve H, Gschaidmeier H, Cohen PS, Huber C, Fischer T. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood, 2005; 105: 335–340. [DOI] [PubMed]
- 22).Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, Tsuchida M, Sugita K, Ida K, Hayashi Y. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood, 2004; 103: 1085–1088. [DOI] [PubMed]
- 23).Kiyoi H, Naoe T. FLT3 in human hematologic malignancies. Leuk Lymphoma, 2002; 43: 1541–1547. [DOI] [PubMed]
- 24).Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer, 2003; 3: 650–665. [DOI] [PubMed]
- 25).Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia, 2003; 17: 1738–1752. [DOI] [PubMed]
- 26).Kottaridis PD, Gale RE, Linch DC. Flt3 mutations and leukaemia. Br J Haematol, 2003; 122: 523–538. [DOI] [PubMed]
- 27).Naoe T, Kiyoi H. Normal and oncogenic FLT3. Cell Mol Life Sci, 2004; 61: 2932–2938. [DOI] [PMC free article] [PubMed]
- 28).Kiyoi H, Yanada M, Ozekia K. Clinical significance of FLT3 in leukemia. Int J Hematol, 2005; 82: 85–92. [DOI] [PubMed]
- 29).Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. Lyon: WHO Press; 2008.
- 30).Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene, 2002; 21: 2555–2563. [DOI] [PubMed]
- 31).Zhao M, Kiyoi H, Yamamoto Y, Ito M, Towatari M, Omura S, Kitamura T, Ueda R, Saito H, Naoe T. In vivo treatment of mutant FLT3-transformed murine leukemia with a tyrosine kinase inhibitor. Leukemia, 2000; 14: 374–378. [DOI] [PubMed]
- 32).Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, Naoe T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene, 2000; 19: 624–631. [DOI] [PubMed]
- 33).Minami Y, Yamamoto K, Kiyoi H, Ueda R, Saito H, Naoe T. Different antiapoptotic pathways between wild-type and mutated FLT3: insights into therapeutic targets in leukemia. Blood, 2003; 102: 2969–2975. [DOI] [PubMed]
- 34).Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, Sargin B, Steffen B, Matsumura I, Kanakura Y, Böhmer FD, Müller-Tidow C, Berdel WE, Serve H.. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood, 2003; 101: 3164–3173. [DOI] [PubMed]
- 35).Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood, 2002; 99: 310–318. [DOI] [PubMed]
- 36).Pandey A, Volkots DL, Seroogy JM, Rose JW, Yu JC, Lambing JL, Hutchaleelaha A, Hollenbach SJ, Abe K, Giese NA, Scarborough RM. Identification of orally active, potent, and selective 4-piperazinylquinazolines as antagonists of the platelet-derived growth factor receptor tyrosine kinase family. J Med Chem, 2002; 45: 3772–3793. [DOI] [PubMed]
- 37).Kelly LM, Yu JC, Boulton CL, Apatira M, Li J, Sullivan CM, Williams I, Amaral SM, Curley DP, Duclos N, Neuberg D, Scarborough RM, Pandey A, Hollenbach S, Abe K, Lokker NA, Gilliland DG, Giese NA. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell, 2002; 1: 421–432. [DOI] [PubMed]
- 38).DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette R, Bruner-Klisovic R, Caligiuri MA, Cooper MR, LeCerf JM, Iyer G, Heinrich MC, Druker BJ. Phase II Evaluation of the tyrosine kinase inhibitor MLN518 in patients with acute myeloid leukemia (AML) bearing a FLT3 internal tandem duplication (ITD) mutation. Blood, 2004; 104: abstract 1792.
- 39).Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, O’Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood, 2005; 105: 986–993. [DOI] [PubMed]
- 40).Meyer T, Regenass U, Fabbro D, Alteri E, Rösel J, Müller M, Caravatti G, Matter A. A derivative of staurosporine (CGP 41 251) shows selectivity for protein kinase C inhibition and in vitro anti-proliferative as well as in vivo anti-tumor activity. Int J Cancer, 1989; 43: 851–856. [DOI] [PubMed]
- 41).Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood, 2005; 105: 54–60. [DOI] [PubMed]
- 42).George DJ, Dionne CA, Jani J, Angeles T, Murakata C, Lamb J, Isaacs JT. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and Trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555). Cancer Res, 1999; 59: 2395–2401. [PubMed]
- 43).Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood, 2002; 99: 3885–3891. [DOI] [PubMed]
- 44).Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood, 2004; 103: 3669–3676. [DOI] [PubMed]
- 45).Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, Erba H, Stuart RK, Baccarani M, Cripe LD, Tallman MS, Meloni G, Godley LA, Langston AA, Amadori S, Lewis ID, Nagler A, Stone R, Yee K, Advani A, Douer D, Wiktor-Jedrzejczak W, Juliusson G, Litzow MR, Petersdorf S, Sanz M, Kantarjian HM, Sato T, Tremmel L, Bensen-Kennedy DM, Small D, Smith BD. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood, 2011; 117: 3294–3301. [DOI] [PMC free article] [PubMed]
- 46).Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, Ishii K, Mori Y, Ozeki K, Minami Y, Abe A, Maeda H, Akiyama T, Kanda Y, Sato Y, Akinaga S, Naoe T. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood, 2009; 114: 1607–1617. [DOI] [PubMed]
- 47).Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res, 2004; 64: 7099–7109. [DOI] [PubMed]
- 48).Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, O’Brien S, Estrov Z, Borthakur G, Thomas D, Pierce SR, Brandt M, Byrd A, Bekele BN, Pratz K, Luthra R, Levis M, Andreeff M, Kantarjian HM. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol, 2010; 28: 1856–1862. [DOI] [PMC free article] [PubMed]
- 49).Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, Schaich M, Ottmann O, Duyster J, Wandt H, Fischer T, Giagounidis A, Neubauer A, Reichle A, Aulitzky W, Noppeney R, Blau I, Kunzmann V, Stuhlmann R, Krämer A, Kreuzer KA, Brandts C, Steffen B, Thiede C, Müller-Tidow C, Ehninger G, Berdel WE. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol, 2013; 31: 3110–3118. [DOI] [PubMed]
- 50).Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol, 2008; 26: 127–132. [DOI] [PubMed]
- 51).Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, Sprankle KG, Patel HK, Levis M, Armstrong RC, James J, Bhagwat SS. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood, 2009; 114: 2984–2992. [DOI] [PMC free article] [PubMed]
- 52).Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood, 2010; 116: 5089–5102. [DOI] [PubMed]
- 53).Weisberg E, Sattler M, Ray A, Griffin JD. Drug resistance in mutant FLT3-positive AML. Oncogene, 2010; 29: 5120–5134. [DOI] [PubMed]
- 54).Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, Small D, Burnett A, Levis M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood, 2011; 117: 3286–3293. [DOI] [PMC free article] [PubMed]
- 55).Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature, 2012; 485: 260–263. [DOI] [PMC free article] [PubMed]
- 56).Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, Salerno S, Shah NP. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood, 2013; 121: 3165–3171. [DOI] [PMC free article] [PubMed]
- 57).Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, Levis M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood, 2014 Jan 2; 123(1): 94–100. [DOI] [PMC free article] [PubMed]