

Abstract
Selection-based recombineering is a flexible and proven technology to precisely modify bacterial genomes at single base resolution. It consists of two steps of homologous recombination followed by selection/counter-selection. However, the shortage of efficient counter-selectable markers limits the throughput of this method. Additionally, the emergence of ‘selection escapees’ can affect recombinant pools generated through this method, and they must be manually removed at each step of selection-based recombineering. Here, we report a series of efforts to improve the throughput and robustness of selection-based recombineering and to achieve seamless and automatable genome engineering. Using the nucleoside kinase activity of herpes simplex virus thymidine kinase (hsvTK) on the non-natural nucleoside dP, a highly efficient, rapid, and liquid-based counter-selection system was established. By duplicating hsvtk gene, combined with careful control of the population size for the subsequent round, we effectively eliminated selection escapes, enabling seamless and multiple insertions/replacement of gene-size fragments in the chromosome. Four rounds of recombineering could thus be completed in 10 days, requiring only liquid handling and without any need for colony isolation or genotype confirmation. The simplicity and robustness of our method make it broadly accessible for multi-locus chromosomal modifications.
Introduction
Recombination-mediated genetic engineering, known as recombineering, is an efficient and flexible method for modifying host genomes, permitting researchers to delete, replace, and insert DNA at any targeted chromosomal site at single base resolution. In particular, the system using phage-based machinery (Lambda RED recombination system [1]) has been widely used because of its simplicity and flexibility. Because of the low frequency of homologous recombination of gene-size fragments (at most 10−4 recombinants per viable cell, even with the aid of the Lambda RED system [2]), two steps of homologous recombination followed by selection/counter-selection [3] are required. One of the issues of this selection-based recombineering is the shortage of convenient counter-selectable markers. As of today, all accessible counter-selectable markers require either solid media [4,5] or special mutant alleles [6–11].
Another problem with counter-selection is that the counter-selectable markers develop mutations, either by PCR amplification or by spontaneous mutations inside the cells, which inactivate the counter-selectable marker genes or generate mutant cells that are immune to the toxic effect of the counter-selectable marker [11]. Once such ‘counter-selection escape’ mutants emerge, they can quickly dominate the cell population and thereby preclude the enrichment of recombinants in subsequent rounds. The yield of recombinants (10−5 to 10−4/cell, depending on the context and length of the homology arms, it could be as low as 10−7/cell) is not much higher than or sometimes even lower than that of the emergent false-positives (10−6 to 10−5/cell). Consequently, experimentalists must manually pick multiple colonies and analyze their genotypes to isolate the correct clones in each round of selection-based recombineering.
With the maturation of synthetic and systems biology, there is a growing demand for genome modification technologies with more scalable and combinatorial nature. Emerging technologies such as Multiplex Automated Genome Engineering (MAGE) [12,13], Transcription Activator-Like Effector Nucleases (TALEN) [14], and Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) [15] are enabling researchers to modify chromosomes at an unprecedented scale. Notably, MAGE takes advantage of the ultra-high efficiency (0.1–0.3 events/cell [12]) in oligonucleotide-directed short-patch recombination to achieve simultaneous editing of multiple targets in a fully automated manner; given its selection-free format, experimentalists do not need to search for reliable selectable markers or continually combat selection escape. The applications of this technology range from the fast-track creation of a semi-rational chromosomal library (∼1010) in a search for isoprenoid hyper-producers [12] and the simultaneous incorporation of T7 promoters into 12 relevant genes [13], to the addition of histidine tags to 38 targeted genes [16] and the simultaneous replacement of hundreds of codons [17–19] spread over the genome. In theory, a conventional two-step selection-based recombineering method could be repeated for multiple cycles as well, if the aforementioned technical problems are solved. Given that two-step selection-based recombineering is a widely used and flexible technology, and given the lack for automatable multiplex genome engineering technology for gene-size fragments [20], we aimed to greatly improve the convenience, rapidity, and robustness of this method to enable the parallel and continuous operation of genome engineering.
In this paper, we describe our efforts to establish a repeatable workflow in which all recombination/selection steps can be rapidly and seamlessly operated using only liquid handling (Fig. 1). First, we report that the promiscuous activity of herpes simplex virus Thymidine kinase (hsvTK) [21] towards the unnatural nucleoside dP [22] can be used as a highly efficient and rapid counter-selectable marker for genome editing. Second, we also demonstrate that by duplicating the hsvtk genes while controlling the number of recombinants carried to the next round, counter-selection escapes can be effectively virtually exterminated from the experiments. This enables the seamless operation of parallel and iterative rounds of gene replacements and insertions, without the need to isolate and/or confirm the recombinants. The entire process is conducted via liquid handling only, making it adaptable for full automation.
Fig 1. The recombineering protocol adapted for multiple rounds.

First, the dP kinase activity of hsvTK is used for rapid and efficient counter-selection in liquid media. The mechanism of action of dP-selection is shown in the left panel. Second, the number of clones to be transferred between the steps is limited by diluting/aliquoting the transformant cultures, to prevent the propagation and subsequent domination of clones with PCR-generated inactivating mutations. Finally, the gene coding for hsvtk was duplicated to drastically reduce the emergence of selection escapes. TMK: thymidylate kinase, NDK: nucleoside diphosphate kinase, DNA pols: DNA polymerases, Km: kanamycin.
Material and Methods
Materials
The 6-(ß-D-2-deoxyribofuranosyl)-3,4-dihydro-8H-pyrimido[4,5-c][1,2]oxazin-7-one (dP) nucleosides [22] were purchased from Berry and Associates (Bishop Circle East, Dexter, MI; cat. PY7270). Oligonucleotides were synthesized by FASMAC Co., Ltd (Kanagawa, Japan). All other chemicals and media were of the highest grade available. Antibiotics were added to the growth medium as required at the following concentrations: 50 μg/mL carbenicillin (Carb), 30 μg/mL chloramphenicol (Cm), and 50 μg/mL kanamycin (Km).
Strains and plasmids
All plasmids used in this study are shown in Table 1. E. coli strain K-12 MG1655 was used throughout this study, although E. coli strain XL10-Gold (Stratagene, La Jolla, CA, USA) was used for the plasmid construction. The plasmid pKD46 [1], which enables the L-arabinose-mediated induction of the Lambda RED recombination system, was transformed into MG1655 and its derivative cells prior to the genome editing.
Table 1. Plasmids used in this study.
| Plasmid name | Genotype | Ori/marker | Source |
|---|---|---|---|
| pMW-hsvtk-cat | P lac-hsvtk-cat | pSC101/Kmr | This study |
| pHK | p T5-hsvtk mod-p L-Km r | ColE1/Ampr | This study |
| pHKH | p T5-hsvtk mod-p L-Km r-p tet-hsvtk | ColE1/Ampr | This study |
| pUC-p L-gfp mut3.1 | p L-gfp mut3.1 | ColE1/Ampr | This study |
| pJ204-p T5-mrfp | p T5-mrfp | ColE1/Ampr | This study |
| pET23d-mrfp | p T7-mrfp | pBR322/Ampr | This study |
| pKD46 | p BAD/AraC-Lambda REDγβα | pSC101/Ampr | [1] |
MG1655-AI (MG1655ΔaraB::T7rnap-tetA) was constructed by replacing its araB gene with a PCR-amplified portion of AraC/pBAD-T7RNAP from BL21-AI (Invitrogen Life Technologies, Carlsbad, CA). MG1655-tdk (MG1655Δtdk::tetRA) was constructed by replacing its thymidine kinase gene (tdk) with a PCR-amplified tetracycline resistant gene cassette (tetRA). MG1655ΔlacZ::hsvtk-cat was constructed as follows: the hsvtk-cat (where cat encodes chloramphenicol acetyltransferase gene), flanked by homology arms targeting the lacZ locus, was PCR-generated using the appropriate primers/template (S1 Table) using VentR DNA polymerase (New England Biolabs). The resultant hsvtk-cat cassette (HC cassette) was electroporated into MG1655 cells, and the transformant cells were enriched in media containing Cm. The resultant MG1655ΔlacZ::hsvtk-cat cells were checked for their dP sensitivity (dP kinase activity) by spotting onto an LB-agar plate containing 1 μM dP.
Construction of the DNA cassettes
Selection cassettes and insertion cassettes were PCR-amplified with primers that added the appropriate homology to the target genomic regions. The primer pairs used to generate each DNA fragment are summarized in S1 Table. Unless otherwise noted, PCRs were performed with KOD DNA polymerase (TOYOBO, Osaka, Japan). PCR products were DpnI–treated and gel-purified to eliminate the template (plasmid or genomic DNA).
Preparation and use of electroporation-competent cells
Electroporation-competent cells were prepared as described by Datsenko and Wanner [1]. Briefly, cells harboring pKD46 were grown in LB medium supplemented with 50 μg/mL Carb and 10 mM arabinose to induce the expression of Lambda RED enzymes. When the OD (λ = 600 nm) of the culture reached 0.4–1.0, the cells were placed on ice and washed twice with ice-cold water and 10% v/v glycerol, followed by resuspension in 20% v/v glycerol to make an electroporation-competent stock.
To the resulting electroporation-competent cells (40 μL), 100 ng of purified PCR fragments was added on ice. The mixture was then subjected to electroporation in 0.1-cm-gap cuvettes (Bio-Rad, Hercules CA) at 1.8 kV in a Gene Pulser electroporation apparatus (Bio-Rad, Hercules CA).
dP-selection and efficiency evaluation
The efficiency of counter-selection using chromosome-encoded hsvtk/dP was evaluated by replacing genome-integrated HC cassette by PCR-amplified 3 kbp fragments containing the lacZ gene flanked by 1 kbp homology arms; 500 ng of purified PCR fragment was electroporated to 40 μL of electroporation-competent MG1655ΔlacZ::hsvtk-cat cells. The resultant transformant culture was resuspended in 1 mL of LB medium and shaken at 37°C for 3 h. The cured cells were then inoculated into fresh LB medium containing dP (1 μM). After shaking for 1–8 h at 37°C, a portion of the culture was plated on LB agar plates with 0.4% wt/v X-Gal and 0.1 mM IPTG and incubated at 37°C. At each time point, the ratio of recombinants/non-recombinants was determined by the number-ratio of blue/white colonies.
Determination of the frequency of escape in dP-selection
Overnight cultures of the MG1655 strain or its derivatives were inoculated into fresh LB plate containing dP (at a final concentration of 0–103 nM) and appropriate antibiotics. The colonies (if any) were allowed to grow on the plate for 12 h at 37°C. The frequency of dP-selection escape was calculated by:
[Number of colonies observed on 1 μM dP plates]/[Number of colonies on the plate without dP]
Mutation frequency of E. coli strain MG1655 in dP-containing medium
Approximately 106 cells of the MG1655 strain harboring pKD46 were inoculated into fresh LB media (2 mL) containing ampicillin (100 μg/mL). Then, dP (at a final concentration of 0–103 nM) was added to the culture. After 6 hours of shaking, a portion (1 mL) of the culture was collected, quickly washed in fresh media, and then plated on LB-agar with or without rifampicin (rif assay [23]). The colonies (if any) were allowed to grow on the plate for 12 h at 37°C. According to the literature [24], there are ten unique nucleotide positions in rpoB that can alone confer resistance to rifanpicin resistance to E. coli. Given this, the mutation frequency of each sample was defined using the following equation. Mutation frequency [mutation/bp] = Rifampicin resistant clones [c. f. u.]/total cell number [c. f. u.] × 10
Sequential operation of gene insertion/replacement
Step 1: The PCR-amplified selection cassette flanked by the appropriate homology arms (100 ng) was electroporated into electroporation-competent MG1655-AI cells (40 μL). The transformants were immediately resuspended in 1 mL of LB-Carb medium for at 30°C for 3 h. Usually the culture contained 101–104 recombinants depending on the homology-arm length and the length of DNA fragment. Thereafter, the culture was serially conducted 10-fold dilution and divided into aliquots in some of which the number of recombinants (Km-resistant cells) was limited to less than 10, where the probability that the aliquote contains false-positive clones are expected to be less than 2% (calculation provided in Discussion). More specifically, the aliquots was inoculated into fresh LB medium (2 mL) with Carb and Km and incubated for 24 h at 30°C. From the visibly turbid culture with highest dilution rate, cells were harvested and inoculated into fresh LB medium containing Carb, Km, and arabinose (10 mM) used for preparing electropotation-competent cell for recombineering as described above.
Step 2: A purified DNA fragment coding mrfp under the control of the T7 promoter and flanked by appropriate homology arms (p T7-mrfp, 100 ng) was added to the prepared electroporation-competent cells (40 μL). The resulting mixture was electroporated and resuspended in 1 mL of LB-Carb medium. After curation (3 h at 30°C), a portion of the culture was inoculated into fresh LB medium containing Carb and dP (1 μM). The aliquot was shaken at 30°C for 18–30 h, until it reached to stationary phase. Harvested cells were inoculated into fresh LB medium containing Carb and arabinose (10 mM) used for preparing electropotation-competent cell for recombineering as described above. This stock was then used in Step 1 of the next round.
Four rounds of recombineering (two-step replacement of the target sequences with p T7-mrfp) were conducted by repeating Step 1 and Step 2, targeting yiiDE, proV, lacZ, and rssB in a sequential manner.
Fluorescence analysis
The fluorescence of the E. coli strains was measured with a Fluoroskan Ascent (Thermo-Labsystems, Helsinki, Finland). The following excitation/emission pairs (in nm) were used: GFPmut3.1, 485/527, mRFP, 584/620. To correct for variations in cell number, the fluorescence intensities were normalized by the cell density (OD600) of the culture measured in transparent 96-well plates with a SpectraMax Plus 384 system (Molecular Devices, Sunnyvale, CA).
Flow cytometric analyses of the cell mixtures, were conducted as follows: immediately after each round of recombineering (or after recovery from the glycerol stock), the cell mixture was grown in LB medium in test tubes (at 37°C at 200 rpm). Then, 20 μL of the overnight cultures was added to 2 mL of fresh LB medium containing 10 mM arabinose and shaken in a test tube at 37°C for 3 h. Approximately 10,000 cells were applied to a MACSQuant VYB flow cytometer (Miltenyi Biotech, Bergisch-Gladbach, Germany) equipped with a 561 nm laser and appropriate filter sets for mRFP (586/15). The data were analyzed by using the MACSQuant Analyzer (Miltenyi Biotech, Bergisch-Gladbach, Germany).
Results
Chromosomal insertion of hsvtk
HsvTK has long been used as a selectable marker in E. coli [25,26]. In this system, selection is performed by adding 2’-deoxy-5-fluorouridine (5FdU) to the culture media. Here, 5FdU was endogenously phosphorylated to 5FdU-MP, a potent inhibitor of thymidylate synthase (thyA). By blocking the de novo biosynthesis of dTMPs [25,26], cell growth is prevented. When dT is exogenously added and hsvTK is expressed, the biosynthesis of dTMPs (and cell growth) is restored via its salvage pathway.
We first attempted to apply this mechanism for the selection of the recombinants. We designed a selection cassette encoding hsvTK and another selectable marker chloramphenicol acetyltransferase (CAT) (HC cassette). Using the PCR primers shown in S1 Table, we generated double-stranded DNA that harbors HC cassette flanked with short homology arms (191 nt and 41 nt in length, respectively) targeted to the lacZ locus. The DNA fragment (2,132 bps) was electroporated into a cell harboring the Lambda RED system, followed by plating on Tryptone agar containing 5FdU/dT. Although this process successfully enriched the recombinants (MG1655-tdk, ΔlacZ::hsvtk-cat), they consisted only a part of the surviving population. We found the resulting recombinants selectively grew in media containing 5FdU/dT (data not shown), indicating that the titer of the thymidine salvage pathway is sufficiently high with single-copy hsvtk. However, we observed the frequent emergence of 5FdU-resistant clones, possibly because many different amino acid substitutions can confer thyA resistance to its inhibitors (5FdU) [27]. In contrast, we could easily isolate the right recombinants using chloramphenicol selection (Cm-selection) without being bothered by false-positive clones.
dP kinase as a counter-selectable marker for genome engineering
We recently reported that hsvTK has efficient kinase activity against the nucleoside analogue dP [21]. When hsvTK is expressed from middle- to high-copy number plasmids, cells efficiently incorporate dP into the genomic DNA. Because P can base-pair with either A or G [22,28], its incorporation results in the destruction of the genetic information of the cell. By adding dP to the growth media, plasmid-borne hsvtk effectively and rapidly kills the host E. coli strain (#1 in Fig. 1). This system (dP-selection) was originally developed to select for the OFF state of the genetic switches and circuits on multicopy plasmids [21]. It has not been tested whether similar killing efficiency can be produced using chromosome-encoded hsvtk.
HC cassette was electroporated to MG1655, followed by Cm-selection. From the resultant transformants (MG1655-tdkΔlacZ::hsvtk-cat), we randomly picked 92 clones and tested for their dP kinase activity. We found that most (89/92) of the clones completely lost their cell viability (not shown), indicating that the activity of chromosome-coded hsvtk is still high enough to cause cell death of the hosts, making it useful for genome engineering. When tried-and-true dP-sensitive clone (MG1655ΔlacZ::hsvtk-cat) was tested on dP-selection plate containing 1 μM dP, 5 × 103 escape colonies were appeared per 3 × 108 colonies on LB plate without dP, thereby yielding selection escape frequency of 2 × 10−5 (Fig. 2A). With this culture condition, the mutation frequency of the cells to be selected (those not expressing hsvTK) was only 10-fold higher than untreated cells (Fig. 2B).
Fig 2. dP/hsvTK counter-selection.

(a) Host-killing efficiency of dP-selection with different dP concentrations for E. coli strain MG1655ΔlacZ::hsvtk-cat harboring pKD46. (b) Mutation frequency of E. coli strain MG1655/pKD46 in the media containing varying concentration of dP. The bar heights show the average of 3 samples, and error bars indicate the standard deviation.
Time required for dP-selection
Another unique feature of the dP-selection is the speed of the selection procedure [21]: on plasmid, selection time could be reduced to as short as 5 min. To determine whether this is also the case in genome engineering, we analyzed the effect of the selection time on the selection efficiency. We attempted to replace the HC cassette in the chromosome of MG1655ΔlacZ::hsvtk-cat cells with lacZ (Fig. 3A). After electroporating a PCR-generated lacZ gene, the transformant culture was split into two and then shaken in the presence or absence of dP (1 μM). A portion of the culture was collected at various time points and plated onto LB plates containing X-gal. At time zero, the amount of LacZ positive (blue) clones was only 10−4 (ca. 100 recombinants/106 total cells/mL). In the culture without dP, this rate was unchanged during the experiment. In the culture containing dP, this fraction drastically increased over time (Fig. 3B). Because dP-based counter-selection sterilize cells rather than inhibit cell growth [21], this selection does not require overnight growth. Indeed, the selection efficiency quickly plateaued approximately 2 hours after dP addition.
Fig 3. Liquid-based counter-selection using dP kinase activity.

(a) Experimental procedure. The selection cassette inserted in lacZ region of E. coli chromosome was replaced back to lacZ by eletroporating PCR-generated lacZ gene. Transformant cells were incubated at 37°C in dP-containing LB medium, and a portion of the culture were plated at each time points (0–8 hrs). The number of recombinants was estimated by counting the number of blue colonies per plate. (b) Ratio of recombinant cells (LacZ +) various periods after adding dP, with (open circles)/without (closed circles) dP treatment (1 μM).
Inactivation of counter-selectable markers by PCR errors
During the creation of MG1655-tdkΔlacZ::hsvtk-cat, three dP-resistant clones emerged (see above). We found that all three clones possessed non-synonymous and likely inactivating mutations within the coding regions of hsvtk (S2 Table). These mutations were likely derived from replication errors in PCR amplification: we found that the frequency of dP-resistant/Cm-resistant clones (which possess HC cassette with an inactivated hsvTK) is dependent on the fidelity of the polymerase used for the PCR amplification of the cassette (S3 Table). Postulating that 25% of nucleotide substitutions result in the deleterious missense/nonsense mutations [29,30], and given the inactive fraction of hsvtk to be 0.8% (S3 Table), nucleotide mutations per hsvtk gene can be calculated to 0.8%/0.25 = 3.2% for HC cassette amplified with VentR DNA polymerase. Given the nucleotide size of hsvtk (1,129 bps) and amplification factor 10, corresponding to ca. 1,000 fold amplification, error rate of VentR DNA polymerase is calculated to be 0.032 [mutation/gene]/10 [amplification factor]/1,129 [bps/gene] = 3 × 10−6. These values are similar to the reported error values for VentR DNA polymerase (2.8 × 10−6 mutations/base/replication [31]). Thus, inactivating mutations are not enriched in hsvtk, indicating that hsvtk is not under strong counter-selection in E. coli. This is in contrast to other counter-selectable markers such as SacB, which is known to be under severe counter-selection [32,33]. Note that the native (cognate) function of hsvTK (dT kinase) is non-toxic; its toxicity is exerted only when dP is added to the media.
False-positive clones generated by errors in chromosomal replication
Bacterial chromosomes are continuously accumulating spontaneous mutations over generations, such that any given gene could be inactivated with a certain frequency. Even starting from tried-and-true dP sensitive clone, we obtained one dP-resistant clone for every 105 cells after the selection step (Fig. 2A). Given 10−5 as then inactive fraction of hsvtk (Fig. 2A) and postulating about 25% of nucleotide substitutions are inactivating [29,30], and nucleotide mutations per hsvtk gene can be calculated to 10−5/0.25 = 4 × 10−5. Considering that the cell pool was grown for 20 generations (106 fold amplification), error rate of chromosome replication is calculated to be 4 × 10−5 [mutation/gene]/20 [amplification factor]/1,129 [bps/gene] = 2 × 10−9, which is close to the reported values (10−10–10−9 [34]). Considering the number of recombinants (10−5–10−4 with the aid of Lambda RED system) that we routinely obtain, dP-resistant clones (10−5) generated by errors in chromosomal replication represent a non-negligible source of false-positives.
Mutational escape impedes the continuous operation of genome engineering
Once chromosomal hsvtk is inactivated by mutations, either by PCR error or by cellular spontaneous mutations, the resultant clone is expected to quickly dominate the population at the subsequent step (dP-selection). To confirm this, we attempted the two gene replacement steps (ΔlacZ::p L-gfp mut3.1 and ΔproV::p T5-mrfp) by using hsvtk-Km r cassette (where Km r encodes a kanamycin resistant gene, so called ‘HK cassette’) (Fig. 4).
Fig 4. Duplication of hsvtk enables a two-round consecutive recombineering.

(a) Killing efficiency of dP-selection against E. coli strain harboring a single hsvtk gene (HK cassette) or duplicated hsvtk (HKH cassette), ND: Not detected. The bar heights show the average of 3 samples, and error bars indicate the standard deviation. (b) The experimental workflow for a two-round consecutive recombineering: lacZ and proV loci were serially replaced with pL-gfp mut3.1 and pT5-mrfp cassette respectively using HK or HKH. All DNA cassettes used here were prepared with 40-bp homology arms to the individual target sites, except the step for replacing lacZ with pL-gfp mut3.1 cassette where 1-kbp homology arms were adopted.
We conducted the first round of recombineering, where we inserted p L-gfp mut3.1 [35] via a deletion coupled insertion of HK cassette, resulting in MG1655ΔlacZ::pL-gfp mut3.1. After this round, 90% (85 of 94 clones) of the resultant population showed fluorescence of GFPmut3.1, whereas the rest of the populations (9 clones or 10% of the entire population) did not (Table 2). Next, we pooled all of the obtained clones without removing these false-positive clones and proceeded to the second round of recombineering (ΔproV::p T5-mrfp). In this round, we obtained numerous non-fluorescent colonies after the first-half step (insertion of the HK cassette followed by kanamycin selection (Km-selection)). After the completion of the procedure for the second-half step (electroporation of the p T5-mrfp cassette followed by dP-selection), the pool was completely dominated by non-fluorescent clones with the Kmr phenotype (Table 2). We picked nine of the non-fluorescent clones in the pool after the first round and sequenced their ΔlacZ::hsvtk-Km r locus (S4 Table). The eight out of nine non-fluorescent clones were selection-escape clones retaining HK cassettes with mutations in hsvtk: four had non-synonymous mutations in the hsvtk gene, three had a deletion of the entire hsvtk gene, and one was lacking the entire HK cassette. We found one non-fluorescent clone that had the gfp mut3.1 gene at the proper position but also contained a stop codon in the same reading frame. We do not know where these mutants were generated. Note that we did not select for the acquisition of fluorescence.
Table 2. Functional and genotype analysis of recombinant pools.
| Round-1 | Round-2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Phenotype (fluorescence) | GFP | + | - | + | + | - | + | |
| RFP | + | - | + | - | ||||
| genotype 1 (PCR) | NT 2 | + | - | NT 2 | + | + | NT 2 | |
| HK cassette | 85(90%) | 1(1%) | 8(9%) | 0(0%) | 0(0%) | 0(0%) | 94(100%) | |
| HKH cassette | 94(100%) | 0(0%) | 0(0%) | 90(96%) | 2(2%) | 2(2%) | 0(0%) | |
After each round of recombineering depicted in Fig. 4B, 94 clones were randomly picked, grown, and analyzed with regard to both phenotype (fluorescence) and in genotype (genomic PCR).
1. Number of clones with PCR bands corresponding to ΔlacZ:: p L-gfp mut3.1 (for round-1) or ΔlacZ::p L-gfp mut3.1 and ΔproV::p T5-mrfp (for round-2). For the sequences of primers used for genotype analysis (P34 and P42 for p L-gfp mut3.1, P35 and P39 for p T5-mrfp), see S1 Table.
2. NT; not tested.
Duplication of the counter-selectable marker
A drastic decrease in the spontaneous mutation rate is known to be difficult to achieve [36]. The inactivation of the hsvtk gene is therefore an inevitable event in a cell population. We designed a new selection cassette that contains two copies of the hsvtk gene. The idea was that dP kinase activity should be maintained unless both of the hsvtk genes are inactivated. Because the frequency of hsvtk gene to be inactivated is 10−6/cell for HK cassette (Fig. 4A), the frequency of inactivating both hsvtk genes should theoretically be 10−12/cell/step (#3 in Fig. 1). Considering the number of transformants (10−5–10−4/round [2] with 40-base homology arms) that we routinely obtain, the fraction of escapee is estimated to be as low as 10−8–10−7 of the population.
We constructed a new selection cassette with two hsvtk genes (HKH cassette, Fig. 4A). Here, it was expected that simple duplication in hsvtk would result in highly frequent homologous recombination between these genes, thereby promoting the appearance of dP-resistant clones. In E. coli, homologous recombination occurs most frequently when there is a contiguous stretch of sequence >25 bp [37–39]. We therefore synthesized the codon-modified hsvtk (hsvtk mod), which has 72% nucleotide identity to the original version. In this design, the longest stretch of successive nucleotides identical to the original hsvtk contained eleven nucleotides (S1 Fig.). Also, we placed the selectable marker (Km r) between the two hsvtk genes. Thus, if inter-hsvtk recombination occurred, it would result in the loss of Km-resistance as well, thereby effectively removing the clone in subsequent rounds of genome engineering. When E. coli strain MG1655ΔlacZ::hsvtk mod-Km r-hsvtk) was tested on dP-selection plate (containing 1 μM dP), no counter-selection escape colonies were appeared per 6 × 109 colonies on LB plate (without dP) (Fig. 4A).
We conducted continuous two-step recombineering with this new HKH cassette (Fig. 4B). This time, all 94 tested clones were fluorescent and Km-sensitive after the first round of Km-/dP-selection (Table 2). This indicates that the cell population was virtually free from selection escape (clones holding a mutated HKH cassette). The cell mixture was directly subjected to the second round of recombination. Even after the second round of recombineering (ΔproV::p T5-mrfp), all (94/94) of the tested clones were double-recombinants (harboring both p L-gfp mut3.1 and p T5-mrfp cassettes) (Table 2). Thus, duplicating the counter-selectable marker gene (hsvtk) significantly reduced the counter-selection escape, allowing us to directly apply the recombinant pool to subsequent rounds of recombineering without colony isolation or genotyping.
Continuous multiple gene replacements with mRFP
Having established a seamless workflow for multi-round selection-based recombineering (Fig. 4B), we attempted to apply the method for the iterative replacement of four selected loci, yiiDE, proV, lacZ, and rssB, to an E. coli chromosome with a PCR-generated fragment harboring mrfp under the T7 promoter (p T7-mrfp) (Fig. 5A). To enable the expression of mRFP, araB locus of MG1655 was replaced with T7 rnap. The resultant strain MG1655-AI was subjected to the four-round recombineering. In each round, the PCR-amplified HKH cassette with homology arms (40 bp in length) targeted to the corresponding sites was electroporated into the cells (MG1655-AI), followed by Km-selection (24 h). p T7- mrfp was electroporated into the resultant cell culture, followed by 18–30 hours of growth in media containing dP. The resultant culture (recombinant pool) was divided into two pools: one was stored as an intermediate recombineering pool for analysis, whereas the other pool was subjected to the next round of recombineering. This procedure was repeated without break for four rounds.
Fig 5. Sequential replacement of four chromosomal loci with DNA fragments encoding p T7-mrfp.
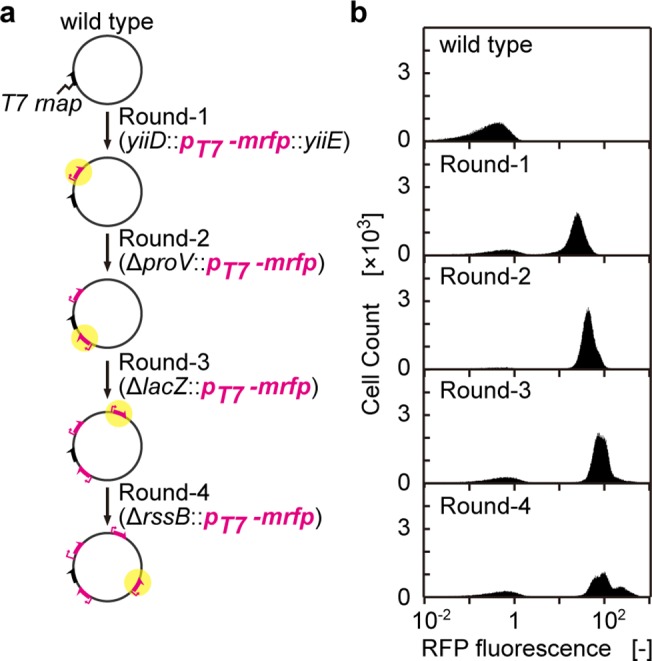
(a) The production flow chart for four-round gene replacement. (b) Flow cytometric analysis of the recombinant pool after each round of recombineering.
At each step of the four-round gene insertions/replacements, we checked the phenotype (RFP fluorescence) of the recombinant pool via flow cytometry (Fig. 5B). The fluorescence signal progressively increased with increasing round number, indicating a step-by-step increase in the copy number of the p T7-mrfp cassette on the chromosome.
The progressive integration of the p T7-mrfp cassettes was also verified by PCR analysis (Fig. 6). After each round of Km- or dP-selection, a portion of the ‘intermediate’ cell mixture was collected and subjected to PCR analysis. In each PCR analysis, a mixture of the four relevant primers (S1 Table) was used for each locus. The primer mix included the common forward primer and three reverse primers designed to anneal to the original target (non-recombinant), HKH cassette, or p T7-mrfp (see the left panel of Fig. 6). In the first round, only the band (218 bp) corresponding to the original sequences (yiiDE) was observable for the starting cell mixture. After the replacement of the sequence with the HKH cassette, we observed a single-banded PCR product (322 bp) corresponding to the HKH cassette inserted. After electroporation of the p T7-mrfp cassette followed by dP-selection, the band (765 bp) corresponding to the p T7-mrfp-inserted genome was observed as a single band. For each of all subsequent three rounds, we observed the complete shift in genotype at the targeted locus (Fig. 6). It should be noted that PCR was conducted directly from the culture, not from individual isolates. Thus, the data shown in Fig. 6 represent the genotype distribution of the entire population. No incorrect bands were detected in any of the eight steps (4 rounds), indicating that the number of clones with incorrect genotypes, including selection escape, was negligible. Indeed, we picked and tested 7 individual clones from the round-4 cell population (S2 Fig.) and could not identify a single clone that gives the incorrect PCR patterns.
Fig 6. Genotyping of the cell population in each step of mrfp replacement in four different loci.

Before, after, and during the 4 successive rounds of gene replacement, all 9 cultures were subjected to competitive PCR analysis using 3 primers annealing to the target, HKH cassette, and p T7-mrfp cassette sequences. The local sequence, location of the primers and expected size (in bp) of the PCR products are presented for the (1) yiiDE, (2) proV, (3) lacZ, and (4) rssB loci. The sequences of the primers used are shown in S1 Table.
We have not invested any effort in shortening the operation time of each step. Nevertheless, one round is completed in two to three days (see the experimental section), depending on the growth rate of the recombinants. In this case (Figs. 5 and 6), four rounds of recombineering were completed in ∼10 days. Also note that all of the steps were conducted using only liquid handling, without the need for a manual procedure for identifying the right recombinants by colony picking and conducting multiple PCR reactions for each colony.
The cellular fluorescence slowly leveled off with the increase in recombineering rounds. Additionally, we observed a significant retardation in the growth rate of cells in later rounds. We do not know whether this retardation came from: it could be possibly ascribed to the metabolic load of over-expression of RFP, to the multiple deletions of four genes, or by accumulated mutations throughout the chromosome. At the fourth round, we observed a two-peaked fluorescence, together with a minor fraction with no fluorescence. We believe that the lower peak of the two-peaked fluorescence represents clones with three functional mrfp and one mrfp inactivated by PCR errors, whereas the non-fluorescent peaks represent clones with inactivation mutations in the reading frame of the T7 RNA polymerase.
Discussion
Rapid advances in the field of synthetic biology are creating a demand for robust, broadly accessible methodologies for manipulating multiple genomic loci in a high-throughput manner. In this study, we described dP kinase activity can be used as highly efficient and rapid counter-selectable marker in genome engineering (Figs. 2 and 3). Because all required procedures can be completed by liquid handling only, different recombineering projects can be conducted in parallel and in multi-well formats.
Our dP-selection is mechanistically based on the lethal mutagenesis [28], which is not generally favored by genome engineers. However, with our protocol (i.e. treatment with 1,000 nM dP), the mutation frequency of the cells to be selected (those not expressing hsvTK) was only 10-fold higher than that of cells not treated with dP (Fig. 2B). Note that this mutation frequency is much lower than that of the mutS mutant strain (100-fold higher mutation frequency than that of wild type strain [40]) used in the MAGE system. We found no detectable increase in the chromosomal mutation frequency of E. coli in the presence of 100 nM dP. Engineering or searching for a more efficient dP kinase (with a lower KM for dP) or over-expression of nucleoside importers [41] could be effective in further decreasing the concentration of dP required for the counter-selection process, thereby also decreasing the mutation rate of the recombinant cells.
The largest obstacle for the continuous/automatable operation of gene-size recombineering is the emergence and propagation of selection escapes generated by inactivating mutations in counter-selectable marker genes. By duplicating the hsvtk genes (using the HKH cassette), the frequency of their appearance could be reduced to <2 × 10−10/cell during cell growth (Fig. 4A). We picked nine dP-resistant clones and sequenced in its hsvtk locus. All clones had mutations in hsvtk (S5 Table). We confirmed that these mutations alone confer dP-resistance to E. coli (S3 Fig.). These results indicate that, very few, if not zero, of the escape events comes from the off-site mutations in dP-selection. This is in sharp contrast to other counter-selection mechanisms: for example, Gregg et al. reported that in tolC-based systems, ‘off-target’ inactivating mutations accounted for significant part of the escape events [11]. They succeeded in lowering the frequency of selection escape not by duplicating the selector tolC cassette, but rather by duplicating the off-site mutational hotspot (tolQRA) [11]. Or, selection stringency could be improved by combinatorial use of different types of counter-selectable markers in tandem to improve the selection stringency [5,6,11].
Although duplication of hsvtk was highly effective in decreasing the frequency of the dP-resistant cassette, we still observed a non-negligible frequency of clones with the dP-resistant cassette. When the HKH cassette was PCR-generated, where 0.2–0.8% of individual hsvtk could receive inactivating mutation (S3 Table), up to 0.4–6 × 10−5 of the genome-integrated HKH cassette could lack dP kinase activity. This is comparable to the rate of recombination (10−5 to 10−4/cell [2]).
The appearance of such parasitic entities has also been an important issue for autonomous replication systems [42–44]. Once they appear, they can eventually dominate the population and inhibit the further evolution of replication systems. Theoretical [45–47] and experimental [44] works have shown that spatial structures such as compartments could effectively repress the propagation of parasites by quarantining them and thereby preventing them from taking over the entire population.
Inspired by this, we split the pool of recombinants and restricted the number of recombinants in each pool to be fed to the next step to be << 100 (#2 in Fig. 1). Throughout this work, we used the PCR-generated cassette to achieve fast-track genome engineering. Even with the highest-fidelity polymerases, 0.2% of the recombinants had inactivated hsvtk (S3 Table). When the number of recombinants per aliquot is set to be 10, the probability of having selection escape would be 2% and 0.004% for HK cassette and HKH cassette, respectively. Although we have not encountered such throughout this work, each aliquot has a certain risk to be contaminated with selection escapes generated by PCR error, and the risk increases with the increase of the pool size. Because recombination efficiency is context dependent per se, the proper dilution ratio could differ every single time. Our recommendation is to prepare the dilution series of the recombinant pool such that several dilutions can be independently subjected to the next round of recombineering in parallel: the contaminated aliquot, if appears, could be easily distinguished from others in the subsequent processes. This way, the passage can be kept free from selection escapes, despite the lower replication fidelity of PCR.
Because PCR-error is much higher compared with genome replication [31], this removal of PCR-generated selection escape is the key. Once PCR-generated selection escapes are removed this way, or if the selection cassettes were directly prepared from sequence-confirmed plasmids, selection escapes (dP-resistant/Km-resistant clones) could be as low as 10−6 and 10−12 for HK cassette and HKH cassette respectively. If all DNA cassettes are prepared from plasmids (PCR-free), population-control procedure (#2 in Fig. 1) could be omitted.
In summary, we have established, for the first time, a repeatable workflow for gene replacement/insertion where all the steps can be seamlessly operated using only liquid handling with a re-designed selection cassette. Given its simplicity and flexibility, we predict that this proven technology will be valuable as a tool for high-throughput and multi-target genome editing projects.
Supporting Information
Identical bases are highlighted with grey-shading.
(TIF)
Seven clones were isolated and individually subjected to PCR analysis using primers annealing to the mrfp gene. The local sequence, location of the primers, and expected size (in bp) of the PCR products are shown for the (1) yiiDE, (2) proV, (3) lacZ, and (4) rssB loci. The sequences of the primers used are shown in S1 Table.
(TIF)
Mutant hsvtk gene identified in dP-selection escapees (S5 Table) were PCR-amplified and inserted into the lacZ locus of MG1655. The resultant recombinants were grown in LB media (0.5 mL) containing Km (50 μg/mL). From each culture, defined number of cells were plated onto LB-Km-agar containing 0 (open bar) or 1 μM (closed bar) of dP. Viability was determined by the colony forming units on each plate. The corresponding clone number in S5 Table is/are also given in parenthesis.
(TIF)
(PDF)
Out of the 92 transformants (chloramphenicol-resistant clones isolated from the pool of MG1655 electroporated with HC cassette), we found three dP-resistant clones. The cassette inserted on chromosome was PCR-amplified using primer P30 and P32 (S1 Table) for the sequence analysis.
(PDF)
The HC cassette was amplified with either VentR DNA polymerase or Phusion DNA polymerase using primer P1 and P2 (S1 Table) to attach homology arms for targeting to the lacZ locus. The resultant DNA fragment was electroporated into E.coli strain MG1655 harboring pKD46 and plated onto an LB-Cm and LB-Cm/dP plate. The frequency of dP resistance (%) was calculated by: 100 × [number of colonies observed on Cm/dP plates] / [number of colonies on the Cm plate] The numbers show the average of 3 samples with standard deviations.
(PDF)
From the first round of two-step recombination using the HK cassette (Table 2), nine clones without GFPmut3.1 fluorescence were individually picked and analyzed in their lacZ (or ΔlacZ::hsvTK-km r) locus. For the sequence of primers used for PCR amplification and sequencing (primers P30, 33, P43, and P44), see S1 Table.
(PDF)
dP sensitive clone was cultured overnight and plated on dP-selection plates. From these plates nine clones were picked and sequenced in their hsvtk coding regions.
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Commission for Development of Artificial Gene Synthesis Technology for Creating Innovative Biomaterial from the Ministry of Economy, Trade and Industry (METI, Japan), the Precursory Research for Embryonic Science and Technology (PRESTO) program of the Japan Science and Technology Agency (JST), and Grant-in-Aid for Scientific Research on Innovative Areas (23108507) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/Japan Society for Promotion of Science (JSPS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Datsenko KA, Wanner BL, One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sawitzke JA, Thomason LC, Costantino N, Bubunenko M, Datta S, Court DL. Recombineering: in vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol. 2007;421: 171–199. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli . Nat Genet. 1998;20: 123–128. [DOI] [PubMed] [Google Scholar]
- 4. Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol. 1991;5: 1447–1457. [DOI] [PubMed] [Google Scholar]
- 5. Li XT, Thomason LC, Sawitzke JA, Costantino N, Court DL. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli . Nucleic Acids Res. 2013;41: e204 10.1093/nar/gkt1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stavropoulos TA, Strathdee CA. Synergy between tetA and rpsL provides high-stringency positive and negative selection in bacterial artificial chromosome vectors. Genomics. 2001;72: 99–104. [DOI] [PubMed] [Google Scholar]
- 7. Bird AW, Erler A, Fu J, Heriche JK, Maresca M, Zhang Y, et al. High-efficiency counterselection recombineering for site-directed mutagenesis in bacterial artificial chromosomes. Nat Methods. 2012;9: 103–109. 10.1038/nmeth.1803 [DOI] [PubMed] [Google Scholar]
- 8. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33: e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong QN, Ng VC, Lin MC, Kung HF, Chan D, Huang JD. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli . Nucleic Acids Res. 2005;33: e59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli . Nucleic Acids Res. 2008;36: e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gregg CJ, Lajoie MJ, Napolitano MG, Mosberg JA, Goodman DB, Aach J, et al. Rational optimization of tolC as a powerful dual selectable marker for genome engineering. Nucleic Acids Res. 2014;42: 4779–4790. 10.1093/nar/gkt1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460: 894–898. 10.1038/nature08187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang HH, Kim H, Cong L, Jeong J, Bang D, Church GM. Genome-scale promoter engineering by coselection MAGE. Nat Methods. 2012;9: 591–593. 10.1038/nmeth.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14: 49–55. 10.1038/nrm3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157: 1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang HH, Huang PY, Xu G, Haas W, Marblestone A, Li J, et al. Multiplexed in vivo His-tagging of enzyme pathways for in vitro single-pot multi-enzyme catalysis. ACS Synth Biol. 2012;1: 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333: 348–353. 10.1126/science.1205822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, et al. Genomically recoded organisms expand biological functions. Science. 2013;342: 357–360. 10.1126/science.1241459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lajoie MJ, Kosuri S, Mosberg JA, Gregg CJ, Zhang D, Church GM. Probing the limits of genetic recoding in essential genes. Science. 2013;342: 361–363. 10.1126/science.1241460 [DOI] [PubMed] [Google Scholar]
- 20. Pal C, Papp B, Posfai G. The dawn of evolutionary genome engineering. Nat Rev Genet. 2014;15: 504–512. 10.1038/nrg3746 [DOI] [PubMed] [Google Scholar]
- 21. Tashiro Y, Fukutomi H, Terakubo K, Saito K, Umeno D. A nucleoside kinase as a dual selector for genetic switches and circuits. Nucleic Acids Res. 2011;39: e12 10.1093/nar/gkq1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin PK, Brown DM. Synthesis and duplex stability of oligonucleotides containing cytosine-thymine analogues. Nucleic Acids Res. 1989;17: 10373–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cupples CG, Miller JH. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A. 1989;86: 5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Severinov K, Soushko M, Goldfarb A, Nikiforov V. Rifampicin region revisited. New rifampicin-resistant and streptolydigin-resistant mutants in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem. 1993;268: 14820–14825. [PubMed] [Google Scholar]
- 25. Dube DK, Black ME, Munir KM, Loeb LA. Selection of new biologically active molecules from random nucleotide sequences. Gene. 1993;137: 41–47. [DOI] [PubMed] [Google Scholar]
- 26. Summers WC, Raksin P. A method for selection of mutations at the tdk locus in Escherichia coli . J Bacteriol. 1993;175: 6049–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawate H, Landis DM, Loeb LA. Distribution of mutations in human thymidylate synthase yielding resistance to 5-fluorodeoxyuridine. J Biol Chem. 2002;277: 36304–36311. [DOI] [PubMed] [Google Scholar]
- 28. Negishi K, Loakes D, Schaaper RM. Saturation of DNA mismatch repair and error catastrophe by a base analogue in Escherichia coli . Genetics. 2002;161: 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bershtein S, Tawfik DS. Ohno's model revisited: measuring the frequency of potentially adaptive mutations under various mutational drifts. Mol Biol Evol. 2008;25: 2311–2318. 10.1093/molbev/msn174 [DOI] [PubMed] [Google Scholar]
- 30. Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proc Natl Acad Sci U S A. 2004;101: 9205–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24: 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 1997;179: 6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizoguchi H, Tanaka-Masuda K, Mori H. A simple method for multiple modification of the Escherichia coli K-12 chromosome. Biosci Biotechnol Biochem. 2007;71: 2905–2911. [DOI] [PubMed] [Google Scholar]
- 34. Drake JW. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A. 1991;88: 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173: 33–38. [DOI] [PubMed] [Google Scholar]
- 36. Drake JW. General antimutators are improbable. J Mol Biol. 1993;229: 8–13. [DOI] [PubMed] [Google Scholar]
- 37. Fujitani Y, Yamamoto K, Kobayashi I. Dependence of frequency of homologous recombination on the homology length. Genetics. 1995;140: 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen P, Huang HV. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics. 1986;112: 441–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lovett ST, Luisi-DeLuca C, Kolodner RD. The genetic dependence of recombination in recD mutants of Escherichia coli . Genetics. 1988;120: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schaaper RM, Dunn RL. Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: the nature of in vivo DNA replication errors. Proc Natl Acad Sci U S A. 1987;84: 6220–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vickers MF, Mani RS, Sundaram M, Hogue DL, Young JD, Baldwin SA, et al. Functional production and reconstitution of the human equilibrative nucleoside transporter (hENT1) in Saccharomyces cerevisiae. Interaction of inhibitors of nucleoside transport with recombinant hENT1 and a glycosylation-defective derivative (hENT1/N48Q). Biochem J. 1999;339 (Pt 1): 21–32. [PMC free article] [PubMed] [Google Scholar]
- 42. Breaker RR, Joyce GF. Emergence of a replicating species from an in vitro RNA evolution reaction. Proc Natl Acad Sci U S A. 1994;91: 6093–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hanczyc MM, Dorit RL. Experimental evolution of complexity: in vitro emergence of intermolecular ribozyme interactions. RNA. 1998;4: 268–275. [PMC free article] [PubMed] [Google Scholar]
- 44. Bansho Y, Ichihashi N, Kazuta Y, Matsuura T, Suzuki H, Yomo T. Importance of parasite RNA species repression for prolonged translation-coupled RNA self-replication. Chem Biol. 2012;19: 478–487. 10.1016/j.chembiol.2012.01.019 [DOI] [PubMed] [Google Scholar]
- 45. Verroust PJ, Adam C, Smith MD, Richard-Lenoble D, Kourilsky O, Morel-Maroger LJ. Circulating immune complexes and C3d in human parasitosis. Kidney Int. 1979;16: 9–14. [DOI] [PubMed] [Google Scholar]
- 46. Bresch C, Niesert U, Harnasch D. Hypercycles, parasites and packages. J Theor Biol. 1980;85: 399–405. [DOI] [PubMed] [Google Scholar]
- 47. Szathmary E, Smith JM. The major evolutionary transitions. Nature. 1995;374: 227–232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identical bases are highlighted with grey-shading.
(TIF)
Seven clones were isolated and individually subjected to PCR analysis using primers annealing to the mrfp gene. The local sequence, location of the primers, and expected size (in bp) of the PCR products are shown for the (1) yiiDE, (2) proV, (3) lacZ, and (4) rssB loci. The sequences of the primers used are shown in S1 Table.
(TIF)
Mutant hsvtk gene identified in dP-selection escapees (S5 Table) were PCR-amplified and inserted into the lacZ locus of MG1655. The resultant recombinants were grown in LB media (0.5 mL) containing Km (50 μg/mL). From each culture, defined number of cells were plated onto LB-Km-agar containing 0 (open bar) or 1 μM (closed bar) of dP. Viability was determined by the colony forming units on each plate. The corresponding clone number in S5 Table is/are also given in parenthesis.
(TIF)
(PDF)
Out of the 92 transformants (chloramphenicol-resistant clones isolated from the pool of MG1655 electroporated with HC cassette), we found three dP-resistant clones. The cassette inserted on chromosome was PCR-amplified using primer P30 and P32 (S1 Table) for the sequence analysis.
(PDF)
The HC cassette was amplified with either VentR DNA polymerase or Phusion DNA polymerase using primer P1 and P2 (S1 Table) to attach homology arms for targeting to the lacZ locus. The resultant DNA fragment was electroporated into E.coli strain MG1655 harboring pKD46 and plated onto an LB-Cm and LB-Cm/dP plate. The frequency of dP resistance (%) was calculated by: 100 × [number of colonies observed on Cm/dP plates] / [number of colonies on the Cm plate] The numbers show the average of 3 samples with standard deviations.
(PDF)
From the first round of two-step recombination using the HK cassette (Table 2), nine clones without GFPmut3.1 fluorescence were individually picked and analyzed in their lacZ (or ΔlacZ::hsvTK-km r) locus. For the sequence of primers used for PCR amplification and sequencing (primers P30, 33, P43, and P44), see S1 Table.
(PDF)
dP sensitive clone was cultured overnight and plated on dP-selection plates. From these plates nine clones were picked and sequenced in their hsvtk coding regions.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.