

Abstract
Fraser syndrome (FS) is a phenotypically variable, autosomal recessive disorder characterized by cryptophthalmus, cutaneous syndactyly and other malformations resulting from mutations in FRAS1, FREM2 and GRIP1. Transient embryonic epidermal blistering causes the characteristic defects of the disorder. Fras1, Frem1 and Frem2 form the extracellular Fraser complex, which is believed to stabilize the basement membrane (BM). However, several cases of FS could not be attributed to mutations in FRAS1, FREM2 or GRIP1, while Fraser syndrome displays high clinical variability, suggesting there is an additional genetic, possibly modifying contribution to this disorder. AMACO, encoded by the VWA2 gene, has a very similar tissue distribution to the Fraser complex proteins in both mouse and zebrafish. Here, we show that AMACO deposition is lost in Fras1 deficient zebrafish and mice and that Fras1 and AMACO interact directly via their CSPG and P2 domains. Knockdown of vwa2, which alone causes no phenotype, enhances the phenotype of hypomorphic Fras1 mutant zebrafish. Together, our data suggest that AMACO represents a novel member of the Fraser complex.
Introduction
Fraser syndrome (FS; OMIM #219000) is a rare, autosomal recessive disorder characterized by cryptophthalmus, cutaneous syndactyly and variable other malformations (Slavotinek and Tifft, 2002). Both the human syndrome and the closely related bleb mouse phenotype are caused by mutations in genes encoding proteins of the extracellular Fraser complex, FRAS1, FREM2 and, in mice, Frem1 (McGregor et al., 2003; Jadeja et al., 2005). The ectodomain of Fras1 is shed from the cell surface and forms a ternary complex with Frem1 and Frem2, which reciprocally stabilize each other at the BM (Kiyozumi et al., 2006). The proteins are characterized by the shared presence of 12 CSPG repeats and single or multiple Calx-β domain(s) (Kiyozumi et al., 2007). In addition mutations in GRIP1, which encodes a cytoplasmic scaffolding protein required for proper Fras1 localization at the BM (Takamiya et al., 2004; Long et al., 2008), result in classical FS (Vogel et al., 2012). Analysis of the mouse bleb mutants revealed transient embryonic epidermal blistering, particularly over the developing eyes and digits, as the likely primary defect leading to the later malformations characteristic of the human disorder (McGregor et al., 2003; Vrontou et al., 2003; Jadeja et al., 2005). The blisters result from a rupture just beneath the lamina densa of the BM and a separation of the dermis from the epidermis (McGregor et al., 2003; Vrontou et al., 2003; Jadeja et al., 2005). Indeed, Fras1, Frem1 and Frem2 have been shown by immuno-gold EM to be localized on the dermal side of the lamina densa at islands normally associated with anchoring fibrils (Dalezios et al., 2007; Petrou et al., 2007a; Petrou et al., 2007b). The Fraser complex associated protein, Frem3, has a similar ultrastructural localization, which, however, is independent of Fras1 (Petrou et al., 2007b).
Despite the identification of mutations in FRAS1, FREM2 and GRIP1 in both mice and humans with FS, a proportion of human cases remain unresolved suggesting that other genes may be involved (van Haelst et al., 2008; Vogel et al., 2012). Furthermore, phenotypic variation in Fraser syndrome points to the existence of common modifier genes (Slavotinek and Tifft, 2002). Mutations in FREM1 were recently shown to result in bifid nose, renal agenesis, and anorectal malformations (BNAR) and Manitoba-oculo-tricho-anal (MOTA) syndromes, two rare conditions with many similar phenotypic traits to FS, although milder (Alazami et al., 2009; Slavotinek et al., 2011). Recent analysis of ENU-induced mutants in zebrafish (Danio rerio) exhibiting transient blistering within the developing fins reveals functional conservation of the FS complex in lower vertebrates and suggests new potential FS-causing genes, such as hemicentin1 (hmcn1), encoding a large extracellular matrix protein (Carney et al., 2010) whose biochemical relationship to the FS complex is unclear.
Via a biochemical candidate approach in zebrafish and subsequent analysis in mice, we have identified a novel member of the Fraser complex, AMACO, encoded by the VWA2 gene. AMACO (VWA2 protein) is a member of the von Willebrand factor A (VWA) domain containing protein superfamily (Whittaker and Hynes, 2002). The protein consists of an N-terminal VWA domain, which is followed by a cysteine-rich domain, an epidermal growth factor (EGF)-like domain carrying elongated O-glucosylated and O-fucosylated glycan chains and two more VWA domains. At the C-terminus another EGF-like domain and a unique domain are present (Sengle et al., 2003; Gebauer et al., 2008). Analysis of mice and zebrafish revealed specific AMACO localization at the BM of multiple tissues during development (Gebauer et al., 2010; Gebauer et al., 2009; Sengle et al., 2003). We demonstrate that AMACO is lost from both zebrafish and mouse mutants that lack Fras1 and that Fras1 and AMACO interact directly. Finally, we show that targeted knockdown of AMACO in zebrafish, which produces no overt phenotype alone, enhances the phenotype severity of hypomorphic Fras1 mutant zebrafish suggesting a supportive role for AMACO in the Fraser complex.
Results
AMACO is reduced or absent in Fras1 mutant zebrafish
It was previously shown that AMACO/vwa2 is expressed during zebrafish development in a pattern very similar to that of the FS genes, fras1, frem1a, frem1b, frem2a, frem2b and frem3 (Carney et al., 2010; Gebauer et al., 2010). We, therefore, analyzed AMACO in our panel of Fraser complex and related mutants: fras1te262/te262, fras1tm95b/tm95b, frem2ata90/ta90, frem1atc280b/tc280b and hmcn1tq207/tq207 (Carney et al., 2010). AMACO and Fras1 are present in the myosepta, the extracellular boundaries between the somites, and the developing caudal fin of 32 hpf wild-type zebrafish (Figure 1a). However, AMACO, was completely lost in null fras1te262/te262 fish (Figure 1b) and strongly reduced in hypomorphic fras1tm95b/tm95b fish both in immunofluorescence analyses (Figure 1b and c) and immunoblots (Figure 1g and h), exactly correlating with Fras1 levels (Figure 1b and c, inset), whereas vwa2 mRNA levels were unaffected in fras1 mutant fish (Supplementary Figure S1). By contrast, AMACO and Fras1 levels were normal in frem2ata90/ta90, frem1atc280b/tc280b and hmcn1tq207/tq207 fish (Figure 1d–h).
Figure 1. AMACO expression is affected in zebrafish models of Fraser syndrome.

Immunofluorescence analysis of whole-mount zebrafish at 32 hpf reveals that (a) AMACO is strongly expressed in the myosepta and developing caudal fin of wild-type zebrafish, very similar to the expression of Fras1 (inset). (b,c) By contrast, AMACO is completely absent from a zebrafish Fras1 null allele (fras1te262/te262) (b), and partially lost from a hypomorphic Fras1 allele (fras1tm95b/tm95b) which exhibits some residual Fras1 expression (c). (d-f) AMACO expression is normal in null alleles for Frem2a (frem2ata90/ta90; d), Frem1a (frem1atc280b/tc280b; e) and Hemicentin1 (hmcn1tq207/tq207; f). Expression of Fras1 is also normal in each of these alleles (inset in d–f). Scale bar = 500 μm. (g,h) Quantitative AMACO immunoblot analysis (Gebauer et al., 2010) in the zebrafish mutants shown in a–f. In each case a pool of 50 fish of a given genotype was extracted to give sufficient material and to minimize effects of individual variations. In g, Ponceau loading control is shown.
AMACO is absent from Fras1bl/bl mice
Next, we analyzed AMACO in Fras1bl/bl mice. During mouse development AMACO is specifically localized at the BMs of skin, lung, heart, tooth germs and kidney (Sengle et al., 2003). Analysis of these regions in E14.5 Fras1bl/bl mice revealed a complete loss of AMACO from the epidermal BM (Figure 2c) when compared to wild-type or Fras1+/bl littermates (Figure 2a and b) even though laminin deposition was normal (Figure 2d–f). Similarly, analysis of a blistered region over the eye of a Fras1bl/bl E14.5 embryo revealed a complete lack of AMACO (Figure 2g) even though laminin deposition demonstrated that a BM was still present as reported previously (Figure 2h; Vrontou et al., 2003). Analysis of other regions, including the eyelid, kidney and developing tooth germs at E14.5 also revealed a complete loss of AMACO deposition (Figure 2i–l and data not shown).
Figure 2. AMACO expression is lost from Fras1 mutant mice.

(a-f) Immunofluorescence analysis of the epidermis of E14.5 wild type (a,d), Fras1+/bl (b,e) and Fras1bl/bl (c,f) littermates reveals a complete loss of AMACO expression from the basement membrane of Fras1bl/bl mice (c), whereas laminin expression is normal (f). (g–l) Similar analysis of other regions demonstrates the loss of AMACO from a blister over the eye of an E14.5 Fras1bl/bl embryo (g) whereas laminin expression was still observed under the detached epidermis (j). Strong expression of AMACO was observed surrounding the developing tooth germs of E14.5 wild-type mice (h) but was completely lost from a Fras1bl/bl littermate (i) despite laminin being normal (k,l). Scale bars = 50 μm. For generation and characterization of the mouse AMACO-P3 antibody see Gebauer et al., 2009.
AMACO and Fras1 co-localize beneath the lamina densa
Previous ultrastructural examinations demonstrated that Fras1, Frem1, Frem2 and AMACO are all localized at the BM zone (Gebauer et al., 2009; Dalezios et al., 2007; Petrou et al., 2007a; Petrou et al., 2007b). To determine co-localization of Fras1 and AMACO, double immunoelectron microscopy labeling was performed on P0 mouse skin using secondary antibodies coupled to different sized gold particles. Both proteins were detected in distinct regions, anchoring plaques, in the dermis close to the BM (Figure 3a and b). These AMACO and Fras1 positive patches often occurred where anchoring fibrils fuse (Figure 3a). Although regions were observed where only one protein was present, mostly AMACO and Fras1 co-localized (Figure 3a and b). Collagen VII/Fras1 double immunoelectron microscopy showed that collagen VII, a major component of anchoring fibrils, occurs in proximity to Fras1 at anchoring plaques, but mostly at the opposite side of the plaque, indicating that the two proteins may not interact directly (Figure 3c).
Figure 3. Fras1 and AMACO co-localize at the basement membrane.

For immuno-EM analysis, newborn mouse skin was immunolabeled enbloc with antibodies directed against mouse AMACO-P3, and mouse Fras1-CSPG (a,b) or human collagen VII and mouse Fras1-CSPG (c). Secondary antibodies conjugated to gold particles of different size (Fras1: 6 nm; AMACO and collagen VII: 10 nm) were detected at anchoring plaques below the lamina densa (LD, black arrowheads). Often anchoring fibrils (AF, open arrowheads) are seen to intersect the anchoring plaques (AP). Collagen VII (arrows) and Fras1 often occur at opposite sides of anchoring plaques (c). (a and c) shows an overview and (b) shows selected anchoring plaques. The scale bar corresponds to 100 nm in (a and c) and 50 nm in (b).
The AMACO fragment P2 binds to Fras1 CSPG repeats
To test for direct interactions between AMACO and the Fras1 ectodomain we employed surface plasmon resonance assays. For AMACO, we generated recombinant full-length protein as well as protein fragments corresponding to AMACO domains P1, P2 and P3 (Figure 4a). Recombinant expression of the full-length Fras1 ectodomain repeatedly failed (data not shown). To overcome this problem, Fras1 cDNA fragments, representing the VWC-, Furin-like-, CSPG-, and Calx-β domains and the unique domain alone (Figure 4a) were cloned for protein production, but only the Furin-like-, CSPG-, and Calx-β domains were well expressed.
Figure 4. AMACO and Fras1 interact directly.

(a) Domain structures of Fras1 and AMACO. (S) signal peptide, (TM) transmembrane domain, the arrow indicates the furin cleavage site. (b, c) Surface plasmon resonance sensorgrams showing binding of different concentrations of soluble analytes to Fras1 CSPG (b) and AMACO P2 (c) coupled onto a chip. Full-length AMACO interacts with Fras1 CSPG, as does the AMACO P2 fragment. AMACO P1 and AMACO P3 do not interact (b). In the reversed orientation Fras1 CSPG interacts with AMACO P2 (c). All proteins were from mouse.
Among these Fras1 fragments, we were only able to immobilize the CSPG domains on the sensor chip. When full-length AMACO was injected as soluble analyte at increasing concentrations (0–400 nM) over the CSPG chip we obtained association and dissociation curves (Figure 4b) which could be fitted with a Langmuir 1:1 binding model and could calculate a dissociation constant (KD) of 75 nM. Of the three different domains (P1, P2, P3), binding to immobilized Fras1 CSPG was only seen with AMACO P2 (KD 151 nM) (Figure 4b). This binding is independent of the two unusual glycan chains on AMACO P2, as mutant forms lacking either one or both glycosylation sites still bound to immobilized Fras1 CSPG with similar KDs between 77 and 108 nM (data not shown).
In the reverse set up (immobilization of AMACO P2 on the sensor chip and injection of the Fras1 CSPG), we could confirm the binding between the AMACO P2 domain and the Fras1 CSPG domain (KD 42 nM; Figure 4c).
Loss of AMACO does not affect zebrafish development
To analyze the role of AMACO/vwa2 during zebrafish development, we generated two translation inhibiting morpholinos, which were injected into the yolk of fertilized eggs. Both morpholinos led to a marked decrease in AMACO protein levels (Figure 5a and b,), while Fras1 appeared largely normal (Figure 5a and b, insets). Nevertheless, vwa2 morphants exhibited normal morphology both at 48 (Figure 5c and d) and 80 hpf (Figure 5g and h), including regions that normally display high AMACO levels (Gebauer et al., 2010) and that are, at least partly, affected in fras1 mutant zebrafish (Carney et al., 2010; Talbot et al., 2012), such as the body fins (Figure 5c and d), the pronephros (data not shown), the somitic myosepta and their connection to the myotomes (Figure 5e and f) and the craniofacial cartilage (Figure 5g and h). This suggests that AMACO per se is dispensable for early zebrafish development.
Figure 5. AMACO deficient zebrafish are phenotypically normal.
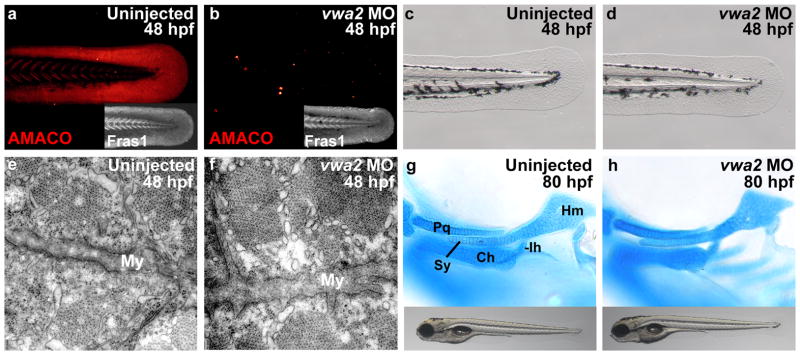
(a–b) Immunofluorecence analysis with antibodies against the zebrafish proteins (Gebauer et al., 2010; Carney et al. 2010) reveals complete loss of AMACO protein in vwa2 morphant (b), whereas levels of Fras1 are normal (inset in b). (c,d) vwa2 morphants display normal fin morphology at 48 hpf. (e,f) Transmission electron microscopy (TEM) analysis at 48 hpf reveals no defect in the structure of the myosepta in vwa2 morphants (f) when compared to wild-type controls (e). (g,h) No abnormalities can be observed in the craniofacial cartilages or general morphology of vwa2 morphants at 80 hpf (h). My = myosepta; Pq, palatoquadrate; Ch, ceratohyal; Ih, interhyal; Hm, hyomandibular; Sy, symplectic.
Antisense morpholino knockdown of AMACO in Fras1 hypomorphic zebrafish results in a more severe phenotype
The loss of AMACO in Fras1 deficient zebrafish and mice suggests that Fras1 is required for AMACO deposition or protein stability. To determine if AMACO in reverse stabilizes Fras1 protein and promotes its function, we injected the vwa2 morpholino into eggs resulting from an in-cross of fras1+/tm95b fish (Figure 6). These fish carry the missense mutation G3816W in Fras1 and exhibit reduced, but detectable levels of Fras1 protein (Figure 1c, Figure 6b) and are therefore believed to represent a hypomorphic allele. When an uninjected in-cross of fras1+/tm95b fish (n=415; 327=wt, 88=mut) was compared to an in-cross of fras1+/te262 (null allele) fish (n=88; 65=wt, 23=mut), fras1tm95b/tm95b fish exhibit a milder caudal fin blistering phenotype as assessed by morphology (Figure 6a). In addition, injection of a missense morpholino into an in-cross of fras1+/tm95b fish (n=389; 308=wt, 81=mut) had no effect on the severity of the caudal fin blistering phenotype (Figure 6a). Injection of vwa2 morpholino into an in-cross of fras1+/tm95b fish (n=500; 400=wt, 100=mut), however, resulted in a more severe phenotype, to a comparable level to that of null fras1te262/te262 fish (Figure 6a). Representative morphologies of the caudal fin at 34 hpf for each condition are shown in Figure 6b. Immunofluorescence analysis of Fras1 revealed high levels in the myosepta and fin of wild-type fish, reduced levels in uninjected and missense morpholino injected fras1tm95b/tm95b fish and a complete absence in fras1te262/te262 fish and fras1tm95b/tm95b fish injected with vwa2 morpholino (Figure 6b), corresponding to the severity of the morphological phenotype. In contrast, laminin levels were unaltered under all conditions (Figure 6b).
Figure 6. vwa2 knockdown in Fras1 hypomorphic zebrafish increases the severity of the phenotype.

(a) Chart depicting the relative severity of the blistering phenotype of Fras1 zebrafish injected with a missense morpholino (5mm) or a vwa2 specific MO. In all cases, mutant embryos were derived from heterozygous in-crosses, segregating in a normal Mendelian ratio. Only homozygotes are considered in the chart. Included are representative images of mild, moderate and severe blistering. Severe blistering was determined by a large number of caudal fin blisters (arrowheads) often extending further anterior within the dorsal region of the caudal fin. Associated extensive blistering of the caudal vein region was also observed (arrows). Moderate blistering involved fewer fin tip blisters of variable sizes and less, although always some associated caudal vein blistering. Mild blistering was determined by a small number of fin tip blisters with no associated caudal vein blistering. Compare to b for a representative wild type fin. (b) Immunolocalization of Fras1 (with zebrafish-specific antibody, Carney et al. 2010) and laminin for each condition.
Discussion
FS is a heterogeneous disorder affecting the development of the skin, eyes, digits and kidneys. Although mutations in FRAS1, FREM2 and GRIP1 have been identified in approximately 95% of all FS patients, additional genetic contributions to this disorder remain likely. The highly variable inter- and intra-familial phenotypic severity of FS also suggests the presence of genetic modifiers (Slavotinek and Tifft, 2002). In this study, we have demonstrated that vwa2, a gene encoding AMACO, a BM associated extracellular protein containing three von Willebrand factor A (VWA) domains, contributes to the Fraser complex in zebrafish and mice.
We could show that AMACO protein is completely absent in zebrafish and mouse mutants lacking Fras1, suggesting that Fras1 is crucial for the deposition or stabilization of AMACO. The vwa2 mRNA expression level in Fras1 mutant zebrafish is, in contrast, completely normal (Supplementary Figure S1), suggesting specific effects on protein stabilization. TEM further supports a direct interaction, as AMACO and Fras1 co-localize at a distance that is consistent with direct binding in islands beneath the lamina densa. Unfortunately, a comprehensive study of the interaction between AMACO and Fras1 was hampered by the fact that the ectodomain, the VWC domains and the unique domain of Fras1 could not be recombinantly expressed in sufficient amounts. However, AMACO fragments covering the whole sequence were expressed and surface plasmon resonance spectroscopy revealed direct binding between AMACO P2 and the CSPG domains of Fras1.
Furthermore, we could demonstrate that knockdown of AMACO, which alone results in no morphological defects, can increase the phenotypic severity in hypomorphic Fras1 zebrafish. Upon AMACO knockdown the hypomorphic fish resemble the complete Fras1 knockout suggesting that, in addition to the requirement of Fras1 for AMACO deposition or stabilization, AMACO can stabilize Fras1. Reciprocal stabilization of Fras1, Frem1, Frem2 and possibly Frem3 has been revealed in mouse and zebrafish (Kiyozumi et al., 2006; Carney et al., 2010), whereas zebrafish Hmcn1, despite its co-expression with the FS genes and the similar phenotype of hmcn1 and fras1 mutants, is dispensable for Fras1 stabilization (Carney et al., 2010). So, although loss of AMACO alone does not cause a tissue integrity phenotype, our results demonstrate that AMACO can also stabilize Fras1 at the BM, potentially in a manner redundant with other Fraser complex proteins.
We currently do not know the nature of these potential redundant factors. For zebrafish Frem2, the Fras1-stabilizing effect was only revealed upon concomitant knockdown of the three closely related proteins, Frem2a, Frem2b and Frem3 (Carney et al., 2010). However, AMACO, although being a member of the VWA domain containing protein superfamily, does not belong to a distinct subfamily such as the matrilins or VWA domain containing collagens. Also, no direct AMACO paralogue has been annotated in the zebrafish genome. Therefore, it seems more likely that AMACO acts in partial functional redundancy with a more distantly related VWA protein. Nevertheless, the essential character of AMACO becomes apparent when Fras1 function is compromised. This indicates that interactions between several members of the Fraser complex are required for stabilization beneath the BM, with AMACO’s function to support this structural complex becoming vital when the multi-protein structure is compromised by reduced levels or activities of other components. Due to these particular genetic features of AMACO and its physical involvement in the Fraser complex both in zebrafish and mouse, VWA2 represents an interesting candidate for human mutation analysis in multigenic scenarios, possibly mediating a genetic predisposition for Fraser syndrome aetiology.
Materials & Methods
Zebrafish husbandry
Embryos were obtained from natural crosses and staged according to Kimmel et al., 1995. The mutant alleles pifte262 (fras1), piftm95 (fras1), neltq207 (hmcn1), blata90 (frem2a) and rfltc280b (frem1a) have been described previously (Carney et al., 2010).
Antisense morpholino knockdown
Morpholino antisense oligonucleotides (Supplement) were designed by and obtained from Gene-Tools (Philomath, OR) and dissolved in distilled water to 1 mM stock solutions. Two different morpholinos covering the ATG translation start codon and the 5′ UTR of vwa2 were used, both resulting in translational inhibition. A five-mismatch morpholino was used as negative control. For injection, stocks were diluted to 0.1 mM in Danieau’s buffer and phenol red (Nasevicius and Ekker, 2000). 0.5 nl of MO solution was injected into embryos at the 1–4 cell stage using glass needles pulled on a Sutter needle puller and a Nanoject injection apparatus (World Precision Instruments).
Mouse lines
E14.5 mouse embryos, generated from an in-cross of Fras1+/bl mice and fixed in 4% paraformaldehyde (PFA), were obtained from Peter Scambler (UCL, London, UK).
Tissue-labeling procedures
For whole-mount immunofluorescence analysis zebrafish were fixed in 4% PFA overnight at 4°C and washed with 1x PBS. Fish were then washed for several hours in dH20, blocked in 10% fetal calf serum (FCS) in 1x PBS, 0.5% Triton-X, incubated in primary antibody in 10% FCS overnight at 4°C, washed extensively in 1x PBS, 0.5% Triton-X, incubated overnight in secondary antibody in 10% FCS at 4°C, washed extensively in 1xPBS, 0.5% Triton-X and re-fixed in 4% PFA. For immunofluorescence analysis on sections, tissue was dehydrated in a graded series of alcohols, cleared in Roti-Histol (Carl Roth) and embedded in paraffin wax. Immunofluorescence analysis was performed using standard protocols. Primary antibodies used were: rabbit anti-zebrafish AMACO (Gebauer et al., 2010), rabbit anti-zebrafish Fras1 (Carney et al., 2010), rabbit anti-mouse AMACO (Gebauer et al., 2009), and rabbit anti-laminin (Sigma Aldrich, L9393). Images were captured on a Zeiss Axiophot, Zeiss Apotome, Zeiss Confocal (LSM710 META) or Leica M165 FC compound microscope.
For whole-mount alcian blue stainings, the embryos were washed in phosphate buffered saline and incubated in 0.1 mg/ml alcian blue in ethanol/acetic acid (4:1) for 2–6 h at 37°C. After clearing overnight by digestion with 50 mg/ml trypsin in 30% sodium tetraborate in water, the specimens were destained in 1% KOH/glycerol, flat-mounted between a slide and a coverslip and photographed using a Zeiss Axiophot microscope equipped with a Nikon digital camera. Whole-mount in situ hybridization was performed as previously described (Gebauer et al., 2010).
Expression of full length AMACO and Fras1 fragments
Mouse cDNA fragments were generated by RT-PCR and cloned with 5′-terminal NheI and 3′-terminal NotI restriction sites using oligonucleotide primers (Supplemental Table 1). The amplified PCR products were inserted into a modified pCEP-Pu vector containing an N-terminal BM-40 signal peptide (Kohfeldt, 1997) followed by an N-terminal One-STrEP-tag (IBA GmbH) upstream of the restriction sites. The expression constructs were transfected into 293 EBNA cells with FuGENE HD (Roche) according to the manufacturer’s instructions. The cells were cultured in the presence of 10% FCS prior to harvest of cells and cell culture supernatant. After filtration and centrifugation of supernatants containing recombinant protein for 1 h at 10,000 × g, these were applied to a Streptactin column (1.5 ml; IBA GmbH) and proteins eluted with 2.5 mM desthiobiotin, 150mM NaCl, 100 mM Tris-HCl, pH 8.0.
Fras1 antibody production
Purified mouse Fras1 CSPG was used for guinea pig immunization. The antiserum was purified by affinity chromatography on a column with antigen coupled to CNBr-activated Sepharose (GE Healthcare). Specific antibodies were eluted with 0.1 M glycine, pH 2.5, and the eluate was neutralized with 1 M Tris-HCl, pH 8.8. Specificity was tested by immunoblot (Supplementary Figure S2). The antibody was only used for immunoelectron microscopy.
Surface plasmon resonance binding assays
Binding analyses were performed using a BIAcore2000 (GE Healthcare). All proteins were from mouse. Expression and purification of AMACO fragments has been described earlier (Gebauer et al., 2009) and purity of the proteins is documented in (Supplementary Figure S3). Fras1 CSPG (2200 RUs), AMACO full-length (2300 RUs), AMACO P1 (2500 RUs), AMACO P2 (2400 RUs), and AMACO P3 (2500 RUs) were covalently coupled to carboxymethyldextran hydrogel 500M sensor chips (XanTec, Düsseldorf, Germany) using the amine coupling kit (GE Healthcare). Binding assays were performed at 25°C in 10 mM Hepes buffer, pH 7.4, containing 0.15 M NaCl, 3 mM EDTA, and 0.005% (v/v) P20 surfactant (HBS-EP buffer). Omission of EDTA and addition of 1mM MgCl2 and CaCl2 did not result in any change of binding events. Equilibrium dissociation constants (KD) were then calculated as the ratio kd/ka. Kinetic constants were calculated by nonlinear fitting of association and dissociation curves (BIAevaluation 4.1 software).
Protein extraction and immunoblot
For protein extraction, 32 hpf wild type and mutant zebrafish larvae (50 each) were homogenized in 500μl 150mM NaCl, 2mM EDTA, 1% Nonidet P-40 and 50mM Tris, pH 7.4, put on ice for 15 min. 1/3 volume of 4x SDS sample buffer (8% (w/v) SDS, 40% (v/v) glycerol, 0.2% (w/v) bromphenol blue, 250mM Tris-HCl, pH 6.8) was added, the samples boiled for 5 min, centrifuged for 10 min and subjected to non-reducing 4–12% (w/v) SDS PAGE. Proteins were transferred to a nitrocellulose membrane and detected using polyclonal antibodies diluted in TBS/5% milk powder. Bands were detected by chemoluminescence immunoassay using a peroxidase-conjugated swine anti rabbit secondary antibody (Dako). Image J was used for quantification of bands.
Electron microscopy
Wild type and morphant zebrafish larvae 48 hpf were anesthetized with 0.08% Tricaine, transferred into 6% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, for immersion fixation and stored in this solution for 3 days at 4°C. Then the larvae were rinsed 2x 15 min in 0.1 M cacodylate buffer, pH 7.2, postfixed 120 min with 1% OsO4 in 0.1 M cacodylate buffer, pH 7.2, at room temperature in the dark, rinsed again, dehydrated with acetone and embedded in araldite CY212 (Durcupan ACM, Fluka). Ultrathin sections were cut at grey interference colour (25–30 nm) with a 35°-diamond knife (Diatome) on an Ultracut E (Leica), stretched with chloroform vapour, mounted on 200 mesh copper grids (5 μm bar thickness) and contrasted 10 min with saturated (2%) uranyl acetate in 70% ethanol and 5 min with 0.2% aqueous lead citrate, pH 11.8. Microscopy was performed with a Zeiss EM109 (80 kV, 500 μm condenser 1 aperture, 200 μm condenser 2 aperture, 30 μm objective aperture) equipped with a temperature-stabilized wide angle YAC-CCD camera at the side entry port (1024×1024 pixel, 12-bit greyscale/pixel; info@trs-system.de). Magnification was calibrated with a cross grating replica (2160 lines/mm, d = 0.463 μm).
Immunoelectron microscopy
Newborn mouse skin was carefully sliced into 1 mm cubes, all including epithelium and dermis. Tissues were immersed in a combination of affinity purified rabbit AMACO-P3 antibody (Gebauer et al., 2009) or collagen VII antibody (Lunstrum et al., 1986) with guinea pig Fras1 CSPG antibody (Supplementary Figure S2) in Dulbecco’s Modified Eagle Media (DMEM) at a ratio of 1:1:4 overnight at 4°C. Tissues were washed in DMEM for 4 hours at 4°C and then immersed in a combination of goat anti-rabbit 10-nm and goat anti-guinea pig 6-nm colloidal gold conjugates in DMEM at a ratio of 1:1:3 overnight at 4°C. The tissues were then rinsed extensively in DMEM, fixed in 1.5% glutaraldehyde/1.5% paraformaldehyde containing 0.5% tannic acid, post-fixed in 1% OsO4, then dehydrated and embedded in Spurr’s epoxy resin. Tissue was oriented so that cross sections of the dermal-epidermal junction would be obtained. Ultrathin sections were contrasted with uranyl acetate and lead citrate and examined using a FEI Tecnai G20 TEM.
Supplementary Material
Acknowledgments
Excellent technical assistance from Evelin Fahle and Petra Müller is gratefully acknowledged. We are very grateful to Peter Scambler for Fras1 (bleb) mutant mice. Work in the laboratories of RW and MP was funded by the Deutsche Forschungsgemeinschaft (WA1338/2-6 and SFB829/B2), work in the laboratory of GS by the Deutsche Forschungsgemeinschaft (SFB829/B12), work in the laboratory of MH by the Deutsche Forschungsgemeinschaft (SFB829/A9), the European Union (Seventh Framework Program, Integrated Project ZF-HEALTH, EC Grant Agreement HEALTH-F4-2010-242048), the US National Institute of General Medical Sciences (GM63904), and an EMBO long-term postdoctoral fellowship to RR. JMG was a member of the International Graduate School in Genetics and Functional Genomics at the University of Cologne.
Abbreviations
- FS
Fraser syndrome
- BM
basement membrane
- BNAR
bifid nose, renal agenesis, and anorectal malformations syndrome
- MOTA
Manitoba-oculo-tricho-anal syndrome
- RU
response unit
- TEM
transmission electron microscopy
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Alazami AM, Shaheen R, Alzahrani F, et al. FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am J Hum Genet. 2009;85:414–18. doi: 10.1016/j.ajhg.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney TJ, Feitosa NM, Sonntag C, et al. Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes. PLoS Genet. 2010;6:e1000907. doi: 10.1371/journal.pgen.1000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalezios Y, Papasozomenos B, Petrou P, et al. Ultrastructural localization of Fras1 in the sublamina densa of embryonic epithelial basement membranes. Arch Dermatol Res. 2007;299:337–43. doi: 10.1007/s00403-007-0763-8. [DOI] [PubMed] [Google Scholar]
- Gebauer JM, Karlsen KR, Neiss WF, et al. Expression of the AMACO (VWA2 protein) ortholog in zebrafish. Gene Expr Patterns. 2010;10:53–59. doi: 10.1016/j.gep.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Gebauer JM, Keene DR, Olsen BR, et al. Mouse AMACO, a kidney and skin basement membrane associated molecule that mediates RGD-dependent cell attachment. Matrix Biol. 2009;28:456–62. doi: 10.1016/j.matbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Gebauer JM, Müller S, Hanisch FG, et al. O-glucosylation and O-fucosylation occur together in close proximity on the first epidermal growth factor repeat of AMACO (VWA2 protein) J Biol Chem. 2008;283:17846–54. doi: 10.1074/jbc.M704820200. [DOI] [PubMed] [Google Scholar]
- Jadeja S, Smyth I, Pitera JE, et al. Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat Genet. 2005;37:520–25. doi: 10.1038/ng1549. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, et al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Kiyozumi D, Sugimoto N, Nakano I, et al. Frem3, a member of the 12 CSPG repeats-containing extracellular matrix protein family, is a basement membrane protein with tissue distribution patterns distinct from those of Fras1, Frem2, and QBRICK/Frem1. Matrix Biol. 2007;26:456–62. doi: 10.1016/j.matbio.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Kiyozumi D, Sugimoto N, Sekiguchi K. Breakdown of the reciprocal stabilization of QBRICK/Frem1, Fras1, and Frem2 at the basement membrane provokes Fraser syndrome-like defects. Proc Natl Acad Sci U S A. 2006;103(32):11981–6. doi: 10.1073/pnas.0601011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohfeldt E, Maurer P, Vannahme C, et al. Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Lett. 1997;414:557–61. doi: 10.1016/s0014-5793(97)01070-3. [DOI] [PubMed] [Google Scholar]
- Long J, Wei Z, Feng W, et al. Supramodular nature of GRIP1 revealed by the structure of its PDZ12 tandem in complex with the carboxyl tail of Fras1. J Mol Biol. 2008;375:1457–68. doi: 10.1016/j.jmb.2007.11.088. [DOI] [PubMed] [Google Scholar]
- Lunstrum GP, Sakai LY, Keene DR, et al. Large complex globular domains of type VII procollagen contribute to the structure of anchoring fibrils. J Biol Chem. 1986;261:9042–8. [PubMed] [Google Scholar]
- McGregor L, Makela V, Darling SM, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003;34:203–8. doi: 10.1038/ng1142. [DOI] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Petrou P, Chiotaki R, Dalezios Y, et al. Overlapping and divergent localization of Frem1 and Fras1 and its functional implications during mouse embryonic development. Exp Cell Res. 2007a;313:910–20. doi: 10.1016/j.yexcr.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Petrou P, Pavlakis E, Dalezios Y, et al. Basement membrane localization of Frem3 is independent of the Fras1/Frem1/Frem2 protein complex within the sublamina densa. Matrix Biol. 2007b;26:652–58. doi: 10.1016/j.matbio.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Sengle G, Kobbe B, Mörgelin M, et al. Identification and characterization of AMACO, a new member of the von Willebrand factor A-like domain protein superfamily with a regulated expression in the kidney. J Biol Chem. 2003;278:50240–9. doi: 10.1074/jbc.M307794200. [DOI] [PubMed] [Google Scholar]
- Slavotinek AM, Baranzini SE, Schanze D, et al. Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J Med Genet. 2011;48:375–82. doi: 10.1136/jmg.2011.089631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek AM, Tifft CJ. Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J Med Genet. 2002;39:623–33. doi: 10.1136/jmg.39.9.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamiya K, Kostourou V, Adams S, et al. A direct functional link between the multi-PDZ domain protein GRIP1 and the Fraser syndrome protein Fras1. Nat Genet. 2004;36:172–77. doi: 10.1038/ng1292. [DOI] [PubMed] [Google Scholar]
- Talbot JC, Walker MB, Carney TJ, et al. fras1 shapes endodermal pouch 1 and stabilizes zebrafish pharyngeal skeletal development. Development. 2012;139:2804–13. doi: 10.1242/dev.074906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haelst MM, Maiburg M, Baujat G, et al. Molecular study of 33 families with Fraser syndrome new data and mutation review. Am J Med Genet A. 2008;146A:2252–57. doi: 10.1002/ajmg.a.32440. [DOI] [PubMed] [Google Scholar]
- Vogel MJ, van Zon P, Brueton L, et al. Mutations in GRIP1 cause Fraser syndrome. J Med Genet. 2012;49:303–6. doi: 10.1136/jmedgenet-2011-100590. [DOI] [PubMed] [Google Scholar]
- Vrontou S, Petrou P, Meyer BI, et al. Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat Genet. 2003;34:209–14. doi: 10.1038/ng1168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.