

Abstract
The biosynthesis of the antibiotic epsilon-poly-lysine (ε-PL) in Streptomyces albulus is performed by polylysine synthase (pls); however, the regulatory mechanism of this process is still unknown. Here, we first obtained the complete genome sequence of S. albulus ZPM, which consists of 9,784,577 bp and has a GC content of 72.2%. The genome houses 44 gene clusters for secondary metabolite biosynthesis, in which 20 gene clusters are involved in the biosynthesis of polyketides and nonribosomally synthesized peptides. High-throughput sequencing was further performed, and genetic variants were identified from pooled libraries consisting of the 30 highest-yield mutants or 30 lowest-yield mutants. More than 350 genetic variants associated with ε-PL yield have been identified. One hundred sixty-two affected proteins, from important metabolic enzymes to novel transcriptional regulators, were identified as being related to ε-PL synthesis. HrdD, one of the affected genes, is a sigma factor that shows the most sensitive response to pH change and contains a non-synonymous mutation (A132V) in mutant strains with lower ε-PL yields. Electrophoretic mobility shift assays showed that the pls gene is likely regulated by transcriptional activator HrdD. The data obtained in this study will facilitate future studies on ε-PL yield improvement and industrial bioprocess optimization.
Streptomyces species are Gram-positive bacteria that predominantly inhabit highly complex and competitive soil environments. The secondary metabolites produced by Streptomyces species have been the sources of antibiotics, parasiticides, herbicides and pharmacologically active substances1. Currently, more than half of the medically important antimicrobial and antitumor agents are provided by Streptomyces species2. ε-poly-lysine (ε-PL) is a homo-poly-amino acid characterized by the peptide backbone between the carboxyl and ε-amino groups of L-lysine3. ε-PL was first isolated from Streptomyces albulus and characterized as a novel peptide antibiotic in the 1970s4. ε-PLs with chain lengths larger than nine L-lysine residues exhibit antimicrobial activities against a wide spectrum of microorganisms, including gram-positive and gram-negative bacteria and phages3,5,6. As ε-PL is safe, biodegradable, and stable at high temperature and low-pH environments3, it has been introduced as a food preservative in many countries3. The application of ε-PL in antiobesity and selective endotoxin-removal action has been reported7. In addition to S. albulus, ε-PL has also been isolated from S. griseofuscus8, Streptomyces sp. M-Z189, Streptomyces sp. GIM810 and Kitasatosporasp PL6-311.
The synthesis of ε-PL in S. albulus is controlled by an unusual non-ribosomal peptide synthase (NRPS), poly-lysine synthase (Pls)12. When the pls gene is disrupted, S. albulus loses its capacity to produce ε-PL12. Consistently, the NRPS Pls is able to utilize L-lysine as a substrate to synthesize ε-PL in vitro12. Additionally, ε-PL can be degraded into lysine monomers by the proteases Pld and PldII in S. albulus5,13. Interestingly, pldII, which encodes the main degradase in S. albulus, is located adjacent to the pls gene, with a 4-bp overlap, and they form a single operon in the genome. The fermentation of S. albulus in media containing only glucose and ammonium sulfate, as carbon and nitrogen sources, respectively, results in ε-PL production3. Therefore, the lysine biosynthesis pathway is essential because L-aspartic acid is the initial building block of ε-PL in vivo. It is noteworthy that pls is not a constitutive gene but is induced ~12 hours after fermentation, when the pH of the medium is lower than 513, which implies that pls expression is strictly regulated and low pH may be a possible inducer. The success of pH control strategies on improving PL yield14 has also implied that pH may have a regulatory function in ε-PL synthesis. Although the biosynthesis and degradation mechanisms of ε-PL have been elucidated, the regulatory mechanism of ε-PL synthesis, particularly in vivo, is still unclear.
In the current study, we first performed whole-genome shotgun sequencing of S. albulus ZPM, which was isolated from the soil and produces the homopolymer antibiotic, ε-PL. Through genome annotation, we characterized a large portion of transcriptional regulators and many secondary metabolite gene clusters. Furthermore, pan-genome analysis revealed that strain-specific genes are highly abundant in S. albulus ZPM compared with other Streptomycetes species. To study the mechanism of the regulation of ε-PL yield, a large mutant library of strains was constructed using both physical and chemical mutagenesis strategies. The genomic DNAs (gDNAs) of the high- and low-yield groups were pooled, and high-throughput DNA sequencing was performed. Single-nucleotide polymorphism (SNP) and Indel calling were subsequently performed, and 208 and 163 SNPs/Indels that were specific to the low- or high-yield groups, respectively, were identified. In total, the amino acid sequences of 162 proteins that were involved in diverse cellular metabolism processes were affected by these genetic variations. In conclusion, our findings enhance the understanding of ε-PL biosynthesis.
Results
Genome analysis of Streptomyces albulus ZPM
The strain S. albulus ZPM, which produces ε-PL, was isolated from the soil of Zi-Peng Mountain (ZPM) west of Hefei, China using the methylene blue screening method15 (Fig. S1A). The pls gene was amplified from the strain's gDNA by PCR and was verified by Sanger sequencing (Fig. S1B). The ε-PL yield of this strain in a shaking flask is ~0.79 g/L, which is 30% higher than the industrial S. albulus 410 strain (~0.6 g/L) under similar circumstances14. Phylogenetic analysis of 16S rRNA revealed that the strain was closest to S. albulus; thus, the stain was designated S. albulus ZPM (Fig. S1C).
The complete genome sequence of S. albulus ZPM revealed a single linear chromosome composed of 9,784,577 bp, with the pZPM234 plasmid and an average G + C content of 72.2%. The 36,995-bp plasmid is homologous to the pNO33 plasmid and contains 37 genes that possibly code for membrane proteins, exonucleases and metabolic enzymes, along with proteins involved in partitioning (Fig. S2). The chromosome has a 139,385-bp terminal inverted repeat (TIR) with a remarkably low GC content of 66.7% at both ends (1 ~ 139385, 9645193 ~ 9784577) (Fig. 1). In total, 9197 genes have been identified in the S. albulus ZPM chromosome, including 9073 protein-coding genes, 18 ribosomal genes in 6 operons in the order 16S-23S-5S and 70 tRNA genes (Table S1). A total of 5816 of the 9073 protein-coding genes (64.1%) have been classified into at least one Cluster of Orthologous Groups (COG) with known or putative function (Fig. S3). In the TIR region, 106 genes have been identified, including 10 genes that are related to recombination, 5 involved in defense and 17 genes involved in secondary metabolite biosynthesis. The replication origin, oriC, contains 18 DnaA box-like sequences and is located at the center of the chromosome (5114740–5116614 bp). The terminal protein (Tp) and telomere-associated protein (Tap), which are encoded by SAZ_8415 and SAZ_8416, respectively, are responsible for telomere replication and are 28.70% and 55.88% identical to the Tap and Tpg proteins of S. griseus, respectively16. The proteins have no detectable similarities with the conserved Tp and Tap proteins of other Streptomyces species, such as S. coelicolor A3 (2)17 and S. avermitilis18.
Figure 1. Circular representation of the S. albulus ZPM chromosome.

Circles 1 and 2, all genes (forward and reverse strands, respectively) are color-coded by function (blue, RNA processing and modification; vlblue, Chromatin structure and dynamics; chrm, Energy production and conversion; churn, Cell cycle control, cell division, chromosome partitioning; lgreen, Amino acid transport and metabolism; vlgreen, Nucleotide transport and metabolism; grey, Carbohydrate transport and metabolism; dblue, Coenzyme transport and metabolism; dyellow, Translation, ribosomal structure and biogenesis; vlred, Transcription; vlyellow, Replication, recombination and repair; lpurple, Cell wall/membrane/envelope biogenesis; black, Posttranslational modification, protein turnover, chaperones; vlorange, Inorganic ion transport and metabolism; lorange, Secondary metabolite biosynthesis, transport and catabolism; dpurple, General function prediction only; vlpurple, Function unknown; lred, Signal transduction mechanisms; dgrey, Intracellular trafficking, secretion, and vesicular transport; vvlgrey, Defense mechanisms); Circle 3, tRNA (red) and rRNA operon (blue); Circle 4, secondary metabolism genes; Circles 5 and 6 (forward and reverse strands), distributions of conserved (red) and strain-specific genes (blue) in the S. albulus ZpM genome compared with 11 other Streptomyces species; Circle 7, GC content; Circle 8, GC bias ([G − C/G + C], green indicates values > 1, dark < 1). The inside scale is numbered clockwise in Mb. The outer scale indicates the core (red) and noncore (gray) chromosomal regions. The origin of replication (Ori) is also indicated.
Comparative genomic analysis shows that the chromosomal region ranging from position 2.0 Mb to position of 8.0 Mb of S. albulus ZPM is conserved and shows significant synteny among Streptomyces species; thus, the 6-Mb chromosomal region is defined as the core region of S. albulus ZPM, whereas the chromosomal regions at both ends are referred to as the variable regions (Fig. 1, Fig. S4). The lengths of the core regions of Streptomyces species range from 5.0 M to 7.5 M, which is proportional to their chromosome lengths (Fig. S4). Compared with S. albulus ZPM, the chromosomal sequences around the oriC appear to have significant genomic synteny with other Streptomyces species, although numerous inversions have been observed around this region (Fig. S4).
Pan-genome analysis reveals a large portion of specific genes in S. albulus ZPM
Pan-genome analyses were performed to analyze the core, lineage- and strain-specific genes of S. albulus ZPM together with those of 11 other Streptomyces species with complete genomes. First, an all-against-all BLASTP search with a cutoff E-value of 1e-5 was performed using sequences of proteins encoded in the genomes. Second, proteins with sequence identity > 50% and alignment coverage >70% were clustered; 93,586 proteins were grouped into 21,245 distinct protein families. The core families shared by the 11 Streptomyces species consisted of 1980 families, in which 43.92–55.60% of the genes of each species were included (Fig. 2A, Table S2). Although the numbers of core genes of the species appeared to be significantly different, the correlation between the core gene count and genome size was very high (R2 = 0.94, p-value = 1.73e-7, Table S2). The lowest and highest expansion rates of the core genes were 1.56 (3105/1980) for S. cattleya and 2.30 (4555/1980) for S. bingchenggensis, respectively, and the differences were also due to distinct genome sizes and gene counts (Table S2). Based on the phylogenetic relationships of the 12 Streptomyces species, lineage-specific genes (LSGs) shared by at least two clades occupied 25.20–39.58% of the genes in each species (Fig. S5). These LSGs contribute to species diversity and confer selective advantages, such as adaptation to different niches and antibiotic resistance. Similarly, the number of LSGs in each species showed strong correlations with the genome size (R2 = 0.87, p-value = 1.06e-5, Table S2).
Figure 2. Pan-genome analysis among Streptomyces species.

(A) Percentages of core (blue), dispensable (red) and specific (green) genes in S. albulus ZPM and 11 other Streptomyces species. The core genes represent genes shared by all 12 Streptomyces species, dispensable genes represent genes shared by at least two Streptomyces species, specific genes represent genes unique to one Streptomyces species. The 12 Streptomyces species are S. avermitilis (SAV), S. albulus ZPM (SAZ), S. bingchenggensis (SBB), S. cattleya (SCN), S. coelicolor (SCO), S. flavogriseus (SFA), S. griseus (SGN), S. hygroscopicus (SJG), S. scabiei (SSB), S. sp. SirexAA-E (SSE), S. venezuelae (SVA) and S. violaceusniger (SVT). (B) Density plot of the core (red) and strain-specific genes (green) along the chromosomes of twelve Streptomyces species. The bin size is 400 kb. The x- and y-axes represent the percentage of the chromosomal length and the proportion of genes in each Streptomyces species, respectively.
Notably, the correlation between the number of specific genes in each species and the genome sizes of 12 Streptomyces species was 0.64, which was lower than that of the core and lineage-specific genes. Interestingly, 2172 of the specific genes were exclusively identified in S. albulus ZPM, which is the highest specific gene number among the 12 Streptomyces species. The ratio between the specific genes and the core genes in S. albulus ZPM is 0.545 (2172/3985), which is even higher than 0.40 (1840/4555) for S. bingchenggensis, which has a 22% larger genome (Fig. 2A, Table S2). Because the specific genes encode proteins involved in supplementary biochemical pathways and proteins whose functions contribute to unique phenotypic traits or adaptation to living niches, the high genome plasticity of S. albulus ZPM suggests that the bacteria have a higher competence for acquiring and adapting new genes in its genome. Density plots show that the specific genes are predominantly located in the variable genome (Fig. 2B). Correspondingly, 66.23% of the specific genes of S. albulus ZPM have been mapped to the ends of the chromosome. Gene ontology (GO) enrichment analysis of the specific genes of S. albulus ZPM revealed that the specific genes were rich in classes of phosphopantetheinyl transferases (GO: 0016740), DNA transposases (GO: 0004803) (FDR < 2.4e-4) and catalytic activity (GO: 0003824) at the molecular function level (Fig. S6). Because phosphopantetheine is an essential prosthetic group for polyketide synthase, this result suggests that S. albulus ZPM might have many specific PKS pathways. Meanwhile, the enrichment of proteins with DNA transposase activity may explain how S. albulus ZPM easily acquires and adapts other genes in its genome.
Gene clusters and the potential for the production of secondary metabolites
Streptomyces are well-known producers of a variety of secondary metabolites, such as antibiotics, antiparasitics, and anticancer agents. Bioinformatics analyses revealed 44 gene clusters for secondary metabolites in S. albulus ZPM (Table 1), which was almost twice the 25 gene clusters of S. coelicolor A3 (2). The total length of these gene clusters is 1,983,954 bp and accounts for 20% of the S. albulus ZPM genome. The distribution of those gene clusters on the chromosome is not uniform: 31 of the 44 gene clusters are located in the variable genome, which indicates that these gene clusters might have been horizontally acquired through evolution. The other 13 gene clusters are in the core region, which contains most of the essential genes.
Table 1. Gene clusters for secondary metabolites in S. albulus ZPM.
| Clusters | Type | From | To |
|---|---|---|---|
| 1 | Nrps-t4pks | 17026 | 87092 |
| 2 | Unknown | 67864 | 109753 |
| 3 | Butyrolactone | 330907 | 341776 |
| 4 | T1pks | 364054 | 412441 |
| 5 | Unknown | 433815 | 474768 |
| 6 | Nrps-t1pks-oligosaccharide | 526610 | 600593 |
| 7 | Unknown | 596797 | 640057 |
| 8 | Unknown | 883066 | 926491 |
| 9 | Lantipeptide | 954336 | 978788 |
| 10 | T1pks | 1103165 | 1150604 |
| 11 | Nrps | 1137227 | 1196791 |
| 12 | Nrps | 1404109 | 1468194 |
| 13 | Nrps-t1pkstransatpks | 1476172 | 1525894 |
| 14 | T1pks-t4pks | 1670949 | 1783018 |
| 15 | Bacteriocin | 1838897 | 1849184 |
| 16 | T2pks | 1896764 | 1939279 |
| 17 | Siderophore | 2770965 | 2782767 |
| 18 | Siderophore | 2865668 | 2880351 |
| 19 | Ectoine | 3020511 | 3031683 |
| 20 | Terpene | 5358015 | 5380444 |
| 21 | Unknown | 6045012 | 6089448 |
| 22 | T4pks-nrps-transatpks | 6538252 | 6638419 |
| 23 | T3pks | 7015396 | 7056448 |
| 24 | Nrps-butyrolactone | 7056728 | 7124818 |
| 25 | Bacteriocin | 7169080 | 7179982 |
| 26 | Oligosaccharide | 7198268 | 7222581 |
| 27 | Lantipeptide | 7528854 | 7553415 |
| 28 | T2pks-oligosaccharide | 7788124 | 7853180 |
| 29 | Nrps-nucleoside | 7848120 | 7894680 |
| 30 | Terpene | 8001726 | 8028454 |
| 31 | pls biosynthesis gene cluster(PBC) | 8283445 | 8326182 |
| 32 | Butyrolactone | 8585464 | 8596462 |
| 33 | Nrps | 8706076 | 8762919 |
| 34 | Lantipeptide | 8781455 | 8823243 |
| 35 | Butyrolactone | 8870590 | 8881612 |
| 36 | Unknown | 9116811 | 9160601 |
| 37 | T1pks-lantipeptide | 9192594 | 9347388 |
| 38 | Terpene | 9329502 | 9379196 |
| 39 | T1pks | 9444073 | 9491961 |
| 40 | Unknown | 9485986 | 9526705 |
| 41 | Lantipeptide | 9545426 | 9574401 |
| 42 | T1pks | 9614130 | 9661965 |
| 43 | Unknown | 9674825 | 9716714 |
| 44 | T4pks-Nrps | 9697486 | 9767552 |
Polyketide synthases (PKSs) and NRPSs are key players in the synthesis of secondary metabolites from primary metabolites. PKSs generate polyketide chains through the oligomerization of small carboxylic acids, whereas NRPSs utilize amino acids as building blocks to form amide or ester bonds. S. albulus ZPM includes 9 PKS clusters, 6 NRPS clusters and 5 hybrid PKS-NRPS clusters (Table 1). Of the 9 PKS gene clusters, S. albulus ZPM contains 4 gene clusters for type I PKSs, 2 gene clusters for type II PKSs, 1 gene cluster for type III PKSs and 1 gene cluster for type I and type IV hybrid PKSs. The pls NRPS gene cluster (PGC) is 42.7 kb in length and consists of 42 proteins. In PGC, the poly-lysine synthase encoded by SAZ_7727 and poly-lysine degradase II encoded by SAZ_7728 form an operon and overlap each other by 4 nt, indicating that translation of the two genes is coordinately regulated. In addition to the 20 PKS/NRPS clusters and 8 clusters of unknown function, the remaining 16 clusters have been predicted to direct the synthesis of secondary metabolites, including terpene, siderophores, lantipeptides and butyrolactone. One of the five lantipeptide gene clusters (cluster 27) of S. albulus ZPM is homologous to a gene cluster (class III lantipeptides) of S. coelicolor A3 (2). The genes of gene cluster 27 of S. albulus ZPM and of Class III lantipeptides of S. coelicolor A3 (2) are located in an unusually low-GC-content region that houses two transposase proteins (SAZ_6981, SAZ_7026) of S. albulus ZPM and one transposase protein (SCO6910) of S. coelicolor A3 (2), respectively, which are essential for horizontal gene transfer (HGT) from other species.
Identification of ε-PL yield-related genetic variants
To identify other genes involved in ε-PL biosynthesis, forward genetics was employed. S. albulus ZPM was used as a starting strain to perform mutagenic breeding using ultraviolet (UV) irradiation and nitrosoguanidine (NTG). One hundred eighty mutant strains with ε-PL yields from 0 g/L to 1.05 g/L were obtained from the original isolate of S. albulus ZPM. More than 70 of the strains had higher ε-PL yields than the original strain, indicating that ε-PL biosynthesis was enhanced by the mutated genes, whereas strains with lower ε-PL yields suggested that the biosynthesis pathway was impaired or even completely blocked by the mutated genes.
To determine the genetic variations related to ε-PL yield, equal amounts of the gDNAs of 30 mutants with the lowest ε-PL yields (group-L) and 30 mutants with the highest ε-PL yields (group-H) were pooled and sequenced using the Illumina HiSeq-2000 platform (Figure 3A, Table S3). In total, 675 and 630 genetic variants (SNP or Indel) were identified in group-L and group-H, respectively. Group-L and group-H share 467 common variants, accounting for 69.2% and 74.1% of the genomes of group-L and group-H, respectively (Table S4). To make the analysis simpler, only 208 group-L-specific variants and 163 group-H-specific variants that were believed to be related to ε-PL yield were analyzed further (Figure 3B).
Figure 3. Identification and analysis of ε-PL yield-related genetic variants.
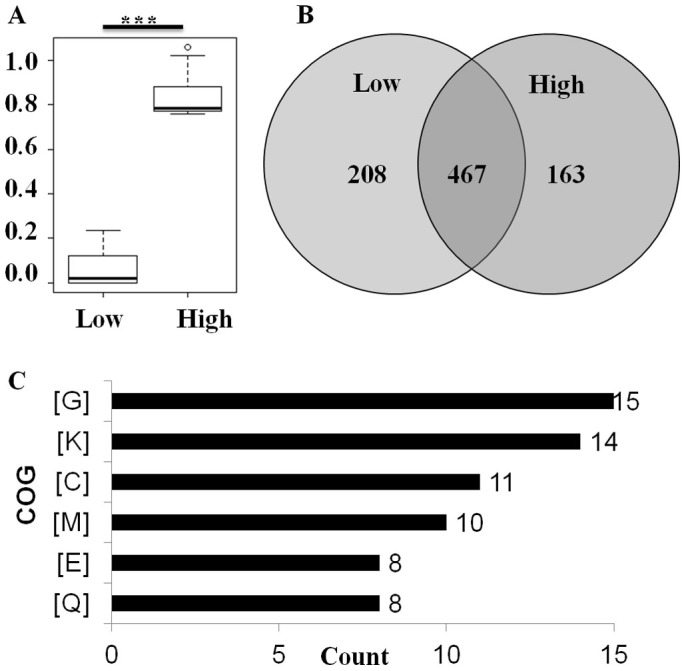
(A) Box plot of the PL yield in the group-L and group-H mutant strains. Significance between the two groups is indicated by asterisks. (B) Venn diagram showing the genetic variants identified from the Low- and High-groups. (C) COG classifications of genes with non-synonymous mutations. [G]: Carbohydrate transport and metabolism; [K]: Transcription; [C]: Energy production and conversion; [M]: Cell wall/membrane/envelope biogenesis; [E]: Amino acid transport and metabolism; and [Q]: Secondary metabolites biosynthesis, transport and catabolism.
For the group-L-specific genetic variants, 94 of 208 (45.2.0%) specific genetic variants were located in the coding region and led to amino acid changes, including 86 non-synonymous SNPs, 5 frameshift deletions/insertions, 2 non-frameshift deletions/insertions, and 1 stop gain/loss (Table 2). The group-H-specific genetic variations had similar distribution. Eighty-four of 163 (51.5%) group-H specific genetic variations were located in the coding regions and led to non-synonymous substitutions. Eighty-two group-L-specific and 52 group-H-specific SNPs/Indels mapped within 300 bp upstream or downstream of the coding regions of genes, which means that these SNPs/Indels might play a role in regulating the transcription of neighboring genes (Table 2). When combining the group-L- and group-H-specific genetic variants, 162 genes had non-synonymous substitutions.
Table 2. Classification of low- and high-specific variants.
| #lib | low-specific | high-specific |
|---|---|---|
| non-coding region | 89 | 57 |
| intergenic | 7 | 5 |
| upstream | 23 | 11 |
| downstream | 28 | 19 |
| upstream; downstream | 31 | 22 |
| coding region | 119 | 106 |
| synonymous SNV | 25 | 22 |
| nonsynonymous SNV | 86 | 81 |
| frameshift deletion | 3 | 1 |
| frameshift insertion | 2 | 2 |
| nonframeshift deletion | 1 | 0 |
| nonframeshift insertion | 1 | 0 |
| stopgain SNV | 1 | 0 |
| stoploss SNV | 0 | 0 |
| Total | 208 | 163 |
ε-PL yield is associated with both primary and secondary metabolism
COG analysis revealed that 66 of the 162 mutated genes could be assigned to at least one COG; in particular, genes from groups related to carbohydrate transport and metabolism, transcription and energy production were significantly enriched (Fig. 3C). In Streptomycetes, primary metabolism significantly influences secondary metabolism by providing precursors and reducing equivalents21. Although L-lysine is the substrate for ε-PL biosynthesis, these results show that ε-PL biosynthesis is not only related to the biosynthesis of L-lysine and ATP generation but is also associated with multiple cellular processes (Table S5).
Carbohydrate transport and metabolism is an essential component of central metabolism. It not only produces precursor metabolites for macromolecule biosynthesis but also generates ATP and other co-factors. In S. albulus ZPM, L-lysine is biosynthesized through the succinyl-diaminopimelic acid (DAP) pathway starting from L-aspartate. In addition to L-aspartate, other substrates, including pyruvate, succinyl-CoA, glutamate, ATP and NADPH are also required to complete the entire L-lysine biosynthesis pathway. L-aspartate is generated from oxaloacetate by the transfer of the amine group of L-glutamate to the keto group of oxaloacetate, whereas transamination of α-ketoglutarate results in glutamate. Pyruvate, α-ketoglutarate and oxaloacetate are key components of cellular metabolism, due to their contributions as substrates or intermediates in the fundamental processes of glycolysis, gluconeogenesis, and the citric acid cycle. Pyruvate kinase is a key enzyme in glycolysis that catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to ADP to yield one molecule of pyruvate and one molecule of ATP. In group-L, an insertion mutation (261_262insCC) in pyruvate kinase (SAZ_6202) causes frameshifting of translation and impairments in the glycolytic pathway and L-lysine biosynthesis, which confirms that the ε-PL yield is correlated with pyruvate production and the downstream tricarboxylic acid cycle (TCA cycle). Non-synonymous SNPs have been identified in other genes involved in glycolysis and the TCA cycle, such as citrate synthase (N357H of SAZ_1229), NAD-glutamate dehydrogenase (P1341T of SAZ_3874), acetyl-CoA acetyltransferase (H185P of SAZ_8129) and 4-hydroxy-2-oxovalerate aldolase (T118P of SAZ_8139). Although the enzymatic activities of these mutated genes could not be determined, we suspect that these mutants most likely perturb the metabolic flux from glucose to L-lysine following the TCA cycle route.
Due to limitations in energy and/or the primary metabolite supply, the synthetic pathways of different secondary metabolites use common primary metabolites as precursors and compete with each other. The precursor L-lysine for ε-PL biosynthesis could be de novo synthesized from L-aspartate or transported from the surrounding environment. In our analysis, 21 mutated transporter genes were identified, including 8 ABC transporters, 6 transporters of the major facilitator superfamily, 6 other transporters and 1 potassium/proton antiporter. These proteins do not transport L-lysine directly but are responsible for content exchange between the extracellular environment and the cytoplasm, which is important for maintaining the stable physiological status of S. albulus ZPM and most likely indirectly impacts the ε-PL yield. L-aspartate is the precursor to several amino acids, including lysine, methionine, threonine, and isoleucine. No SNP has been identified in aspartokinase (SAZ_5045), which is the key enzyme of the lysine biosynthesis pathway. However, methionine synthase (SAZ_8322) with an R203M substitution, which is involved in the superpathway of lysine, threonine and methionine biosynthesis and catalyzes the formation of L-methionine from L-homocysteine, has been identified in group-L. Interestingly, non-synonymous substitutions have been identified in 2 polyketide synthase genes (SAZ_8963, SAZ_8662) and 1 NRPS-type-I PKS fusion protein (SAZ_1118); in these cases, oligomerization of small carboxylic acids catalyzed by these synthases might be disturbed, resulting in perturbation of the ε-PL yield through metabolic networks.
Transcriptional activator HrdD binds with the promoter of pls gene in vitro
Because ε-PL biosynthesis is induced by the acid condition (pH < 5) in S. albulus ZPM, we suspected that a sigma factor (σ factor) that responds to environmental pH change will guide RNA polymerase to bind the promoter and to initiate transcription of the pls gene. HrdD, encoded by SAZ_4132, is a sigma factor that shows the most sensitive response to pH change19 and contains a non-synonymous mutation (A132V) in mutant strains with lower ε-PL yields. To test whether HrdD binds to the promoter of the pls gene, electrophoretic mobility shift assays (EMSAs) were performed. 3 nM of DNA fragments of 500 bp that corresponded to the pls promoter (pls-p500) were incubated with his-tagged HrdD protein, and clear band shifts were observed when the concentration of HrdD was higher than 0.2 μM (Fig. 4A, lanes 3–4). To exclude the possibility of unspecific binding between HrdD to pls-p500, competition assay was done by incubating his-tagged HrdD with pls-p500 DNA fragments labeled with fluorescein isothiocyanate (FITC) and also with unlabeled cold pls-p500 DNA substrates (specific DNA) or the DNA fragments of green fluorescent protein gene (nonspecific DNA substrates) (Fig. 4B). The unlabeled cold pls-p500 DNA substrates competitively inhibited the binding of HrdD to the FITC-labeled pls-p500 DNA substrates (Fig. 4B, lanes 7–9). However, the non-specific DNA fragments, even up to 20 times excessive DNA substrates, have negligible effect on the binding of HrdD to the labeled pls-p500 DNA substrates (Fig. 4B, lanes 10–12). The competition assay definitively proved that HrdD is able to specifically bind to the promoter of pls gene and might monitor the pls expression and initiates ε-PL biosynthesis in response to pH changes in vivo.
Figure 4.
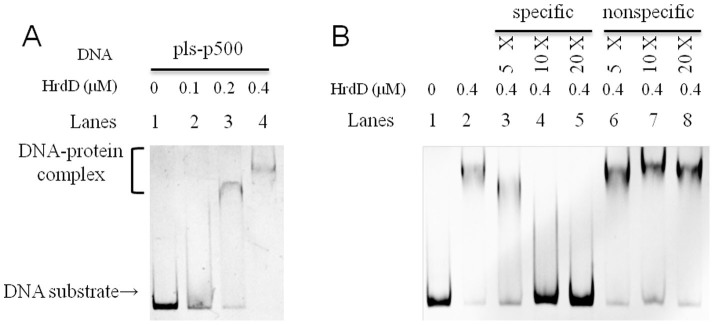
(A) EMSA assays of HrdD and the putative promoter of the pls gene. The recombinant HrdD protein was co-incubated with 30 nM 500-bp (lanes 1–4) upstream promoter DNA of the pls gene and were assayed on a 5% native PAGE gel. The final concentrations of the His-tag HrdD proteins in each mixture were 0, 0.1, 0.2 and 0.4 μM, as indicated in the second line. (B) In the competition assay, 3 nM of DNA substrates corresponding to the putative promoter of the pls gene (pls-p500) were labeled with fluorescein isothiocyanate (FITC) and were incubated with 0.4 μM his-tagged HrdD protein to compete with either excessive unlabeled pls-p500 DNA substrates or excessive nonspecific DNA substrates (lanes 7–9) derived from the gene coding green fluorescent protein(lanes 10–12).
Discussion
In the past decades, much effort has been made to improve ε-PL yield, including genome shuffling and chemical mutagenesis. Although some industrial strains with higher yields have been obtained, the underlying regulatory mechanism of ε-PL biosynthesis is still unknown. In this study, we report the genome sequence of S. albulus ZPM, which is the first complete genome sequence of S. albulus without gaps or ambiguous regions. Currently, the genome sequences of two other S. albulus strains, S. albulus CCRC 1181420 and S. albulus PD-121, not only contain contaminants from other species but also miss tens of thousands of nucleotides (data not shown). The high-quality complete sequence of S. albulus ZPM allows us to perform comparative genomic analysis of Streptomycetes species and identify genes associated with ε-PL biosynthesis by genome sequencing of S. albulus ZPM mutants.
Genome analysis showed that 162 proteins, from important metabolic enzymes to novel transcriptional regulators, were identified as related to ε-PL synthesis, which suggests that ε-PL biosynthesis is not only dependent on the supply of the primary metabolites L-lysine and ATP but also on multiple cellular processes. Meanwhile, our study shows at least 14 non-synonymous substitutions in the genes of transcription factors, including RNA polymerase alpha subunit A (SAZ_4270), the principal sigma factor HrdD (SAZ_4132) and the ECF subfamily RNA polymerase sigma factor (SAZ_4218, SAZ_5788). Previous studies have shown that the secondary metabolite yields of industrial strains could be enhanced by global transcription machinery engineering (gTME). For example, the application of gTME to Saccharomyces cerevisiae improved glucose/ethanol tolerance and increased biofuel production22,23. Thus, the transcription factors identified in this study might impact ε-PL yield in a similar manner.
Methods
Enzymes, plasmids and reagents
Restriction enzymes, DNA polymerase, dNTPs and all antibiotics were purchased from TaKaRa Biotech. PCR primers were synthesized by Sangon Biotech Company. All other reagents were purchased from Sigma unless specified.
Strains and media
The original S. albulus ZPM strain was isolated from the soil of Zi-Peng Mountain, Hefei, China. The spore suspension of S. albulus was treated with UV (20 W) irradiation for 30–65 seconds at a 20-cm distance, followed with 0.5 g/L NTG for 30 minutes at 30°C and 180 rpm. All strains of S. albulus were cultured on SGB agar plates or in M3G broth at 30°C. E. coli strains were cultured in LB medium.
Genome sequencing, assembly, annotation and analysis
gDNA was extracted using the UltraClean® Microbial RNA Isolation Kit following the manual, and Multiplex Identifier (MID)-tagged paired-end and mate-pair libraries were generated using the NextFlex DNA-seq preparation kit following the manufacturer's instructions. The libraries were pooled, and paired-end sequencing (2 × 100 bp) was performed using a Hiseq 2000/2500 instrument. Raw sequence data were processed using the manufacturer's software and quality-filtering algorithms. After demultiplexing, quality control and adapter trimming, 205,282,144 paired-end reads, 17,428,002 mate-paired (3–5 kb) reads and approximately 890 × coverage reads were obtained (we used a 10-Mb genome size to calculate the genome coverage of the sequencing output). The genome assembly of these reads was performed with SOAPdenovo224 using filtered reads (Q > 20) and resulted in 9.7 Mb of sequence data in 9 contigs, in which the 36-kb contiguous contig represented the complete sequence of the plasmid pZPM234 (Fig. 1B). The order and orientations of the other 8 contigs were determined using the mate-pair reads, and the internal gaps were filled by additional genomic sequencing data from mutants using GapFiller25 and were then manually checked. Open reading frames (ORFs) were predicted with Glimmer3.0226 and Prodigal.v227. The programs were trained with ~400,000 ORFs from the completely sequenced Streptomycetes genes that were available in public databases. We also used FramPlot29, BLAST30 (National Center for Biotechnology Information BLAST package; ftp://ftp.ncbi.nih.gov/), and HmmPfam31 to confirm the protein-coding genes predicted by Glimmer and Prodigal. CDS annotation was based on the BLASTP program with the NR, CDD and COG databases. Blast2GO was also used to identify GO annotations of the proteins28. S. albulus ZPM was annotated with GO terms, enrichment analysis was subsequently performed for specific and core genes, and Fisher's Exact test was performed with a P-value cutoff of 0.01. Phosphopantetheine binding (GO: 0031177) and DNA integration (GO: 0015074) were significantly enriched in the specific genes, with corrected P-values by False Discovery Rate (FDR) control of 2.0e-17 and 2.4e-4, respectively. tRNA and rRNA were predicted by tRNA-Scan29 and rRNAmmer30, respectively. Pair-wise alignments between S. albulus ZPM genome and other Streptomycetes genomes were performed using the Nucmer or Promer programs of the MUMmer package. The metabolic pathway was constructed based on the Kyoto Encyclopedia of Genes and Genomes (KEGG), and COG functional classification for all of the nonsynonymous variants was performed.
Analytical method for determining the ε-PL concentration
To measure the ε-PL concentration by the method of Itzhaki14, a 0.1 mM phosphate buffer (prepared by adjusting the pH of a 15.6 g/l NaH2PO4·2H2O solution to 6.6 with a 35.8 g/l Na2HPO3·12 H2O solution) and a 0.1 mM methyl orange solution (Sigma) were prepared. Then, 1.9 ml of phosphate buffer and 2.0 ml of methyl orange solution were added to 0.1 ml of the supernatant. The mixtures were then vigorously reacted on a reciprocal shaker at 30°C and then centrifuged. The optical density of the resulting supernatant was measured at 465 nm, and the ε-PL concentration was calculated from the calibration curve.
Library pooling and high-throughput sequencing
One hundred eighty mutant strains were obtained using multiple mutagenesis strategies. Of these strains, 30 that produced the lowest yield of ε-PL and 30 that produced the highest yield of ε-PL were chosen to be the low (L) and high (H) groups, respectively. For each strain, the gDNA was extracted with the UltraClean Microbial DNA Isolation Kit and checked by gel electrophoresis, and the DNA concentration was determined using a NanoDrop 2500. For each group, equal amounts of gDNA from each of the 30 strains were pooled, resulting in two DNA mixtures. Then, the DNA mixtures were fragmented using the Bioruptor standard module, and paired-end DNA sequencing libraries with an insert size of ~300 bp were constructed using the NextFlex DNA-seq Library Kit as described in the manufacturer's manuals. DNA sequencing was performed on an Illumina HiSeq 2000 sequencer according to standard protocols. The Illumina base-calling pipeline was used to process the raw fluorescent images and to call sequences. Raw reads were cleaned using in-house scripts, and low-quality reads from the paired-end sequencing were discarded.
SNP calling and functional annotation
After de-duplication and quality filtering, 19,762,419 and 16,918,601 high-quality read pairs (2 × 100) were obtained for group-L and group-H, respectively. These short reads were aligned to the S. albulus ZPM reference genome sequence using Bowtie231, and SNP-calling was performed. Based on the alignment, 92.5% and 95.1% of the reads were aligned exactly 1 time, and the overall alignment rates were 98.3% for group-L and 98.8% for group-H, respectively. Pileups were generated using samtools32 and were directly piped to VarScan233 to perform pooled-library SNP calling. Low-quality read alignments (MAPQ < 10) and/or bases (BaseQ < 20) were discarded from the pileup. The minimal coverage (--min-coverage), minimal variant frequency (--min-var-freq) and strand filter (--strand-filter) of VARSCAN2 were set to be 50, 0.03, and true, respectively. Functional annotation of the SNPs, including determining whether the SNPs caused protein coding changes and identifying the affected amino acids, was performed using ANNOVAR34.
Cloning, expression and purification of the recombinant proteins
S. albulus ZPM genes were amplified from gDNA using PCR primers. The corresponding genes were cloned into the pET28a over-expression vector to produce recombinant vectors, and protein expression and purification were performed according to the previously described procedure35. The elutions were dialyzed overnight and stored at −80°C, and the protein concentrations were measured using Coomassie Brilliant Blue assays36.
Electrophoretic mobility shift assay (EMSA)
The binding of transcriptional regulators to the putative promoter (500 bp) of the pls gene was examined by EMSAs as described35. The HrdD coding sequence was amplified from the gDNA of S. albulus ZPM using specific primers and was cloned into pET28(a) expression vector. The DNA substrates of the putative pls promoter (pls-p500) were obtained by PCR and used in the EMSA assays. A 500 bp DNA segment of gfp gene (gfp-500) coding green fluorescent protein(GFP) was used as negative control. 3 nM of the DNA fragments was incubated at 4°C for 30 min or 1 hr with various amounts of proteins in a total volume of 25 μl of EMSA buffer consisting of 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, and 50 mM NaCl. Then, the mixtures were directly subjected to 5% native PAGE containing 0.5 × Tris-borate-EDTA (TBE) buffer, and electrophoresis was performed at 200 V at 4°C or in an ice bath until the bromophenol blue dye reached the bottom of the gel. The gels were then stained with ethidium bromide for ~5 minutes, and the images were acquired using a Gel Doc XR Snapper (Bio-Rad). In the competition assay, 3 nM pls-p500 DNA fragments labeled with fluorescein isothiocyanate (FITC) were incubated with 0.4 μM his-tagged HrdD protein together with 5, 10 and 20 times excessive DNA substrates of unlabeled pls-p500 or gfp-500. After 1 hr incubation and further electrophoresis, the image was acquired with Typhoon Scanner (GE healthcare).
Accession numbers
The whole-genome sequencing projects described in this study have been deposited at GenBank under accession number CP006871.
Author Contributions
Q.W. designed the experiments. L.W., C.G. and N.T. performed the experiments. L.W., C.G., S.H. and Q.W. analyzed the results. L.W., C.G. and Q.W. wrote the manuscript.
Supplementary Material
Supplementary file
Acknowledgments
This study was supported by the National Science and Technology Major Project (2013ZX10004605-001-002), the Fundamental Research Funds for the Central Universities (Grants WK2070000034, WK2070000019).
References
- Berdy J. Thoughts and facts about antibiotics: where we are now and where we are heading. J Antibiot (Tokyo) 65, 385–395 (2012). [DOI] [PubMed] [Google Scholar]
- Liu G., Chater K. F., Chandra G., Niu G. & Tan H. Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol Mol Biol Rev 77, 112–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T. & Nagasawa T. epsilon-Poly-L-lysine: microbial production, biodegradation and application potential. Appl Microbiol Biotechnol 62, 21–26 (2003). [DOI] [PubMed] [Google Scholar]
- Shima S. & Sakai H. Polylysine Produced by Streptomyces. Agric Biol Chem 41, 1807–1809 (1977). [Google Scholar]
- Shima S., Matsuoka H., Iwamoto T. & Sakai H. Antimicrobial action of epsilon-poly-L-lysine. J Antibiot (Tokyo) 37, 1449–1455 (1984). [DOI] [PubMed] [Google Scholar]
- Hamano Y. et al. Biological function of the pld gene product that degrades epsilon-poly-L-lysine in Streptomyces albulus. Appl Microbiol Biotechnol 72, 173–181 (2006). [DOI] [PubMed] [Google Scholar]
- Hamano Y. Occurrence, biosynthesis, biodegradation, and industrial and medical applications of a naturally occurring epsilon-poly-L-lysine. Biosci Biotechnol Biochem 75, 1226–1233 (2011). [DOI] [PubMed] [Google Scholar]
- Li S. et al. Isolation and characterization of a novel epsilon-poly-L-lysine producing strain: Streptomyces griseofuscus. J Ind Microbiol Biotechnol 38, 557–563 (2011). [DOI] [PubMed] [Google Scholar]
- Chen X. et al. Optimization of medium for enhancement of epsilon-poly-L-lysine production by Streptomyces sp. M-Z18 with glycerol as carbon source. Bioresour Technol 102, 1727–1732 (2011). [DOI] [PubMed] [Google Scholar]
- Liu S. R., Wu Q. P., Zhang J. M. & Mo S. P. Production of epsilon-poly-L-lysine by Streptomyces sp using resin-based, in situ product removal. Biotechnol Lett 33, 1581–1585 (2011). [DOI] [PubMed] [Google Scholar]
- Ouyang J. et al. Production of epsilon-poly-L-lysine by newly isolated Kitasatospora sp. PL6-3. Biotechnol J 1, 1459–1463 (2006). [DOI] [PubMed] [Google Scholar]
- Yamanaka K., Maruyama C., Takagi H. & Hamano Y. epsilon-Poly-L-lysine dispersity is controlled by a highly unusual nonribosomal peptide synthetase. Nat Chem Biol 4, 766–772 (2008). [DOI] [PubMed] [Google Scholar]
- Yamanaka K. et al. Mechanism of epsilon-Poly-L-Lysine Production and Accumulation Revealed by Identification and Analysis of an epsilon-Poly-L-Lysine-Degrading Enzyme. Appl Environ Microbiol 76, 5669–5675 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahar P., Iwata T., Hiraki J., Park E. Y. & Okabe M. Enhancement of epsilon-polylysine production by Streptomyces albulus strain 410 using pH control. J Biosci Bioeng 91, 190–194 (2001). [DOI] [PubMed] [Google Scholar]
- Nishikawa M. & Ogawa K. Distribution of microbes producing antimicrobial epsilon-poly-L-lysine polymers in soil microflora determined by a novel method. Appl Environ Microbiol 68, 3575–3581 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi Y. et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190, 4050–4060 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley S. D. et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 (2002). [DOI] [PubMed] [Google Scholar]
- Ikeda H. et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21, 526–531 (2003). [DOI] [PubMed] [Google Scholar]
- Kim Y. J. et al. Acidic pH shock induces the expressions of a wide range of stress-response genes. BMC Genomics 9, 604 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd A., Swanevelder D., Featherston J. & Rumbold K. Draft Genome Sequence of Streptomyces albulus Strain CCRC 11814, an Epsilon-Poly-L-Lysine-Producing Actinomycete. Genome Announc 1, e00696-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. et al. Genome Sequence of Streptomyces albulus PD-1, a Productive Strain for Epsilon-Poly-L-Lysine and Poly-L-Diaminopropionic Acid. Genome Announc 2, e00297-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper H., Moxley J., Nevoigt E., Fink G. R. & Stephanopoulos G. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science 314, 1565–1568 (2006). [DOI] [PubMed] [Google Scholar]
- Steensels J. et al. Improving industrial yeast strains: exploiting natural and artificial diversity. FEMS Microbiol Rev 38, 947–995 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M. & Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol 13, R56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A. L., Bratke K. A., Powers E. C. & Salzberg S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- Lowe T. M. & Eddy S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucl Acids Res 35, 3100–3108 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D. C. et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M. Y. & Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C. H., Yang M. & He Z. G. Characterization of a Novel ArsR-Like Regulator Encoded by Rv2034 in Mycobacterium tuberculosis. Plos One 7, e36255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file