

Abstract
Tyrosinemia type 1 (HT1) is an inborn error of tyrosine catabolism caused by fumarylacetoacetase deficiency. Biochemically, this results in accumulation of toxic metabolites including succinylacetone. Clinically, HT1 is characterized by severe liver, kidney, and neurological problems. Treatment with NTBC and dietary restriction of tyrosine and phenylalanine have strongly improved outcome, but impaired neurocognitive development has been reported. Whether impaired neurocognitive outcome results from high blood tyrosine or low blood phenylalanine concentrations is currently unknown. In this report, two HT1 newborns, diagnosed by neonatal screening, are presented. The first patient showed low phenylalanine concentrations, growth retardation, neurological impairments, and skin problems, clearly improving after institution of phenylalanine supplementation (~30 mg/kg/day) at age 6 months, while both blood phenylalanine and tyrosine concentrations increased. In the second patient, phenylalanine supplementation (~20 mg/kg/day) was initiated as soon as low phenylalanine concentrations were observed at age 19 days. On this regimen, blood phenylalanine concentrations increased, and hypophenylalaninemia was less frequently observed than in the first patient, whereas blood tyrosine concentrations tended to increase. Clinically, no growth, neurological, or skin problems have been observed. The combination of knowledge obtained from these cases suggests that hypophenylalaninemia rather than hypertyrosinemia during the first months of life may impair neurocognitive development in young HT1 infants. Phenylalanine supplementation should really be considered in HT1 patients with consistently low blood phenylalanine concentrations during the first months of life. However, the minimal phenylalanine concentrations acceptable and the optimal phenylalanine supplementation regimen require further investigation.
Introduction
Clinically, tyrosinemia type I (HT1; OMIM 276600, fumarylacetoacetase deficiency) is characterized by liver failure, hepatocellular carcinoma, renal tubulopathy, and porphyria-like syndrome (de Laet et al. 2013). Implementation in neonatal blood spot screening (NBS) and treatment with 2-(2-nitro-4-trifluoromethyl-benzyl)-1,3-cyclohexanedione (NTBC) to prevent accumulation of toxic metabolites by inhibiting tyrosine catabolism upstream from the primary enzymatic defect has largely improved clinical outcome (Larochelle et al. 2012; Mayorandan et al. 2014).
Unfortunately, however, neurocognitive deficits are observed, which may be related to HT1 itself, NTBC treatment, increased tyrosine concentrations, or low phenylalanine concentrations (De Laet et al. 2011; Masurel-Paulet et al. 2008; Thimm et al. 2012; Bendadi et al. 2014). NTBC increases tyrosine concentrations. Therefore, tyrosine intake is limited by severe natural protein restriction and supplementation of a synthetic amino acid mixture devoid of tyrosine and phenylalanine, as tyrosine is primarily derived from hydroxylation of dietary phenylalanine. This results in both high tyrosine and low phenylalanine concentrations (De Laet et al. 2011; Daly et al. 2012; Wilson et al. 2000), with dietary treatment being primarily monitored based on tyrosine concentrations. In phenylketonuria, too strict dietary treatment, resulting in “low” phenylalanine concentrations, has shown to be related to impaired growth and development (Teissier et al. 2012; Rouse 1966; Smith et al. 1990; Pode-Shakked et al. 2013). In tyrosinemia type II, high tyrosine concentrations are associated with mental retardation. Together, this raises the question whether current dietary treatment creates a tyrosine toxicity or a phenylalanine deficiency that may impair growth and brain development (De Laet et al. 2011; Daly et al. 2012; Wilson et al. 2000) and whether phenylalanine supplementation is beneficial or only increases tyrosine concentrations (Daly et al. 2012; Wilson et al. 2000).
This study reports on blood phenylalanine and tyrosine concentrations as well as on growth and development of two HT1 infants detected by NBS, who received phenylalanine supplementation because of very low phenylalanine concentrations during the first months of life.
Patients and Methods
Patient 1 and patient 2 are both children of healthy, Dutch, non-consanguineous parents with an uneventful fetal development and a normal birth process. HT1 was detected by NBS. Treatment was started with NTBC (1 mg/kg) and dietary restriction of phenylalanine and tyrosine, aiming at a total protein intake of 2.5 g/kg. In below, both patients are described until phenylalanine supplementation was introduced.
Patient 1 (girl) was diagnosed in 2009 (NBS, SA 4.7 μmol/L; DNA, homozygous for FAH c.554-1G>T) at day 7. High tyrosine levels above the upper target of 400 μmol/L (de Laet et al. 2013) led to further decrease of natural protein intake to a minimum of 0.5 g/kg/day (Fig. 1a, b). On this regimen, blood tyrosine concentrations decreased. Also, blood phenylalanine concentrations decreased, while blood concentrations of other essential amino acids remained well within the normal range (valine, 167–373 μmol/L; isoleucine, 39–121 μmol/L; leucine, 82–220 μmol/L; methionine, 19–42 μmol/L; histidine, 72–108 μmol/L; threonine, 138–473 μmol/L; tryptophan, 56–93 μmol/L; and lysine, 146–277 μmol/L). During the subsequent months, she first developed “eczema” (Fig. 2). Later, impaired growth was observed, while BMI was undisturbed (Fig. 1a, b). Thereafter, she showed increasing myoclonus periorally and in the extremities. Electroencephalographic and myographic recordings at day 100 were abnormal demonstrating cortical myoclonus. Development of motor milestones seemed to be impaired despite adjustments by either increasing or decreasing the natural protein intake and tyrosine concentrations varying from some 215–609 μmol/L. In this period, prealbumin decreased from 0.19 g/L at day 9 to values ranging from 0.12 to 0.15 g/L. Because of persisting low blood phenylalanine concentrations (<20 μmol/L), phenylalanine supplementation was started at day 191.
Fig. 1.
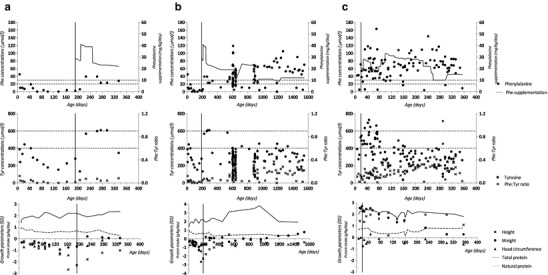
Phenylalanine concentrations and supplementation (upper), tyrosine concentrations and ratios of blood phenylalanine-tyrosine concentrations (middle), and growth parameters as well as protein intake (lower) in patient 1 during the first year (a) and the first 4 years of life (b) and in patient 2 during the first year of life (c). The vertical line indicates the start of phenylalanine supplementation
Fig. 2.

Patient 1 – eczema-like skin eruptions developed at 5 months of age on the whole body, as depicted on the back of the head (a) and buttock (b). After 3 days of phenylalanine supplementation, the skin problem had largely been improved (c)
Patient 2 (boy) was diagnosed in 2013 (NBS, SA 5.1 μmol/L; DNA, FAH c.554-1G>T/c.674T>G) at day 7. Because of low blood phenylalanine concentrations (<20 μmol/L) and the previous experiences with patient 1, phenylalanine supplementation was started at day 19 (Fig. 1c), without clinical signs suggesting phenylalanine deficiency. At day 11, a prealbumin concentration of 0.17 g/L was measured.
NTBC Treatment and Protein Intake
For both patients, NTBC treatment was regularly adjusted based on body weight, targeting at a dose of 1 mg/kg/day.
Both patients received breast milk combined with an amino acid-based tyrosine-free and phenylalanine-free powdered infant formula (Tyr Anamix Infant, Nutricia Advanced Medical Nutrition) during the first 3 months (patient 1) and during the first 5 months (patient 2). Thereafter, breast milk was replaced by a regular infant formula. Additional feedings started at around 6 months (patient 1) and 5 months (patient 2).
Phenylalanine Supplementation
In patient 1, at day 191, ~30 mg/kg/day phenylalanine supplementation was started, which was increased to ~40 mg/kg/day at day 209 and reduced to ~20 mg/kg/day at day 247 (Fig. 1a, b).
In patient 2, phenylalanine supplementation of ~20 mg/kg/day was started at day 19. Due to low blood phenylalanine concentrations (5–48 μmol/L), phenylalanine supplementation was increased to ~25 and ~30 mg/kg/day at day 85 and 99, respectively. Thereafter, dosage has frequently been adjusted (ranging from ~10 to ~30 mg/kg/day), especially based on blood phenylalanine concentrations (Fig. 1c).
Biochemical Analyses
Phenylalanine and tyrosine concentrations were measured in plasma samples as well as in dried blood spots on filter paper. Plasma samples were drawn at the outpatient clinic, not taking into account the time of blood sampling. Actual measurement was performed with standard techniques on a Biochrom 20 or 30 analyzer (Pharmacia Biotech, Cambridge, UK). Blood spots were taken at home at variable times of the day. In dried blood spots, phenylalanine and tyrosine were quantified using high-performance liquid chromatography tandem mass spectrometry. If both plasma and dried blood spot analyses were available from a single time point, results from plasma samples were used.
Assessment of Developmental Milestones
Assessment of developmental milestones was performed at regular intervals at the child health center according to Dutch guidelines. In The Netherlands, developmental milestones are evaluated in children from 4 weeks to 5 years of age according to the Van Wiechen neurodevelopmental assessment (Schlesinger-Was 1985). This assessment is based on milestones and consists of five domains: gross motor skills, fine motor skills, personality, social behavior, and communication skills.
Assessment of Spontaneous Motor Repertoire
The most reliable method to evaluate the integrity of the central nervous system of young infants is to assess the quality of their spontaneous motor repertoire (Einspieler and Prechtl 2005). Around the age of 3 months, the infant’s motor repertoire is characterized by so-called fidgety movements (FMs) that are highly predictive of neurological outcome (Prechtl et al. 1997). From patient 2, video recordings were made during an outpatient visit at the age of 10 weeks and 1 day and at the age of 14 weeks and 1 day. The recordings were made during a period of active wakefulness with the infant lying supine and lasted 10 min (Einspieler and Prechtl 2005). Certified author MMH analyzed the recordings off-line according to Prechtl’s method (Einspieler et al. 1997) and blinded for the outcome. First, the quality of FMs was labeled as normal, abnormal (exaggerated speed, amplitude, and jerkiness), or absent (no FMs observed). Additionally, a more detailed analysis was performed based on the motor optimality score (MOS) by using the Motor Optimality List (Bruggink et al. 2009). The MOS is composed of FM quality (12 points if normal, 4 points if abnormal, and 1 point if absent) plus 4 other items including age adequacy, normality of movement patterns, normality of postural patterns, and quality of the concurrent motor repertoire (4 points if normal, 2 points if nonoptimal, and 1 point if abnormal). The MOS may range from 5 (low optimality) to 28 (high optimality).
Neurocognitive Assessment
Neurocognitive assessment was performed by a qualified clinical psychologist. Verbal and nonverbal IQ were assessed separately. The SON-R (2.5–7) assessment was used for measurement of nonverbal IQ (Tellegen et al. 1998). Verbal performance was assessed using the General Language Index of the WPPSI-III-NL (Hurks et al. 2013).
Results
Clinical Outcome
In patient 1, subsequently to the institution of phenylalanine supplementation at day 191, all clinical parameters improved. Within some days after initiation of phenylalanine supplementation, eczema disappeared (Fig. 2). This was soon followed by resolution of cortical myoclonus as well as improvement of growth parameters (Fig. 1a, b). In patient 2, skin, growth, and development of motor milestones have all been normal.
Developmental Milestones
In patient 1, at 1 month of age, developmental milestones were all reached. At 3, 9, 12, and 15 months of age, fine motor skills, personality, social behavior, and communication skills were normal. However, development of gross motor skills seemed to be impaired without the tendency of progression. In patient 2, developmental milestones were normal at all ages at which developmental milestones were measured, being at age 1, 2, 3, 6, 9, and 12 months.
Neurocognitive Development
In patient 1, neuropsychological assessment was performed at age 4 years and 1 month. IQ score according to the nonverbal SON-R (2.5–7) assessment was 77 (80% CI: 63–79). Verbal performance on the General Language Index of the WPPSI-III-NL was 77 (95% CI: 69–93). These scores correspond to a mental development of children at 3 years of age.
Patient 2 was too young for a detailed neuropsychological assessment to be applied. Instead, video assessment of spontaneous movements was performed. At both recordings, we observed normal FMs with an MOS of 26. The infant only scored nonoptimal on the item concurrent motor repertoire with the movements having a monotonous and a slightly stiff appearance.
Blood Phenylalanine and Tyrosine Concentrations
Median phenylalanine and tyrosine concentrations as well as the incidence of hypophenylalaninemia (blood phenylalanine <30 μmol/L or <20 μmol/L) and hypertyrosinemia (blood tyrosine >400 μmol/L or >600 μmol/L) before and after initiation of phenylalanine supplementation are presented in Table 1.
Table 1.
Phenylalanine and tyrosine concentrations at diagnosis and before and after institution of phenylalanine supplementation
| Patient 1 | Patient 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Additional Phe/day | Diagnosis | 8–364 | Before 8–190 | After 191–1,547 | Diagnosis | 8–364 | 8–190 | Before 8–18 | After 19–364 |
| Phe (μmol/L); median (range) | 45 | 9 (0–39) | 6 (0–19) | 43 (0–119) | 42 | 64 (4–163) | 58 (5–163) | 43 (5–71) | 65 (4–163) |
| Phe <30 (μmol/L) | 88% (15/17) | 100% (10/10) | 33% (32/97) | 9% (9/97) | 10% (6/61) | 33% (1/3) | 9% (8/94) | ||
| Phe <20 (μmol/L) | 71% (12/17) | 100% (10/10) | 15% (15/97) | 6% (6/97) | 5% (3/61) | 33% (1/3) | 5% (5/94) | ||
| Tyr (μmol/L); median (range) | 388 | 292 (133–609) | 253 (152–444) | 298 (132–609) | 391 | 331 (73–728) | 384 (200–728) | 324 (274–526) | 333 (73–728) |
| Tyr >400 (μmol/L) | 24% (4/17) | 10% (1/10) | 11% (11/97) | 31% (30/97) | 49% (30/61) | 33% (1/3) | 31% (29/94) | ||
| Tyr >600 (μmol/L) | 12% (2/17) | 0% (0/10) | 2% (2/97) | 5% (5/97) | 8% (5/61) | 0% (0/3) | 5% (5/94) | ||
| Phe: Tyr ratio; median (range) | 0.12 | 0.03 (0.00–0.08) | 0.02 (0.00–0.05) | 0.15 (0.00–0.38) | 0.11 | 0.21 (0.01–1.07) | 0.16 (0.02–0.35) | 0.13 (0.02–0.16) | 0.21 (0.01–1.07) |
Phe phenylalanine, Tyr tyrosine
Incidence of hypophenylalaninemia and hypertyrosinemia are defined as the percentage of blood samples with phenylalanine concentrations <30 μmol/L or <20 μmol/L and tyrosine concentrations >400 μmol/L or >600 μmol/L, respectively
In patient 1, phenylalanine concentrations could not be quantitated in three samples
Overall, during the first 6 and 12 months of life, median blood phenylalanine concentrations were lower, and hypophenylalaninemia was observed more frequently in patient 1 than in patient 2. Median blood tyrosine concentrations, however, were comparable for both patients, and hypertyrosinemia was observed as frequently in both patients during the first 12 months of life. During the first 6 months of life, median blood tyrosine concentrations were even higher, and hypertyrosinemia was observed more frequently in patient 2 than in patient 1. Ratios of blood phenylalanine-tyrosine concentrations during the first 12 months of life tended to increase with increasing age in patient 2, while this increasing trend was not observed in patient 1.
From the day after diagnosis until day 190 (patient 1) and day 18 (patient 2), in both patients, hypophenylalaninemia was observed more frequently than hypertyrosinemia. Especially in patient 2, phenylalanine supplementation increased median blood phenylalanine concentrations and reduced the incidence of hypophenylalaninemia, while also increasing median blood tyrosine concentrations and the incidence of hypertyrosinemia. Irrespective of phenylalanine supplementation, between 40 and 120 days of age, both in patient 1 (without supplementation) and in patient 2 (with supplementation), blood tyrosine concentrations tended to decrease.
Discussion
The most important findings of this report are that (1) hypophenylalaninemia rather than hypertyrosinemia during the first months of life was observed to correlate with impaired growth, cortical myoclonus, neurological deficits, and skin problems and that (2) phenylalanine supplementation resolved these clinical problems despite causing a higher incidence of hypertyrosinemia.
Blood of both patients was collected at variable times of the day. However, blood phenylalanine concentrations in HT1 have previously been reported to show a large diurnal variation, consistently being lower in the afternoon (Daly et al. 2012), while previous reports showed that detection of deficient intake of essential amino acids can best be detected by comparing an overnight fasting and postprandial blood sample (Ozalp et al. 1972). Further studies may therefore best include a differential overnight fasting to postprandial measurement.
High blood tyrosine is often regarded as the cause of neurocognitive problems in HT1 (Thimm et al. 2011, 2012). However, these two cases support the hypothesis that hypophenylalaninemia rather than hypertyrosinemia could be the most important (De Laet et al. 2011; Bendadi et al. 2014). Patient 1 with the lowest incidence of hypertyrosinemia but highest incidence of hypophenylalaninemia showed skin problems, poor growth, and impairment in motor and cognitive development. In contrast, patient 2 with early preventive phenylalanine supplementation showed a higher incidence of hypertyrosinemia, normal skin, growth, developmental milestones, and quality of spontaneous movements. In addition, the start of phenylalanine supplementation in patient 1 immediately improved growth and resolved eczema and cortical myoclonus.
These cases also show that blood phenylalanine concentrations may not immediately increase in response to phenylalanine supplementation as all supplemented phenylalanine is first used for protein synthesis, leaving phenylalanine concentrations in blood unchanged, thereby supporting the idea that phenylalanine had indeed been rate limiting for protein synthesis. Consistent with this idea, concentrations of other essential amino acids (results not shown) as well as tyrosine all decreased after phenylalanine had been initiated, and catch-up growth was observed.
The fact that, before phenylalanine supplementation had been initiated, both height and weight were affected and weight-for-height remained normal supports the idea that a qualitative rather than a quantitative malnutrition had been present. Also, in both patients, prealbumin concentrations had increased after phenylalanine supplementation. This all suggests that low phenylalanine rather than high tyrosine – at least during the first months of life – may impair growth and neurocognitive outcome in HT1 infants.
This idea is supported by the early experiences with treatment of phenylketonuria in which phenylalanine deficiency during the first months of life has been reported to impair growth and intellectual development and to induce eczema (Rouse 1966). Further circumstantial evidence might be that these problems at least had not been reported before the era of NTBC. This might well be due to a lack of focus, as preventing liver failure and hepatocellular carcinoma was the primary and ultimate target of treatment in those days. However, it could also be that developmental delay and skin problems were indeed not present, for not only tyrosine concentrations were high but also blood phenylalanine concentrations were rather high than low in those patients (Couce et al. 2011). Alternatively, NTBC – at its own – may have resulted in these issues as well, but NTBC dose did not vary substantially between and in our patients.
In conclusion, these cases urge to consider the danger of low phenylalanine concentrations with respect to neurocognitive outcome in HT1 patients and to treat accordingly. Further studies including a larger patient population are certainly required. The exact minimal phenylalanine level – as the maximal tyrosine level – is a matter of further investigation. Also, the optimal phenylalanine supplementation regimen to prevent both phenylalanine deficiency and tyrosine toxicity requires additional research.
Take-Home Message
Phenylalanine supplementation should really be considered in HT1 patients with consistently low blood phenylalanine concentrations during the first months of life.
Compliance with Ethical Guidelines
Conflicts of Interest
Esther van Dam has received advisory board fees from Merck Serono.
Margreet van Rijn has received research grants, consultancy fees, and advisory board fees from Merck Serono and Nutricia Research; speaker’s honoraria from Merck Serono, Nutricia Research, and Orphan Europe; and expert testimony fees from Merck Serono.
Terry G.J. Derks has received research grants from Sigma Tau and Vitaflo and speaker’s honoraria form Nutricia and Vitaflo.
Francjan J. van Spronsen has received research grants, advisory board fees, and/or speaker’s honoraria from Merck Serono, Merck (the Netherlands), Nutricia Research, and Vitaflo.
Danique van Vliet, Gineke Liefaard-Venema, Marrit M. Hitzert, Roelineke J. Lunsing, and M. Rebecca Heiner-Fokkema declare that they have no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000. This study was waived from approval from the responsible committee on human experimentation. Informed consent was obtained from all patients for which identifying information is included in this article.
Details of the Contributions of the Authors
Danique van Vliet drafted the initial manuscript, revised the manuscript, and approved the final manuscript as submitted.
Esther van Dam performed the dietary follow-up of the second patient, drafted the initial manuscript, and approved the final manuscript as submitted.
Margreet van Rijn performed the dietary follow-up of both patients, critically reviewed the manuscript, and approved the final manuscript as submitted.
Terry G.J. Derks performed the treatment and monitoring of both patients, cowrote the manuscript, and approved the final manuscript as submitted.
Gineke Venema-Liefaard performed the dietary follow-up of the first patient, critically reviewed the manuscript, and approved the final manuscript as submitted.
Marrit M. Hitzert performed assessment of spontaneous motor repertoire in patient 2, critically reviewed the manuscript, and approved the final manuscript as submitted. Roelineke J. Lunsing performed was responsible for neurological follow-up of both patients, critically reviewed the manuscript, and approved the final manuscript as submitted.
M. Rebecca Heiner-Fokkema was responsible for the biochemical analyses, critically reviewed the manuscript, and approved the final manuscript as submitted.
Francjan J. van Spronsen was responsible for the diagnosis of HT1, performed the treatment and monitoring of both patients, cowrote the manuscript, and approved the final manuscript as submitted.
Footnotes
Competing interests: None declared
Contributor Information
Francjan J. van Spronsen, Email: f.j.van.spronsen@umcg.nl
Collaborators: Johannes Zschocke, Matthias Baumgartner, K Michael Gibson, Marc Patterson, and Shamima Rahman
References
- Bendadi F, de Koning TJ, Visser G, et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr. 2014;164:398–401. doi: 10.1016/j.jpeds.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Bruggink JL, Einspieler C, Butcher PR, Stremmelaar EF, Prechtl HF, Bos AF. Quantitative aspects of the early motor repertoire in preterm infants: do they predict minor neurological dysfunction at school age? Early Hum Dev. 2009;85:25–36. doi: 10.1016/j.earlhumdev.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Couce ML, Dalmau J, del Toro M, Pintos-Morell G, Aldamiz-Echevarria L, Spanish Working Group on Tyrosinemia Type 1 (2011) Tyrosinemia type 1 in Spain: mutational analysis, treatment and long-term outcome. Pediatr Int 53:985–989 [DOI] [PubMed]
- Daly A, Gokmen-Ozel H, MacDonald A, Preece MA, Davies P, Chakrapani A, McKiernan P. Diurnal variation of phenylalanine concentrations in tyrosinaemia type 1: should we be concerned? J Hum Nutr Diet. 2012;25:111–116. doi: 10.1111/j.1365-277X.2011.01215.x. [DOI] [PubMed] [Google Scholar]
- De Laet C, Munoz VT, Jaeken J, et al. Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol. 2011;53:962–964. doi: 10.1111/j.1469-8749.2011.04048.x. [DOI] [PubMed] [Google Scholar]
- de Laet C, Dionisi-Vici C, Leonard JV et al (2013) Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 8:8-1172-8-8 [DOI] [PMC free article] [PubMed]
- Einspieler C, Prechtl HF. Prechtl’s assessment of general movements: a diagnostic tool for the functional assessment of the young nervous system. Ment Retard Dev Disabil Res Rev. 2005;11:61–67. doi: 10.1002/mrdd.20051. [DOI] [PubMed] [Google Scholar]
- Einspieler C, Prechtl HF, Ferrari F, Cioni G, Bos AF. The qualitative assessment of general movements in preterm, term and young infants–review of the methodology. Early Hum Dev. 1997;50:47–60. doi: 10.1016/S0378-3782(97)00092-3. [DOI] [PubMed] [Google Scholar]
- Hurks PP, Hendriksen JG, Dek JE, Kooij AP. Normal variability of children's scaled scores on subtests of the Dutch Wechsler Preschool and Primary scale of Intelligence – third edition. Clin Neuropsychol. 2013;27:988–1003. doi: 10.1080/13854046.2013.797502. [DOI] [PubMed] [Google Scholar]
- Larochelle J, Alvarez F, Bussieres JF, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Quebec. Mol Genet Metab. 2012;107:49–54. doi: 10.1016/j.ymgme.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al (2008)NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis 31: 81-87. [DOI] [PubMed]
- Mayorandan S, Meyer U, Gokcay G, et al. Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis. 2014;9:107. doi: 10.1186/s13023-014-0107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozalp I, Young VR, Nagchaudhuri J, Tontisirin K, Scrimshaw NS. Plasma amino acid response in young men given diets devoid of single essential amino acids. J Nutr. 1972;102:1147–1158. doi: 10.1093/jn/102.9.1147. [DOI] [PubMed] [Google Scholar]
- Pode-Shakked B, Shemer-Meiri L, Harmelin A, et al. Man made disease: clinical manifestations of low phenylalanine levels in an inadequately treated phenylketonuria patient and mouse study. Mol Genet Metab. 2013;110(Suppl):S66–S70. doi: 10.1016/j.ymgme.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Prechtl HF, Einspieler C, Cioni G, Bos AF, Ferrari F, Sontheimer D (1997)An early marker for neurological deficits after perinatal brain lesions. Lancet 349: 1361-1363. [DOI] [PubMed]
- Rouse BM. Phenylalanine deficiency syndrome. J Pediatr. 1966;69:246–249. doi: 10.1016/S0022-3476(66)80327-X. [DOI] [PubMed] [Google Scholar]
- Schlesinger-Was EA. Child development examinations in well-child clinics for infants and young children. Tijdschr Kindergeneeskd. 1985;53:105–113. [PubMed] [Google Scholar]
- Smith I, Beasley MG, Ades AE. Intelligence and quality of dietary treatment in phenylketonuria. Arch Dis Child. 1990;65:472–478. doi: 10.1136/adc.65.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier R, Nowak E, Assoun M, et al. Maternal phenylketonuria: low phenylalaninemia might increase the risk of intra uterine growth retardation. J Inherit Metab Dis. 2012;35:993–999. doi: 10.1007/s10545-012-9491-0. [DOI] [PubMed] [Google Scholar]
- Tellegen PJ, Winkel M, Wijnberg-Williams BJ, Laros J. Snijders-Oomen Nonverbal Intelligence Test. SON-R. 1998;2:5–7. [Google Scholar]
- Thimm E, Herebian D, Assmann B, Klee D, Mayatepek E, Spiekerkoetter U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol Genet Metab. 2011;102:122–125. doi: 10.1016/j.ymgme.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Thimm E, Richter-Werkle R, Kamp G, et al. Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC. J Inherit Metab Dis. 2012;35:263–268. doi: 10.1007/s10545-011-9394-5. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Van Wyk KG, Leonard JV, Clayton PT. Phenylalanine supplementation improves the phenylalanine profile in tyrosinaemia. J Inherit Metab Dis. 2000;23:677–683. doi: 10.1023/A:1005666426079. [DOI] [PubMed] [Google Scholar]