

Abstract
Hepatocellular adenomas (HCAs) are a common complication in patients with glycogen storage disease type I (GSD I). In this series, we report regression of HCAs in a cohort of patients who achieved metabolic control with strict dietary therapy. A retrospective review of the clinical records for all patients with GSD I was performed at our institution. All available imaging studies were reviewed in patients with reported regression of HCAs in the medical record. The charts of 163 patients with GSD Ia and 42 patients with GSD Ib were reviewed, and HCAs were documented in 47 subjects (43 Ia/4 Ib). After review of all available imaging studies, eight patients met criteria of being followed with both magnetic resonance imaging and ultrasound and were found to show evidence of regression of HCAs. In these individuals, regression of the HCAs occurred once metabolic control was obtained, as determined by decreasing levels of serum triglyceride levels. The average triglyceride level in all patients prior to regression of HCAs was 753 mg/dL (SD ± 293). The average serum triglyceride level in all patients at the time of regression of HCAs was 340 mg/dL (SD ± 164). These findings suggest that strict dietary therapy may cause regression of HCAs. If HCAs are documented in a patient with suboptimal metabolic control, intensive medical therapy may be an alternative to surgical intervention in some individuals.
Introduction
Glycogen storage disease type I (GSD I) is an autosomal recessive inborn error of metabolism caused by defects in the glucose-6-phosphatase complex. All endogenous glucose synthesis is impaired in affected individuals, which leads to metabolic derangements including hypertriglyceridemia, hyperuricemia, and hyperlactatemia. Disordered glucose metabolism results in marked hepatomegaly. Hepatocellular adenoma (HCA) formation occurs frequently in patients with GSD I and can be seen in up to 70% of patients greater than the age of 25 (Wang et al. 2011; Lee 2002; Rake et al. 2002a; Weinstein and Wolfsdorf 2002). The management of HCA has been a source of debate due to the risk of hepatocellular carcinoma, and invasive procedures including embolization, ablation, surgical resection, and transplantation have been proposed as methods for treating growing or suspicious lesions (Rake et al. 2002b; Davis and Weinstein 2008; Reddy et al. 2009). We report regression of HCAs in several patients who experienced metabolic improvement after presenting with suboptimal metabolic control.
Methods
Institutional review board approval and informed written consent was obtained from patients participating in an observational study at the University of Florida Glycogen Storage Disease Program. Subjects have been followed in a longitudinal and prospective manner. A retrospective review of the clinical records for all patients with GSD I was performed. Longitudinal records were available from 205 patients of whom 163 patients had GSD Ia and 42 patients had GSD Ib. The diagnosis for GSD I was established biochemically or by assessment of enzymatic testing from a liver biopsy specimen, and the diagnosis was confirmed with DNA analysis. As part of routine clinical care, patients discovered to have HCAs were subsequently monitored with serial imaging through either magnetic resonance imaging (MRI) or ultrasonography. In some patients, contrast enhanced computed tomography (CT) was also performed. The MR protocol used at our institution for evaluation of liver lesions involves imaging the patient on either a 1.5 or 3.0 Tesla MR unit and obtaining pre-contrast axial in and out of phase T1-weighted images, pre-contrast axial T1-weighted image, and pre-contrast coronal T1- and T2-weighted images. A hepatocellular-specific MR contrast agent, gadoxetate disodium, is then administered intravenously. Axial post-contrast T1-weighted images are then obtained after 25 s, 60 s, 90 s, 3 min, and 20 min. A coronal T1-weighted image is also obtained 20 min after contrast administration. A post-contrast fat-saturated T2-weighted axial image and diffusion weighted imaging are also included in the protocol.
The total number of patients with documented HCAs was 47 (43 GSD Ia/4 GSD Ib) (Tables 1 and 2). The diagnosis of HCA was made based on imaging appearance. Biopsy of the liver lesions was not performed in this group of patients since it is no longer standard of care for this population. The lesions identified on imaging are presumed to represent HCAs given their characteristic imaging appearance and location within the liver. Ultrasound and CT appearance can vary depending on intratumoral hemorrhage and lipid content; however, HCAs tend to be homogenous and hyperechoic with ultrasound. HCAs with CT tend to be isodense to background liver on non-contrast studies and are hyperdense after administration of contrast during the arterial phase and hypodense on delayed phase. Lesions are typically heterogeneous on MRI and demonstrate no significant uptake of hepatocellular specific contrast agents. None of the lesions identified were thought to represent focal fatty deposition or focal nodular hyperplasia. Optimal metabolic control is defined as triglyceride level less than 200 mg/dL, lactate less than 2.2 mmol/L, and serum glucose greater than 75 mg/dL. The clinical records, laboratory data, and all radiographic imaging for these patients were reviewed.
Table 1.
Patients with HCAs that did not regress
| Age | Sex | GSD type | DNA analysis | Age at HCA discovery (years) | Average triglyceride level (mg/dL) | Number of HCAs | Largest HCA size (cm) | HCA change in last 5 years | Avg. follow-up (years) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 20 | M | GSD Ia | 35X/Q347X | 16 | 584 ± 297 | 1 | 0.8 | Stable | 5 |
| 2 | 21 | F | GSD Ia | R83C | 16 | 325 ± 110 | 2 | 1.0 | Stable | 6 |
| 3 | 10 | M | GSD Ia | R83C/E110K | 3 | 289 ± 65 | 1 | 1.3 | Stable | 6 |
| 4 | 47 | M | GSD Ia | R83C/Q347X | 19 | 546 ± 118 | 3 | 1.3 | Stable | 6 |
| 5 | 15 | M | GSD Ia | R83C/G270V | 9 | 295 ± 69 | 1 | 1.4 | Stable | 5 |
| 6 | 14 | M | GSD Ib | 464del10ins3C/1211delCT | 6 | 146 ± 41 | 2 | 1.8 | Stable | 8 |
| 7 | 59 | F | GSD Ia | R170Q/Q347X | 56 | 250 ± 61 | 2 | 3.1 | Stable | 1 |
| 8 | 32 | M | GSD Ia | R83C | ?? | 1,348 ± 953 | 3 | 3.2 | Stable | 7 |
| 9 | 25 | M | GSD Ib | G339C/1042delCT | 23 | 143 ± 25 | 1 | 3.3 | Stable | 2 |
| 10 | 30 | F | GSD Ia | R83C | 16 | 905 ± 299 | 15 | 3.6 | Stable | 15 |
| 11 | 40 | F | GSD Ia | R83C | Teenager | 314 ± 48 | 3 | 3.8 | Stable | 7 |
| 12 | 43 | F | GSD Ia | R83C/212X | 13 | 1,004 ± 113 | 4 | 8.0 | Stable | 12 |
| 13 | 18 | M | GSD Ia | R83C/Q242X | 8 | 890 ± 500 | 4 | 2.5 | Possible regression of adenomas; however, no MR for confirmation | 9 |
| 14 | 20 | M | GSD Ib | 1211delCT/C381 + 1G > T | 15 | 79 ± 25 | 2 | 1.3 | Decrease in size with possible resolution, but no MR for confirmation | 4 |
| 15 | 36 | F | GSD Ia | R83C/Q347X | 31 | 474 ± 129 | 2 | 3.5 | Largest lesion stable, questionable disappearance of smaller lesion | 8 |
| 16 | 38 | M | GSD Ia | R83C | 19 | 342 ± 86 | 2 | 2.2 | Largest lesion stable, smaller 9 cm lesion has developed | 7 |
| 17 | 16 | F | GSD Ia | T108I | 12 | 259 ± 147 | 4 | 1.3 | Largest increased by 0.5 cm | 3 |
| 18 | 38 | F | GSD Ia | ?? | 19 | 501 ± 145 | 5 | 3.9 | Largest increased by 0.7 cm | 12 |
| 19 | 43 | M | GSD Ia | R83C/G188S | ?? | 340 ± 107 | >20 | 5.5 | Largest increased by 1 cm | 5 |
| 20 | 18 | F | GSD Ia | Q347X | 13 | 551 ± 359 | >20 | 4.9 | Largest increased by 1 cm | 6 |
| 21 | 12 | F | GSD Ib | G68R/Q248X | 6 | 189 ± 39 | 4 | 4.0 | Largest grew by 1.3 cm | 6 |
| 22 | 41 | M | GSD Ia | A124T/Q347X | 37 | 341 ± 137 | 8 | 4.0 | Largest increased by 1.7 cm (previously had a 13.5 cm lesion which was resected) | 4 |
| 23 | 28 | M | GSD Ia | Q347X | 15 | 349 ± 91 | >20 | 4.8 | Largest increased by 2 cm with new lesions developing | 4 |
| 24 | 27 | M | GSD Ia | Q347X | 16 | 237 ± 116 | 6 | 4.0 | Largest increased by 2 cm | 12 |
| 25 | 35 | M | GSD Ia | Q347X/Q242X | 25 | 498 ± 102 | 4 | 5.6 | Largest increased by 2 cm | 3 |
| 26 | 24 | F | GSD Ia | 35X/D38V | 14 | 479 ± 107 | 7 | 5.6 | Increase in size by 2.5 cm | 5 |
| 27 | 39 | F | GSD Ia | R83C | 11 | 888 ± 302 | 8 | 4.2 | Largest increased by 3 cm with new lesions developing | 13 |
| 28 | 29 | F | GSD Ia | R83C | 16 | 729 ± 77 | >20 | 6.1 | Largest increased by 4 cm with new lesions developing | 6 |
| 29 | 46 | M | GSD Ia | R83C | 15 | 404 ± 140 | >20 | 17.4 | Increase by 6 cm | 2 |
| 30 | 33 | F | GSD Ia | T108I | 19 | 232 ± 55 | 1 | Prior 14 cm lesion | Lesion resected, no new lesions | 11 |
| 31 | 28 | F | GSD Ia | R83C | 12 | 947 ± 365 | 1 | Prior 4.3 cm lesion | Lesion resected, no new lesions | 7 |
| 32 | 30 | M | GSD Ia | Q347X/Q242X | 21 | 315 ± 113 | 8 | 6.5 | Right hepatectomy, otherwise stable | 2 |
| 33 | 19 | M | GSD Ia | 158delC | 10 | 632 ± 135 | 12 | 6.3 | Largest resected. Next dominant lesion growth by 4 cm | 8 |
| 34 | 23 | M | GSD Ia | R83C/Q347X | 13 | 1,082 ± 301 | 6 | 3.6 | Largest resected, other adenomas small with slow growth | 9 |
| 35 | 23 | M | GSD Ia | R83C/R170Q | 17 | 596 ± 353 | 6 | 6.0 | Largest resected after it grew by 5 cm, other adenomas small with slow growth | 9 |
| 36 | 22 | M | GSD Ia | R83C | 10 | 555 ± 198 | >20 | 5.3 | Largest resected, other adenomas continue to grow with new lesions appearing | 9 |
| 37 | 56 | F | GSD Ia | ??/Q347X | 45 | 725 ± 387 | 3 | 1.7 | Prior liver transplant, no new lesions | 5 |
| 38 | 41 | F | GSD Ia | R83C | 19 | 202 ± 3 | 4 | 7.1 | Unknown | 1 |
| 39 | 29 | M | GSD Ia | R83C | 18 | 1,044 ± 514 | 2 | 2.1 | Unknown | 0 |
?? unknown
Table 2.
Patients with regression of HCA
| Age | Sex | GSD Type | DNA analysis | Age at HCA discovery (years) | Age at Time of HCA regression (years) | # of HCAs | Size of Largest HCA (cm) | Size of HCA after regression | Duration of follow-up since beginning of regression (months) | Duration it took for HCAs to completely resolve (months) | Duration of follow-up since complete resolution (months) | Total duration of follow-up (months) | Average triglyceride level prior to HCA regression in mg/dL | Average triglyceride level since beginning of HCA regression in mg/dL | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 18 | M | GSDIa | R83C/130X | 5 | 9 | 14 | 1.5 | Complete resolution | 86 | 65 | 23 | 131 | 345 ± 68 | 211 ± 59 |
| 2 | 31 | F | GSDIa | R83C/R295C | 27 | 28 | 4 | 1.9 | Complete resolution | 29 | 7 | 24 | 152 | 957 ± 276 | 444 ± 140 |
| 3 | 34 | F | GSDIa | Q347X | 21 | 30 | 2 | 1.9 | Complete resolution | 60 | 19 | 41 | 81 | 591 ± 131 | 207 ± 22 |
| 4 | 32 | F | GSDIa | R83C | 12 | 23 | 5 | 2.0 | Complete resolution | 123 | 23 | 100 | 147 | 961 ± 403 | 294 ± 84 |
| 5 | 23 | M | GSDIa | W77R/G118S | 15 | 17 | 4 | 3.1 | Complete resolution | 57 | 36 | 21 | 57 | 814 ± 243 | 718 ± 169 |
| 6 | 35 | M | GSDIa | R83C/R295C | 22 | 30 | 2 | 3.3 | Complete resolution | 64 | 34 | 40 | 73 | 835 ± 184 | 312 ± 66 |
| 7 | 31 | F | GSDIa | Q347X | 19 | 25 | >10 | 2.6 | 1.5 cm (most small lesions resolved) | 41 | NA | NA | 60 | 314 ± 84 | 187 ± 14 |
| 8 | 34 | F | GSDIa | R83C | 29 | 30 | 5 | 13.2 | 9.1 cm | 49 | NA | NA | 49 | 1,205 ± 852 | 343 ± 110 |
| Average values | 63.6 | 30.7 | 41.5 | 93.8 | 753 ± 293 | 340 ± 164 | |||||||||
Results
There were ten patients (all with GSD Ia) who experienced regression of their HCAs. Eight of these patients were included in this report as they met criteria of having clear regression of their HCAs and have been followed with both MRI and ultrasonography (Table 2). Regression was defined as a minimum of 10% decrease in size or complete resolution of the lesion. Representative images demonstrating regression of HCAs are displayed in Figs. 1, 2, 3, and 4. In the individuals with documented regression, shrinkage of the HCAs occurred once improved metabolic control was obtained with strict dietary therapy as determined by decreasing levels of serum triglycerides. Dietary therapy included uncooked cornstarch given in a slurry of water or artificially sweetened fluid and is given at 3–5 h intervals during the day and 4–6 h intervals overnight. Each patient’s optimal schedule was based on metabolic and clinical monitoring. Intake of fructose, galactose, and sucrose was restricted to 2.5 g of non-utilizable sugar per meal. Of note, other laboratory values were followed in this group of patients including total serum cholesterol, serum glucose, uric acid, AST/ALT, and lactate. However, these biochemical parameters did not correlate as well with long-term metabolic control compared with serum triglycerides because uric acid can be affected by use of medication; serum glucose and lactate concentrations are short-term markers that can fluctuate rapidly. There were no confounding variables in these patients that could influence triglyceride levels, such as use of oral contraception, pregnancy, medications, or supplements.
Fig. 1.
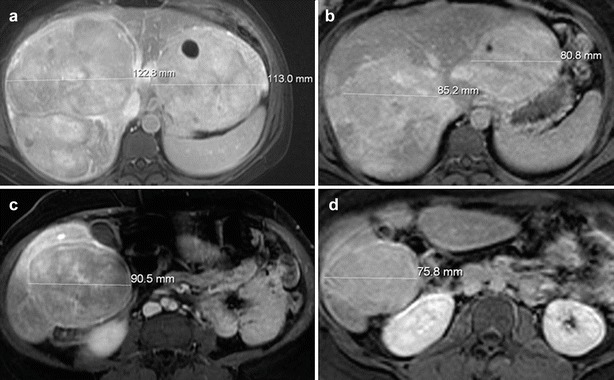
Post-contrast (gadoxetate disodium) MRI of a patient with GSD I with HCAs. (a) Fat-saturated T1-weighted post-contrast axial MR image of the superior liver obtained after intravenous bolus injection of gadoxetate disodium demonstrates multiple large lesions in the liver consistent with HCAs, two of the lesions are measured. (b) Fat-saturated T1-weighted post-contrast axial MR image of the superior liver obtained after intravenous bolus injection of gadoxetate disodium in the same patient 4 years later demonstrates decrease in size of the multiple HCAs, two of which are measured for comparison. (c) Fat-saturated T1-weighted post-contrast axial MR image of the inferior liver obtained after intravenous bolus injection of gadoxetate disodium in the same patient at the same time as the image in (a) demonstrates a large HCA involving the inferior right hepatic lobe (lesion is measured). (d) Fat-saturated T1-weighted post-contrast axial MR image of the inferior liver obtained after intravenous bolus injection of gadoxetate disodium in the same patient four years later, at the same time as the image in (b), demonstrates significant decrease in size of the HCA, measured for comparison
Fig. 2.
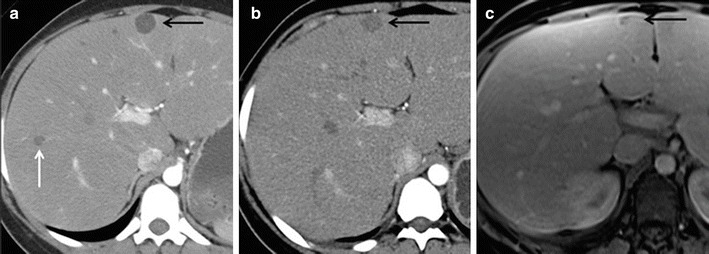
Computed tomography (CT) and MRI of a patient with GSD I with HCAs. (a) Axial CT image of the liver after administration of iodinated contrast demonstrates two hypodense lesions within the liver representing HCAs (black and white arrows). (b) Axial CT image of the liver after administration of iodinated contrast in the same patient 18 months later demonstrates decrease in size of the HCA in segment 4 of the liver (black arrow). The lesion in the right hepatic lobe, white arrow in (a), has completely resolved. (c) Axial fat-saturated T1-weighted post-contrast MR image of the liver obtained after intravenous bolus injection of gadoxetate disodium in the same patient 8 years later demonstrates continued decrease in size of the segment 4 HCA (black arrow)
Fig. 3.

MRI of a patient with GSD I with HCAs. (a) Coronal non-contrast T2-weighted MR image of the abdomen in a patient with GSD I demonstrates a mildly hyperintense lesion (white arrow) in the left lobe of the liver inferior to the diaphragm consistent with a HCA. (b) Coronal non-contrast fat-saturated T2-weighted MR image of the abdomen in the same patient 4 years later demonstrates significant decrease in size of the HCA in the left hepatic lobe (black arrow)
Fig. 4.

CT and MRI of a patient with GSD I with HCAs. (a) Axial CT image of the abdomen with oral contrast and without intravenous contrast in a patient with GSD I demonstrates a hypodense lesion in the inferior right hepatic lobe consistent with a HCA (black arrow). (b–d) Axial non-contrast T1-weighted MR image (b), axial post-contrast T1-weighted MR image (c), and axial non-contrast T2-weighted MR image (d) at the same level as (a) in the same patient 14 months later demonstrate complete resolution of the inferior right hepatic lobe HCA
The average serum triglyceride concentration in this group prior to regression of HCAs was 753 mg/dL (SD ± 293). In contrast, the average serum triglyceride level in all patients at time of regression of HCAs was 340 mg/dL (SD ± 164) (Table 2). Each patient’s average serum triglyceride concentration prior to regression presented in the table was calculated by averaging all their serum triglyceride concentration values prior to documented HCA regression on imaging. Each patient’s average serum triglyceride concentration since beginning of regression presented in the table was calculated by averaging all their serum triglyceride concentration values starting at the time imaging first demonstrated regression and during the follow-up period since regression was first reported. In six of the eight patients, complete regression of the HCAs was noted. The average total duration of patient follow-up was 93.8 months, while duration of follow-up since the beginning of HCA regression was 63.6 months. For the six patients with complete HCA resolution, the mean duration to disappearance of the lesions was 30.7 months from the beginning of adenoma shrinkage.
In contrast, there were eight patients who showed a significant increase in the size of their HCAs (>2 cm). None of these of patients ever achieved optimal metabolic control, and all of these patients struggled with dietary compliance with the prescribed treatment regimen. Of note, pharmacologic therapy was used in all of these patients to lower their triglycerides, but chronic hyperlactatemia remained due to the suboptimal metabolic control.
Discussion
HCA formation occurs frequently in patients with GSD I. Proposed mechanisms of HCA formation include hormonal stimulation, oxidative stress from disordered fatty acid metabolism, and proto-oncogenic activation (Wang et al 2011; Bianchi 1993). Complications of HCAs include local compression and hemorrhage (Rake et al. 2002a, b; Weinstein and Wolfsdorf 2002; Davis and Weinstein 2008). Another complication is the transformation of a HCA into hepatocellular carcinoma (Bianchi 1993; Kudo 2001; Limmer et al. 1988), which may occur in as much as 11% of patients (Coire et al. 1987).
While the risk of malignant transformation may be less than previously reported due to improvements in medical care, an aggressive approach toward managing the lesions has been favored. Once HCAs are detected, ultrasound or MRI is recommended every 3–6 months (Rake et al. 2002a, b), and intervention has been recommended when lesions are rapidly growing or reach 5 cm in diameter (Ault et al. 1996). HCA requires special attention during pregnancy due to risk of increased growth and rupture, especially when lesions are greater than 5 cm (Cobey and Salem 2004). It is appropriate to closely observe patients with HCA during pregnancy if lesions are less than 5 cm with frequent ultrasound surveillance every 6–12 weeks (Bröker et al. 2012). Due to the risk of adenoma growth during pregnancy, some investigators have proposed performing surgery, radiofrequency ablation, or embolization prior to pregnancy (Cobey and Salem 2004; Bröker et al. 2012), but intensive medical treatment offers another option for managing these patients.
Surgical resection of growing lesions has been successfully performed, but the procedure is not without risk due to the associated metabolic instability during surgery (Reddy et al. 2007). Radiofrequency ablation and embolization have also been utilized in treating HCAs. These methods are less invasive than surgery with decreased morbidity and mortality and have the ability to treat multifocal disease. However, these methods can be limited based on the location of the lesions (ablation), may need multiple treatments, may show inadequate response due to heat sink effect (ablation), and decreased efficacy with large (>5 cm) tumors (McDaniel et al. 2013; Rhim et al. 2008; Deodhar et al. 2011). Liver transplant cures the primary hepatic enzyme defect, but does not cure the disease (Davis and Weinstein 2008; Reddy et al. 2009; Maheshwari et al. 2012; Labrune 2002). The kidneys in people with GSD I are not normal, and most recipients of a liver transplant have progressed to renal failure (Matern et al. 1999). Medical management of growing HCAs has not been emphasized. We report successful stabilization and regression of HCAs with improved metabolic control, which may eliminate the need for surgical intervention in certain individuals.
The pathogenesis of HCA development is likely multifactorial. Prior work by Wang et al. (2011) demonstrates that poor metabolic control may play a role in HCA formation. In a case control cohort study, they found a statistically significant difference in the serum triglyceride concentration at the time of HCA discovery compared with age-matched control subjects with a mean triglyceride concentration of 737 mg/dL (SD ± 422) in the case group and 335 mg/dL (SD ± 195) in the control group. At the time of HCA presentation, our patient population had an average triglyceride level of 753 mg/dL (SD ± 293) and an average triglyceride level of 340 mg/dL (SD ± 164) at the time of HCA regression. Because some of the subjects in this study with HCAs are members of the study population that comprised the case–control study by Wang et al. (2011), it is not a surprise that the triglycerides in the HCA group prior to regression were similar. However, it is interesting that the triglyceride levels after regression of HCAs were similar to the control group without HCAs. Another observation that was made is that some patients had regression of HCAs even though triglyceride levels were not in the normal range. The goal with dietary therapy is to get triglyceride levels within normal range; however, reduction of triglyceride levels, even if suboptimal, may be sufficient for regression of HCAs. Within our group of patients, complete resolution occurred in patients with smaller lesions (<3.3 cm) whereas the larger lesions showed decrease in size. This suggests that intensive dietary therapy and optimization of biochemical control is more likely to result in complete resolution of HCAs before the HCAs become large. Genetic and chromosomal alterations may also play a role in HCA formation and progression, such as chromosomal aberration on chromosome 6 with simultaneous gain of 6p and loss of 6q. This may also explain why some HCAs increase in size in some patients with GSD I despite improving metabolic control (Kishnani et al. 2009).
There are a limited amount of cases in the literature documenting disappearance or reduction in the size of HCAs. Parker et al. 1981 demonstrated two cases of disappearance of HCAs and a third case of reduction in the size of a HCA in GSD type Ia patients with strict dietary therapy. In contrast to this prior report which depended upon Tc-99 m sulfur colloid liver-spleen scans, all of our patients were followed with serial MRI and ultrasound imaging over several years. In the cohort followed by the European Study on Glycogen Storage Disease Type I, three cases demonstrated remission of the HCAs, but no further details about these cases were reported (Rake et al. 2002a, b).
These findings suggest that if HCAs are documented in a patient with suboptimal metabolic control, intensive medical therapy should be considered before recommending surgical excision or liver transplantation. These observations add to the body of evidence showing that optimal metabolic control may delay, prevent, or reverse complications in patients with GSD I.
Acknowledgement
This research was supported by philanthropic support provided by the following funds managed through the University of Florida Office of Development: Scott Miller GSD Program Fund and the GSD Dream Fund. This work was also supported in part by the NIH/NCATS Clinical and Translational Science Award UL1 TR000064 granted to the University of Florida.
Abbreviations
- CT
Computed tomography
- GSD
Glycogen storage disease
- HCA
Hepatocellular adenoma
- MRI
Magnetic resonance imaging
Take Home Message
Hepatocellular adenomas can regress after metabolic control is achieved suggesting intensive medical therapy may be an alternative to surgical intervention in some individuals with GSD I.
Compliance with Ethics Guidelines
Conflict of Interest
Richard D. Beegle, Laurie M. Brown, and David A. Weinstein declare that they have no conflicts of interest.
Financial Disclosure
Richard D. Beegle, Laurie M. Brown, and David A. Weinstein have no financial relationships relevant to this article to disclose. The authors confirm independence from the sponsors. The authors confirm the content has not been influenced by the sponsors. The content does not financially affect the sponsors.
Other Relationships
There are no other relationships that present a potential conflict of interest.
Ethics Approval
Institutional review board approval and informed written consent was obtained from patients participating in an observational study at the University of Florida Glycogen Storage Disease Program.
IRB Approval Number: 533–2005
IRB Approval Name: Correlation of Markers of Metabolic Control with Long-Term Complications in Glycogen Storage Disease
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study. There is no identifying information about patients in this article.
Animal Rights
This article does not contain any studies with animal subjects performed by any of the authors.
Details of the Contributions of Individual Authors
Richard Beegle: Involved in design of the project and analysis and interpretation of the data. Involved in interpretation of all radiological images. Drafted the revised manuscript and approved the final manuscript as submitted.
Laurie Brown: Involved in conception and design as well as data interpretation. Involved in data collection and review of medical charts. Coordinated all regulatory aspects of studies. Assisted in drafting the revised manuscript and approved the final manuscript as submitted.
David Weinstein: Involved in conception and design of studies, supervised the clinical and laboratory investigations, and involved in data collection, data interpretation, and analysis. Editor of manuscript and approved the final manuscript as submitted. Guarantor of the study and controlled decision to publish.
Footnotes
Competing interests: None declared
Contributor Information
David A. Weinstein, Email: weinsda@peds.ufl.edu
Collaborators: Johannes Zschocke, Matthias Baumgartner, K Michael Gibson, Marc Patterson, and Shamima Rahman
References
- Ault GT, Wren SM, Ralls PW, Reynolds TB, Stain SC. Selective management of hepatic adenomas. Am Surg. 1996;62:825–829. [PubMed] [Google Scholar]
- Bianchi L. Glycogen storage disease I and hepatocellular tumours. Eur J Pediatr. 1993;152(Suppl 1):534–536. doi: 10.1007/BF02072092. [DOI] [PubMed] [Google Scholar]
- Bröker ME, Ijzermans JN, van Aalten SM, de Man RA, Terkivatan T (2012) The management of pregnancy in women with hepatocellular adenoma: A plea for an individualized approach. Int J Hepatol. doi:10.1155/2012/725735 [DOI] [PMC free article] [PubMed]
- Cobey FC, Salem RR. A review of liver masses in pregnancy and a proposed algorithm for their diagnosis and management. Am J Surg. 2004;187:181–191. doi: 10.1016/j.amjsurg.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Coire CI, Qizilbash AH, Castelli MF. Hepatic adenomata in type Ia glycogen storage disease. Arch Pathol Lab Med. 1987;111:166–169. [PubMed] [Google Scholar]
- Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure. Pediatr Transplant. 2008;12:137–145. doi: 10.1111/j.1399-3046.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- Deodhar A, Brody LA, Covey AM, Brown KT, Getrajdman GI. Bland embolization in the treatment of hepatic adenomas: preliminary experience. J Vasc Interv Radiol. 2011;22:795–799. doi: 10.1016/j.jvir.2011.02.027. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Chuang TP, Bali D, et al. Chromosomal and genetic alteration in human hepatocellular adenomas associated with type Ia glycogen storage disease. Hum Mol Genet. 2009;18:4781–4790. doi: 10.1093/hmg/ddp441. [DOI] [PubMed] [Google Scholar]
- Kudo M. Hepatocellular adenoma in type Ia glycogen storage disease. J Gastroenterol. 2001;35:65–66. doi: 10.1007/s005350170157. [DOI] [PubMed] [Google Scholar]
- Labrune P (2002) Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatr 161(Suppl):S53–S55 [DOI] [PubMed]
- Lee PJ. Glycogen Storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr. 2002;161(Suppl 1):S45–S49. doi: 10.1007/s00431-002-1002-0. [DOI] [PubMed] [Google Scholar]
- Limmer J, Fleig WE, Leupold D, Bittner R, Ditschuneit H, Beger HG. Hepatocellular carcinoma in type I glycogen storage disease. Hepatology. 1988;8:531–537. doi: 10.1002/hep.1840080317. [DOI] [PubMed] [Google Scholar]
- Maheshwari A, Rankin R, Segev DL, Thuluvath PJ. Outcomes of liver transplantation for glycogen storage disease: a matched-control study and a review of literature. Clin transplant. 2012;26:432–436. doi: 10.1111/j.1399-0012.2011.01549.x. [DOI] [PubMed] [Google Scholar]
- Matern D, Starzl TE, Arnaout W, et al. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr. 1999;158(Suppl 2):S43–S48. doi: 10.1007/PL00014320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel JD, Kukreja K, Ristagno RL, Yazigi N, Nathan JD, Tiao G. Radiofrequency ablation of a large hepatic adenoma in a child. J Pediatr Surg. 2013;48:E19–E22. doi: 10.1016/j.jpedsurg.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Parker P, Burr I, Slonim A, Ghishan FK, Greene H. Regression of hepatic adenomas in type Ia glycogen storage disease with dietary therapy. Gastroenterology. 1981;81:534–536. [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type 1: Diagnosis, management, clinical course and outcome. Results of the European study on glycogen storage disease type I (ESGSD I) Eur J Pediatr. 2002;161(Suppl):S20–S34. doi: 10.1007/BF02679990. [DOI] [PubMed] [Google Scholar]
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Guidelines for management of glycogen storage disease type I – European study on glycogen storage disease type I (ESGSD I) Eur J Pediatr. 2002;161(Suppl):S112–S119. doi: 10.1007/BF02680007. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Kishnani PS, Sullivan JA, et al. Resection of hepatocellular adenoma in patients with glycogen storage disease type Ia. J Hepatol. 2007;47:658–663. doi: 10.1016/j.jhep.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Reddy SK, Austin SL, Spencer-Manzon M, et al. Liver transplantation for glycogen storage disease type Ia. J Hepatol. 2009;51:483–490. doi: 10.1016/j.jhep.2009.05.026. [DOI] [PubMed] [Google Scholar]
- Rhim H, Lim HK, Kim YS, Choi D. Percutaneous radiofrequency ablation of hepatocellular adenoma: initial experience in 10 patients. J Gastroenterol Hepatol. 2008;23:e422–e427. doi: 10.1111/j.1440-1746.2007.05177.x. [DOI] [PubMed] [Google Scholar]
- Wang DQ, Fiske LM, Carreras CT, Weinstein DA. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr. 2011;159:442–446. doi: 10.1016/j.jpeds.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type IA glycogen storage disease. Eur J Pediatr. 2002;161(Suppl):S35–S39. doi: 10.1007/BF02679991. [DOI] [PubMed] [Google Scholar]