

Abstract
Glutaric acidemia type I (GA-I) is a treatable autosomal recessive disorder of lysine, hydroxylysine, and tryptophan metabolism caused by glutaryl-CoA dehydrogenase (GCDH) deficiency. Presentation and progression of disease are variable ranging from asymptomatic carrier state to catastrophic encephalopathy. GA-I usually presents before age 18 months, usually triggered by childhood infection, with mild or severe acute encephalopathy, striatal degeneration, and movement disorder, most often acute dystonia. At a presymptomatic stage diagnosis is suggested clinically by macrocephaly, radiologically by widened Sylvian fissures and biochemically by the presence of excess 3-hydroxyglutaric acid and glutaric acid in urine. Treatment consists of lysine-restricted diet and carnitine supplementation, specific diet restrictions, as well as symptomatic and anticatabolic treatment of intercurrent illness. Presymptomatic diagnosis and treatment are essential to prognosis. We report the case of 16-year-old macrocephalic female with late-onset GA-I and unusual paucisymptomatic presentation with fainting after exercise and widespread white matter signal changes at MRI. She was compound heterozygote for a novel mutation (IVS10-2A>G) affecting splicing at GCDH and a common missense mutation (c. 1240C>T; p.Arg402Trp, R402W). Interestingly, the site of the novel mutation is the nucleotide position of a common mutation found almost exclusively in patients of Chinese/Taiwanese origin (IVS10-2A>C).
Introduction
Glutaric aciduria type I (GA-I, OMIM 231670) is a rare but treatable autosomal recessive inborn error of lysine, hydroxylysine, and tryptophan metabolism caused by mutations in the glutaryl-CoA dehydrogenase gene (GCDH) resulting in GCDH deficiency (Goodman et al. 1975; Koeller et al. 1995, 2004; Strauss et al. 2003). GCDH is a homotetramer mitochondrial matrix enzyme that catalyzes the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA and carbon dioxide (Strauss et al. 2003). Clinical presentation with acute encephalopathic crises in infancy or early childhood (typically between 6 and 18 months) often triggered by febrile illness resulting in striatal degeneration and severe dystonic–dyskinetic movement disorder is common, but insidious-onset, late-onset, or slowly progressive cases have also been described (Strauss et al. 2003; Kölker et al. 2011; Kyllerman et al. 1994). In presymptomatic patients, macrocephaly at birth or developing in the first months of life, wide Sylvian fissures and expansion of anterior frontotemporal CSF spaces are characteristic findings (Strauss et al. 2003; Kölker et al. 2011; Kyllerman et al. 1994; Gitiaux et al. 2008; Oguz et al. 2005). Urine organic acid analysis usually shows increased levels of glutaric acid (GA), 3-hydroxyglutaric acid (3OHGA), glutaryl-CoA, glutarylcarnitine, and glutaconic acids, although glutaric acid and dicarboxylic carnitines can be completely normal in some patients. Increased urinary excretion of glutaric acid may also occur in short-gut syndrome, intestinal infections, and the inborn errors of metabolism glutaric aciduria type 2 (GA-II, also known as multiple acyl-CoA dehydrogenase deficiency, MADD), glutaric aciduria type 3 (GA-III), α-aminoadipic acidemia, and protein lipoylation defects, but these can be differentiated biochemically by a distinct metabolite profile in urinary organic acid analysis and the absence of 3OHGA excretion (Merinero et al. 1995; Pineda et al. 1998; Nyhan et al. 1999; Knerr et al. 2002; Gordon 2006; Navarro-Sastre et al. 2011). Thus increased urinary concentrations of 3OHGA is the most sensitive and specific biochemical marker of GA-I (Al-Dirbashi et al. 2011). The diagnosis is confirmed by GCDH enzyme assay in fibroblasts or leucocytes and molecular genetic analysis (Strauss et al. 2003; Kölker et al. 2011). Prenatal or newborn screening and presymptomatic evaluation of asymptomatic siblings are essential for early diagnosis and treatment and prevention of striatal necrosis and permanent neurological sequelae (Strauss et al. 2003; Kölker et al. 2011). Currently, treatment consists of lysine-restricted diet and carnitine supplementation and prompt symptomatic and anticatabolic treatment of intercurrent illness (Kölker et al. 2006a, 2012).
Since the initial description of GA-I by Goodman et al., more than 450 cases have been documented in the literature (Goodman et al. 1975). Prevalence has been variably estimated between 1:100,000 and 1:30,000 in different studies and populations (Strauss et al. 2003; Kölker et al. 2006a, 2011). Incidence is highest in certain genetically homogeneous communities such as the Old Order Amish of Lancaster County, Pennsylvania (1 in 300–400 newborns), and the aboriginal Ojibway–Cree Indians of Northern Canada (1 in 300 newborns) where common mutations in homozygous state are often found (Morton et al. 1991; Haworth et al. 1991). The GCDH gene is located on chromosome 19p13.2, is 7 kb large, contains 11 exons and 10 introns, and codes for a polypeptide product of 438 amino acids, from which the functional GCDH is derived after cleavage of the 44-amino acids N-terminal targeting sequence following import into the mitochondrial matrix(Koeller et al. 1995; Schwartz et al. 1998). More than 150 pathogenic mutations have been identified of which the most frequent mutation in Caucasians is R402W (c.1204C>T) found in up to 10–20% of alleles, while the IVS10-2A>C mutation is found almost exclusively in patients of Chinese/Taiwanese origin (Strauss et al. 2003; Kölker et al. 2006a, 2011; Busquets et al. 2000a; Tang et al. 2000; Shu et al. 2003).
We hereby report the case of a paucisymptomatic 16-year-old Greek female originating from the Aegean island of Lesvos opposite the Minor Asian coast who presented with a fainting episode. Macrocephaly, characteristic neuroimaging findings and urine organic acid analysis suggested the diagnosis of GA-I. Molecular genetic testing confirmed that she was compound heterozygote for the common R402W (c.1204C>T) mutation and the novel mutation IVS10-2A>G (c.1244A>G) at the same nucleotide position as the IVS10-2A>C mutation previously reported exclusively in Far Eastern populations (Busquets et al. 2000a; Tang et al. 2000; Shu et al. 2003).
Case Report
This 16-year-old female patient was admitted to a regional hospital after fainting during physical exercise. She was conceived by IVF with the nonconsanguineous parents as sperm and egg donors and born by cesarean section after 38 weeks of gestation. Psychomotor development was delayed with gait and speech acquired at approximately 2 years of age. She was macrocephalic with occipitofrontal circumference greater than the 98th percentile for chronologic age. She reportedly had mild learning difficulties in writing, spelling, and arithmetic during elementary and secondary school; however she proceeded normally through the state school system, with good grades, despite being considered somewhat “slow” by her mother in relation to her younger sibling. At age 16, she was hospitalized because of nonconvulsive syncope after physical strain. Routine biochemistry, including thyroid status, was normal except for an elevated CRP. Evaluation by Holter electrocardiogram and transthoracic echocardiography was normal. Magnetic resonance imaging (MRI) of the brain revealed periventricular white matter hyperintensities, enlargement of frontotemporopolar CSF spaces, and characteristic widening of Sylvian fissure bilaterally (“bat-wing sign”), while MR angiography was normal. Physical examination revealed macrocephaly, brachydactyly, and mild foot clonus bilaterally. The rest of neurologic and gross neuropsychologic examination were unremarkable. Fundoscopy, EEG, EMNG, and SSEPs were all normal. Diagnosis of glutaric aciduria type I (GA-I) was confirmed by biochemical and molecular genetic studies. Treatment with low protein diet and 3 g/day of carnitine supplementation was initiated and the patient has remained stable without progression of MRI findings at one year follow-up (Kölker et al. 2006a, 2012).
Materials and Methods
Diagnosis of GA-I was supported by blood electrospray-ionization tandem mass spectrometry (ESI-MS/MS) for acylcarnitines and urine gas chromatography–mass spectrometry (GC/MS) for organic acids. Genomic DNA was obtained from fresh blood. PCR conditions and primers were used as previously described for the assessment of mutations in the GCDH gene (Busquets et al. 2000b; Biery et al. 1996). The mutations were detected by Sanger sequencing of all exons of the GCDH gene. The numbering of cDNA nucleotides (c.) follows the recommendations of the Nomenclature Working Group and starts with 1 at the first nucleotide of the initiator codon; thus c. numbering values are 36 nucleotides smaller than those used in older publications on GA1 mutations (Antonarakis 1998).
Results
Plasma acylcarnitine and urine organic acid analysis revealed a typical metabolite profile with total carnitine deficiency in plasma, elevated levels of plasma and urinary C5 dicarboxylic carnitine (C5DC, glutarylcarnitine), increased urinary excretion of 3OH glutaric acid (3OHGA), and markedly increased urinary excretion of glutaric acid (GA) (Table 1). Glutaconic acid values were not measured.
Table 1.
Blood and urine biochemistry results
| Metabolite | Result (μmol/L) | Normal (μmol/L) |
|---|---|---|
| Free carnitine (C0) | 5.90 | 5.00–75.00 |
| Glutaryl carnitine (C5DC) | 1.47 | 0.00–0.10 |
| Total carnitine | 12.19 | 25.0–75.0 |
| Glutaric acid (GA) | 968.1 | 0.0–3.8 |
| 3-Hydroxyglutaric (3OHGA) | 36.8 | 0.0–4.6 |
Magnetic resonance imaging (MRI) of the brain revealed several findings associated with glutaric aciduria I, such as (1) enlargement of temporopolar CSF spaces (Fig. 1a,i,j), (2) widening of Sylvian fissures (“bat-wing sign”) (Fig. 1b,d), (3) widened mesencephalic cistern (Fig. 1c,j,l), (4) periventricular and deep white matter hyperintensities (Fig. 1d–h), and (5) abnormal hyperintensity on T2WI of splenium of corpus callosum (Fig. 1f), dentate nucleus (Fig. 1j), and substantia nigra (Fig. 1k).
Fig. 1.
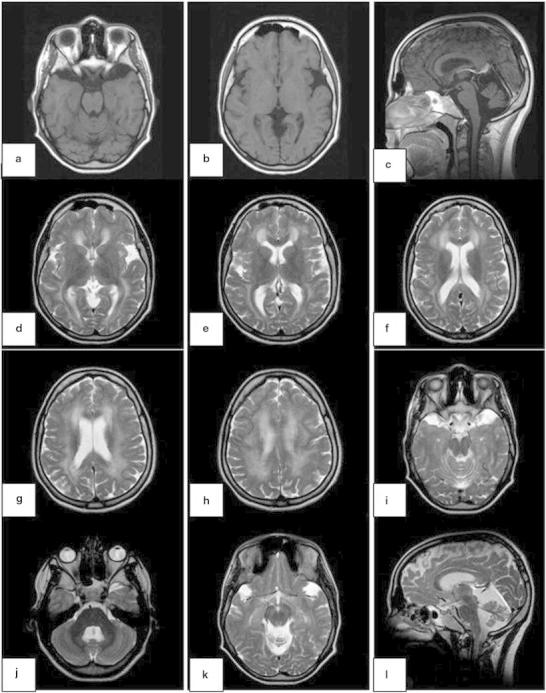
Magnetic resonance imaging of the brain revealed enlargement of temporopolar CSF spaces (a, i, j), widening of Sylvian fissures (“bat-wing sign”) (b, d), widened mesencephalic cistern (c, j, l), periventricular and deep white matter hyperintensities (d–h), and abnormal hyperintensity on T2WI of the splenium of corpus callosum (f), dentate nucleus (j), and substantia nigra (k)
Molecular genetic studies with direct sequencing of all exons of the GCDH gene detected the common c.1204C>T missense mutation in exon 10 (substitution of Arg by Trp at codon 402, R402W) and the novel c.1244A>G transversion (IVS10-2A>G), causing an alteration in splicing.
Discussion
Glutaric aciduria type I is an autosomal recessive neurometabolic disease that is classified among the cerebral organic acid disorders, a subgroup of organic acid disorders, where the central nervous system (CNS) is predominantly and severely affected (Strauss et al. 2003; Kölker et al. 2011, 2013). It is an inborn error of lysine, hydroxylysine, and tryptophan metabolism (Strauss et al. 2003; Kölker et al. 2011). Its pathophysiological basis is glutaryl-CoA dehydrogenase deficiency that results in accumulation of organic acids, such as GA and 3OHGA, that share structural and functional similarities with glutamate, in the CNS where they are believed to exert neurotoxic, gliotoxic, and myelinotoxic effects by several pathomechanisms, involving excitotoxic neuronal and oligodendroglial injury, dysregulation of mitochondrial energy production, and oxidative stress (Strauss et al. 2003; Kölker et al. 2008, 2011). Indeed, in GA-I brain GA has been found to exceed plasma and CSF levels by up to two orders of magnitude even in patients with normal or low GA levels in urine, a counterintuitive finding best explained by the “trapping hypothesis” enunciated by Kölker et al. (Strauss et al. 2003; Kölker et al. 2006b, 2011; Merinero et al. 1995; Pineda et al. 1998; Nyhan et al. 1999; Sauer et al. 2006, 2011).
More than 150 pathogenic mutations (missense, nonsense, and intronic variants) in the GCDH gene have been so far documented (Strauss et al. 2003; Kölker et al. 2006a, 2011, 2012). The disease exhibits a remarkable clinical variability, but apart from a covert correlation between residual enzymatic activity as determined by the so-called “severe” or “mild” mutations and biochemical phenotype, no other genotypic–phenotypic relationship has been established (Busquets et al. 2000b; Mühlhausen et al. 2003; Funk et al. 2005; Gallagher et al. 2005; Christensen et al. 2004). For example, the missense R402W (p.Arg402Trp) mutation in exon 10, which is the commonest worldwide, accounting for up to 20% of all mutations in different studied populations, when expressed in a prokaryotic system leads to only 3% residual GCDH activity, while the A421V (p.Ala421Val) missense mutation in exon 11, which is prevalent in the Old Order Amish of Pennsylvania, results in 40% residual enzyme activity in the same model (Busquets et al. 2000a, b; Biery et al. 1996). Homozygosity or compound heterozygosity of “severe” mutations is reflected in the typically abnormal urinary organic acid profile, while heterozygosity of at least one “mild” mutation may result in normal or mildly affected metabolic profile (Mühlhausen et al. 2003; Funk et al. 2005; Gallagher et al. 2005; Christensen et al. 1997, 2004). Although according to one meta-analysis (n = 115) GA in urine was increased in 97% of cases, it has been postulated that patients with GA-I can be categorized biochemically in high and low excretors of GA and 3OHGA and that “severe” mutations, such as R402W (p.Arg402Trp) or A293T (p.Ala293Thr), are most frequent in high excretors, while “mild” mutations such as V400M (p.Val400Met) or R227P (p.Arg227Pro) are only found in low excretors, because compound heterozygosity with at least one “mild” mutation has a rescue effect on both residual GCDH activity and metabolite excretory profile (Busquets et al. 2000b; Biery et al. 1996; Mühlhausen et al. 2003; Funk et al. 2005; Gallagher et al. 2005; Christensen et al. 1997, 2004; Bjugstad et al. 2000). However, despite the characterization of at least two genetically and biochemically distinct patient groups, no correlation has been found between residual GCDH activity or urinary organic acid profile and clinical phenotype and thus between severity of genetic lesion and clinical picture (Busquets et al. 2000b; Bjugstad et al. 2000). This is well exemplified by the often described discordant phenotypes of homozygote siblings or even twins (Zafeiriou et al. 2000).
There is another apparent phenotypic dichotomy that relates to the clinical presentation of GA-I. Typically, GA-I presents in infancy after a precipitating illness with acute metabolic encephalopathy that in a matter of days results in striatal necrosis with stroke-like characteristics and a severe irreversible neurological syndrome variably encompassing axial hypotonia, generalized dystonia and other dyskinetic/hyperkinetic movement disorders, spasticity, developmental regression, seizures, and ultimately dystonic tetraparesis (Strauss et al. 2003; Kölker et al. 2006a, 2011; Kyllerman et al. 1994; Bjugstad et al. 2000). According to a meta-analysis (n = 115), GA-I onset before 24 months of age occurs in 87% of cases, with a critical susceptibility time window between 6 and 9 months, when about 25% of infantile acute encephalopathic cases debut and the probability of a poor outcome is highest (Bjugstad et al. 2000). In this study, 8.7% of patients were asymptomatic/paucisymptomatic without disease progression apart maybe from macrocephaly present at birth or developing in the first months of life and neuroimaging findings that remained undetected (Bjugstad et al. 2000). Most of these cases are in fact presymptomatic rather than asymptomatic and encompass at-risk relatives detected by screening or subjects that have not yet had an encephalopathic event but received a diagnosis of GA-I on neuroradiological grounds. In any case, an acute encephalopathic crisis with striatal necrosis is the major prognostic factor of morbidity and mortality in GA-I (Kölker et al. 2011; Bjugstad et al. 2000). However, in a minority of cases, the onset is insidious with gradual disease progression and the absence of an acute encephalopathic episode or major pathological changes of basal ganglia in imaging, but, as a rule, presence of other hallmark GA-I clinical and radiological signs, such as macrocephaly and widening of Sylvian fissures (Strauss et al. 2003; Kölker et al. 2011).
Late-juvenile or adult-onset GA-I is extremely rare. There have been scant reports in the medical literature, and in fact, some cases reported as “adult-onset GA-I” were simply childhood-onset GA-I diagnosed in adulthood (Prevett et al. 1996; Corral I et al. 2001; Bähr et al. 2002; Twomey et al. 2003; Fernandez-Alvarez et al. 2003; Külkens et al. 2005; Sonmez et al. 2007; Harting et al. 2009; Chen J et al. 2011). According to the few reports of truly adult-onset GA-I (onset at the age of 18 years or older), (1) the patients were paucisymptomatic or asymptomatic at the time of diagnosis, (2) the disease followed either an acute encephalopathic or a non-encephalopathic clinical course, and (3) in all cases supratentorial, diffuse, symmetric U-fiber-sparing leukoencephalopathy involving periventricular and deep white matter was seen in cranial MRI. Bähr et al. reported the case of a 19-year-old woman without macrocephaly, who presented with recurrent headaches, mild ophthalmoparesis, and hyperreflexia (Bähr et al. 2002). Fernandez-Alvarez et al. described the case of an adolescent girl without macrocephaly and a history of postural hand tremor that by the age of 19 had developed focal dystonia and orofacial dyskinesia (Fernandez-Alvarez et al. 2003). Külkens et al. reported the interesting case of a 15-year-old boy with macrocephaly, psychomotor retardation, progressive vertigo, and intermittent severe diffuse headaches often induced by physical exercise and relieved by rest (Külkens et al. 2005). Sonmez et al. described the case of a 20-year-old man with a 6-month history of recurrent headaches and hyperactive muscle stretch reflexes (Sonmez et al. 2007). Finally, a Chinese patient has been described with a purported ischemic cerebral stroke in young adulthood, whereby the correct diagnosis of GA-I was later made (Chen et al. 2011). More remarkable was a case reported by Külkens et al. of a 66-year-old man macrocephalic since infancy with a history of severe intermittent headaches since the age of 35 and a complex progressive neuropsychiatric syndrome since the age of 50 encompassing hand tremor, seizures, dementia, and aggressive behavior with acoustic and visual hallucinations (Külkens et al. 2005). In most of these atypical late-onset cases, diagnosis was incidental and suggested by MRI findings and metabolic screening. By contrast, Prevett et al. reported the case of a young lady with mild psychomotor retardation, gait disturbance, and orofacial dyskinesia presenting at 7 years of age, while Corral et al. described two siblings with infantile onset of acute encephalopathy and dystonia at age 9 and 16 months, respectively, that were eventually diagnosed with GA-I in adulthood, at ages 29 and 24, respectively, after an expanded diagnostic procedure with MRI and urine organic acid analysis, but did not respond to treatment because of advanced disease (Prevett et al. 1996; Corral et al. 2001).
White matter involvement is common in GA-I with an incidence of 56% (Strauss et al. 2003; Kölker et al. 2011; Bjugstad et al. 2000). MRI findings in GA-I associated white matter disease or leukoencephalopathy are diffuse periventricular and deep white matter signal changes appearing as hypointensities in T1W and hyperintensities in T2W and FLAIR images, with low apparent diffusion coefficient (ADC) values and restricted diffusion in DW images. Less frequently high signal on T2W can involve the corpus callosum or medial lemniscus (Kölker et al. 2011; Oguz et al. 2005). Magnetic resonance spectroscopy (MRS) of affected CNS white or gray matter areas may reveal decreased N-acetylaspartate/creatine, slightly increased or normal choline/creatine, and increased myoinositol/creatine ratios, corresponding to neuroaxonal damage, delayed myelination or demyelination, and astrocytosis, respectively (Oguz et al. 2005). Neuropathology demonstrated extensive spongiform leukoencephalopathy at many sites in white matter at the level of cerebral cortex, centrum semiovale, internal capsule, central tegmental tracts or brachium of inferior colliculus without significant reactive astrocytosis, and marked spongiform myelinopathy in myelinated fascicles of putamen and other nuclei such as thalamus, substantia innominata, or inferior colliculus, while arcuate fibers and cortical grey matter were spared (Funk et al. 2005). White matter involvement in GA-I may be found in both early-onset acute encephalopathic and late-onset or progressive degenerative clinical subtypes and may precede striatal damage or be an incidental neuroradiological finding during screening of presymptomatic relatives (Strauss et al. 2003; Kölker et al. 2011; Funk et al. 2005). Leukoencephalopathy is not an age-specific or clinical subtype-specific manifestation in GA-I, as most children with the disease have white matter abnormalities, but it seems to be correlated with disease duration (Bähr et al. 2002). This conclusion is supported by studies of Gcdh-deficient mice, where the biochemical phenotype was similar to GA-I patients but there was no striatal pathology and no development of dystonia (Koeller et al. 2004). Yet, in this mouse model spongiform myelinopathy, found in white matter or cortex, increased with age and was similar to the spongiform leukoencephalopathy of GA-I patients (Koeller et al. 2004).
Leukoencephalopathy, albeit of different etiopathogenesis, extent and clinical characteristics, is an important feature of most cerebral organic acid disorders, including glutaric aciduria type I, aspartoacylase deficiency (or Canavan disease), L-2-hydroxyglutaric aciduria, and D-2-hydroxyglutaric aciduria (Kölker et al. 2006b, 2013; Sauer et al. 2006, 2011). Notably, macrocephaly is also a frequent and prominent sign of the first three (Strauss et al. 2003; Kölker et al. 2011). It has been hypothesized that accumulation of organic acids with neurotoxic and myelinotoxic potential might hamper CNS myelination, causing hypomyelination, dysmyelination, delayed myelination, or demyelination, during a critical period of development of the nervous system (Kölker et al. 2006b, 2008, 2011, 2013; Sauer et al. 2006, 2011). This is believed to be mediated by several direct or indirect mechanisms affecting immature and mature oligodendrocytes or neurons, such as excitotoxicity via AMPA/kainate and NMDA receptors, impairment of mitochondrial function and oxidative phosphorylation, generation of reactive oxygen species, deregulation of glutamergic and GABAergic neurotransmission, and cytokine-induced amplification of cellular damage (Strauss et al. 2003; Kölker et al. 2006b, 2008, 2011, 2013; Sauer et al. 2006, 2011; Funk et al. 2005).
The late-onset case of GA-I presented here is remarkable for the following reasons: Few well-documented late-onset GA-I case reports have been published heretofore (Bähr et al. 2002; Twomey et al. 2003; Fernandez-Alvarez et al. 2003; Külkens et al. 2005; Sonmez et al. 2007; Harting et al. 2009; Chen et al. 2011). Our patient was asymptomatic at 16 years of age, apart from macrocephaly and mild psychomotor delay since infancy, and the diagnosis was made incidentally after a fainting episode during physical exercise without encephalopathy. Despite the paucisymptomatic status of this patient, MRI showed white matter disease. Molecular genetic analysis of the GCDH revealed compound heterozygosity for the common R402W (p.Arg402Trp) mutation and a novel mutation, IVS10-2A>G, at a nucleotide position previously known to be mutated (IVS10-2A>C) exclusively in patients of Chinese/Taiwanese origin (Busquets et al. 2000a; Tang et al. 2000; Shu et al. 2003). Based on the results of urine organic acid analysis, the patient being a high excretor of GA and 3OHGA, we could only deduce a genotype–biochemical phenotype correlation, and we hypothesize that the novel pathogenic mutation in the GCDH gene described in this case report (c.1244A>G transversion; IVS10-2A>G) does not alleviate the effect of the common R402W mutation on residual GCDH activity and metabolite excretory profile. The novel mutation described expands the spectrum of mutations causally linked to the disease and asserts the importance of splicing defects in disease pathogenesis.
Given the fact that GA-I is a treatable inborn metabolic disorder, the clinical case reported herein emphasizes the importance of including GA-I in the differential diagnosis of leukoencephalopathy discovered in adolescence and early adulthood, especially if combined with macrocephaly and characteristic MRI findings. It also serves to highlight that thorough biochemical evaluation with chromatographic analysis of urine organic acids in asymptomatic or paucisymptomatic children or adolescents with macrocephaly will prevent diagnostic delay in GA-I and may prove vital for prognosis (Heringer et al. 2010). Although our case escaped the severe dystonic encephalopathy associated with GA-I, this is a rare occurrence. Thus, early diagnosis of GA-I and prompt implementation of appropriate dietary, therapeutic, and preventive measures can dramatically improve prognosis for this treatable inborn error of metabolism (Heringer et al. 2010).
Compliance with Ethics Guidelines
Conflict of Interest
All the authors of this chapter declare that there are no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the patient included in this study. Proof that informed consent has been obtained is available upon request.
Animal Studies
This article does not contain any studies with animal subjects performed by the any of the authors.
Author Contributorship Statement
Matthew J Fraidakis: conception and design, analysis, and interpretation of data, writing/drafting of the manuscript, final approval of article.
Chrissa Liadinioti: data acquisition.
Argyris Dinopoulos: data acquisition, analysis, and interpretation of data.
Matilda Papathanassiou: data acquisition, analysis, and interpretation of data.
Judit Garcia-Villoria: data acquisition, analysis, and interpretation of data.
Antonia Ribes: data acquisition, analysis, and interpretation of data.
Roser Pons: conception and design, analysis, and interpretation of data, final approval of article.
Leonidas Stefanis: conception and design, analysis, and interpretation of data, final approval of article.
Footnotes
Competing interests: None declared
Contributor Information
M. J. Fraidakis, Email: mjfraidakis@bioacademy.gr
Collaborators: Johannes Zschocke, Matthias Baumgartner, K Michael Gibson, Marc Patterson, and Shamima Rahman
References
- Al-Dirbashi OY, Kölker S, Ng D, et al. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J Inherit Metab Dis. 2011;34:173–180. doi: 10.1007/s10545-010-9223-2. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, the Nomenclature Working Group Recommendations for a nomenclature system for human gene mutations. Hum Mutat. 1998;11:1–3. doi: 10.1002/(SICI)1098-1004(1998)11:1<1::AID-HUMU1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Bähr O, Mader I, Zschocke J, et al. Adult onset glutaric aciduria type I presenting with a leukoencephalopathy. Neurology. 2002;59:1802–1804. doi: 10.1212/01.WNL.0000036616.11962.3C. [DOI] [PubMed] [Google Scholar]
- Biery BJ, Stein DE, Morton DH, et al. Gene structure and mutations of glutaryl-coenzyme A dehydrogenase: impaired association of enzyme subunits that is due to an A421V substitution causes glutaric acidemia type I in the Amish. Am J Hum Genet. 1996;59:1006–1011. [PMC free article] [PubMed] [Google Scholar]
- Bjugstad KB, Goodman SI, Freed CR. Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type 1. J Pediat. 2000;137:681–686. doi: 10.1067/mpd.2000.108954. [DOI] [PubMed] [Google Scholar]
- Busquets C, Coll MJ, Ribes A. Evidence of a single origin for the most frequent mutation (R402W) causing glutaryl-CoA dehydrogenase deficiency: identification of 3 novel polymorphisms and haplotype definition. Hum Mutat. 2000;15:207. doi: 10.1002/(SICI)1098-1004(200002)15:2<207::AID-HUMU15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Busquets C, Merinero B, Christensen E, et al. Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct. Pediatr Res. 2000;48:315–322. doi: 10.1203/00006450-200009000-00009. [DOI] [PubMed] [Google Scholar]
- Chen J, Wang ZX, Zhang JL et al (2011) [Mutation analysis of GCDH gene in eight patients with glutaric aciduria type I]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 28:374–378. [Article in Chinese] [DOI] [PubMed]
- Christensen E, Ribes A, Busquets C, et al. Compound heterozygosity in the glutaryl-CoA dehydrogenase gene with R227P mutation in one allele is associated with no or very low free glutarate excretion. J Inherit Metab Dis. 1997;20:383–386. doi: 10.1023/A:1005390214391. [DOI] [PubMed] [Google Scholar]
- Christensen E, Ribes A, Merinero B, et al. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:861–868. doi: 10.1023/B:BOLI.0000045770.93429.3c. [DOI] [PubMed] [Google Scholar]
- Corral I, Martínez Castrillo JC, Martínez-Pardo M, et al (2001) [Glutaric aciduria type I: diagnosis in adulthood and phenotypic variability]. Neurologia 16:377–380. [Article in Spanish] [PubMed]
- Fernandez-Alvarez E, Garcia-Cazorla A, Sans A, et al. Hand tremor and orofacial dyskinesia: clinical manifestation of glutaric aciduria type I in a young girl. Mov Disord. 2003;18:1076–1079. doi: 10.1002/mds.10442. [DOI] [Google Scholar]
- Funk CB, Prasad AN, Frosk P, et al. Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain. 2005;128:711–722. doi: 10.1093/brain/awh401. [DOI] [PubMed] [Google Scholar]
- Gallagher RC, Cowan TM, Goodman SI, et al. Glutaryl-CoA dehydrogenase deficiency and newborn screening: retrospective analysis of a low excretor provides further evidence that some cases may be missed. Mol Genet Metab. 2005;86:417–420. doi: 10.1016/j.ymgme.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Gitiaux C, Roze E, Kinugawa K, et al. Spectrum of movement disorders associated with glutaric aciduria type 1: a study of 16 patients. Mov Disord. 2008;23:2392–2397. doi: 10.1002/mds.22313. [DOI] [PubMed] [Google Scholar]
- Goodman SI, Markey SP, Moe PG, et al. Glutaric aciduria; a ‘new’ disorder of amino acid metabolism. Biochem Med. 1975;12:12–21. doi: 10.1016/0006-2944(75)90091-5. [DOI] [PubMed] [Google Scholar]
- Gordon N. Glutaric aciduria types I and II. Brain Dev. 2006;28:136–140. doi: 10.1016/j.braindev.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Harting I, Neumaier-Probst E, Seitz A, et al. Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain. 2009;132:1764–1782. doi: 10.1093/brain/awp112. [DOI] [PubMed] [Google Scholar]
- Haworth JC, Dilling LA, Seargeant LE. Increased prevalence of hereditary metabolic diseases among native Indians in Manitoba and northwestern Ontario. Can Med Ass J. 1991;145:123–129. [PMC free article] [PubMed] [Google Scholar]
- Heringer J, Boy SP, Ensenauer R, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol. 2010;68:743–752. doi: 10.1002/ana.22095. [DOI] [PubMed] [Google Scholar]
- Knerr I, Zschocke J, Trautmann U, et al. Glutaric aciduria type III: a distinctive non-disease? J Inherit Metab Dis. 2002;25:483–490. doi: 10.1023/A:1021207419125. [DOI] [PubMed] [Google Scholar]
- Koeller DM, DiGiulio KA, Angeloni SV, et al. Cloning, structure, and chromosome localization of the mouse glutaryl-CoA dehydrogenase gene. Genomics. 1995;28:508–512. doi: 10.1006/geno.1995.1182. [DOI] [PubMed] [Google Scholar]
- Koeller DM, Sauer S, Wajner M, et al. Animal models for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2004;27:813–818. doi: 10.1023/B:BOLI.0000045763.52907.5e. [DOI] [PubMed] [Google Scholar]
- Kölker S, Garbade SF, Greenberg CR, et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res. 2006;59:840–847. doi: 10.1203/01.pdr.0000219387.79887.86. [DOI] [PubMed] [Google Scholar]
- Kölker S, Sauer SW, Surtees RA, et al. The aetiology of neurological complications of organic acidaemias–a role for the blood–brain barrier. J Inherit Metab Dis. 2006;29:701–704. doi: 10.1007/s10545-006-0415-8. [DOI] [PubMed] [Google Scholar]
- Kölker S, Sauer SW, Hoffmann GF, et al. Pathogenesis of CNS involvement in disorders of amino and organic acid metabolism. J Inherit Metab Dis. 2008;31:194–204. doi: 10.1007/s10545-008-0823-z. [DOI] [PubMed] [Google Scholar]
- Kölker S, Christensen E, Leonard JV, et al. Diagnosis and management of glutaric aciduria type I-revised recommendations. J Inherit Metab Dis. 2011;34:677–694. doi: 10.1007/s10545-011-9289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kölker S, Boy SP, Heringer J, et al. Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I – a decade of experience. Mol Genet Metab. 2012;107:72–80. doi: 10.1016/j.ymgme.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Kölker S, Burgard P, Sauer SW, et al. Current concepts in organic acidurias: understanding intra- and extracerebral disease manifestation. J Inherit Metab Dis. 2013;36:635–644. doi: 10.1007/s10545-013-9600-8. [DOI] [PubMed] [Google Scholar]
- Külkens S, Harting I, Sauer S, et al. Late-onset neurologic disease in glutaryl-CoA dehydrogenase deficiency. Neurology. 2005;64:2142–2144. doi: 10.1212/01.WNL.0000167428.12417.B2. [DOI] [PubMed] [Google Scholar]
- Kyllerman M, Skjeldal OH, Lundberg M, et al. Dystonia and dyskinesia in glutaric aciduria type I: clinical heterogeneity and therapeutic considerations. Mov Disord. 1994;9:22–30. doi: 10.1002/mds.870090105. [DOI] [PubMed] [Google Scholar]
- Merinero B, Perez-Cerda C, Font LM, et al. Variable clinical and biochemical presentation of seven Spanish cases with glutaryl-CoA-dehydrogenase deficiency. Neuropediatrics. 1995;26:238–242. doi: 10.1055/s-2007-979763. [DOI] [PubMed] [Google Scholar]
- Morton DH, Bennett MJ, Seargeant LE, et al. Glutaric aciduria type I: a common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am J Med Genet. 1991;41:89–95. doi: 10.1002/ajmg.1320410122. [DOI] [PubMed] [Google Scholar]
- Mühlhausen C, Christensen E, Schwartz M, et al. Severe phenotype despite high residual glutaryl-CoA dehydrogenase activity: a novel mutation in a Turkish patient with glutaric aciduria type I. J Inherit Metab Dis. 2003;26:713–714. doi: 10.1023/B:BOLI.0000005604.90621.e2. [DOI] [PubMed] [Google Scholar]
- Navarro-Sastre A, Tort F, Stehling O, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyhan WL, Zschocke J, Hoffmann GF, et al. Glutaryl-CoA dehydrogenase deficiency presenting as 3-hydroxyglutaric aciduria. Mol Genet Metab. 1999;66:199–204. doi: 10.1006/mgme.1998.2794. [DOI] [PubMed] [Google Scholar]
- Oguz KK, Ozturk A, Cila A. Diffusion-weighted MR imaging and MR spectroscopy in glutaric aciduria type I. Neuroradiology. 2005;47:229–234. doi: 10.1007/s00234-005-1350-3. [DOI] [PubMed] [Google Scholar]
- Pineda M, Ribes A, Busquets C, et al. Glutaric aciduria type I with high residual glutaryl-CoA dehydrogenase activity. Dev Med Child Neurol. 1998;40:840–842. doi: 10.1111/j.1469-8749.1998.tb12362.x. [DOI] [PubMed] [Google Scholar]
- Prevett MC, Howard RS, Dalton RN, et al. Glutaric aciduria type 1 in adulthood. J Neurol Neurosurg Psychiatry. 1996;60:352–353. doi: 10.1136/jnnp.60.3.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer SW, Okun JG, Fricker G, et al. Intracerebral accumulation of glutaric and 3-hydroxyglutaric acids secondary to limited flux across the blood–brain barrier constitute a biochemical risk factor for neurodegeneration in glutaryl-CoA dehydrogenase deficiency. J Neurochem. 2006;97:899–910. doi: 10.1111/j.1471-4159.2006.03813.x. [DOI] [PubMed] [Google Scholar]
- Sauer SW, Opp S, Hoffmann GF, et al. Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain. 2011;134:157–170. doi: 10.1093/brain/awq269. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Christensen E, Superti-Furga A, et al. The human glutaryl-CoA dehydrogenase gene: report of intronic sequences and of 13 novel mutations causing glutaric aciduria type I. Hum Genet. 1998;102:452–458. doi: 10.1007/s004390050720. [DOI] [PubMed] [Google Scholar]
- Shu SG, Tsai CR, Chen LH, et al. Type I glutaric aciduria: phenotypes and genotypes in 5 Taiwanese children. J Formos Med Assoc. 2003;102:729–732. [PubMed] [Google Scholar]
- Sonmez G, Mutlu H, Ozturk E, et al. Magnetic resonance imaging findings of adult-onset glutaric aciduria type I. Acta Radiol. 2007;48:557–559. doi: 10.1080/02841850701280874. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Puffenberger EG, Robinson DL, et al. Type I glutaric aciduria, part 1: natural history of 77 patients. Am J Med Genet. 2003;121:38–52. doi: 10.1002/ajmg.c.20007. [DOI] [PubMed] [Google Scholar]
- Tang NL, Hui J, Law LK, et al. Recurrent and novel mutations of GCDH gene in Chinese glutaric acidemia type I families. Hum Mutat. 2000;16:446. doi: 10.1002/1098-1004(200011)16:5<446::AID-HUMU14>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Twomey EL, Naughten ER, Donoghue VB, et al. Neuroimaging findings in glutaric aciduria type 1. Pediatr Radiol. 2003;33:823–830. doi: 10.1007/s00247-003-0956-z. [DOI] [PubMed] [Google Scholar]
- Zafeiriou DI, Zschocke J, Augoustidou-Savvopoulou P, et al. Atypical and variable clinical presentation of glutaric aciduria type I. Neuropediatrics. 2000;31:303–306. doi: 10.1055/s-2000-12943. [DOI] [PubMed] [Google Scholar]