

Abstract
Objective:
To ascertain the incidence and clinical implications of agenesis of the corpus callosum (ACC) in spinal open neural tube defects (SONTD).
Methods:
All cases of SONTD registered at the Spina Bifida Clinic in King Khalid University Hospital, Riyadh, Saudi Arabia between 1995 and 2010 were retrospectively reviewed, and mid-sagittal MRI of the corpus callosum (CC) area was analyzed in each case. Neurodevelopmental outcome was classified as poor in children with seizures, severe neurodevelopmental impairment, or death.
Results:
Thirty-eight patients (45.8%) with ACC were identified among 83 cases with SONTD. Patients’ age ranged between one and 16 years. Total ACC was found in 10 patients, partial ACC in 25, and in 3 patients, the CC was hypoplastic. Active hydrocephalus was an associated finding in 9 out of 10 patients with total ACC, 22 out of 25 with partial ACC, and in all patients with hypoplasia of the CC. Thirteen patients (34.2%) had normal intellectual function, whereas 24 patients presented with learning disability, epilepsy, or poor intellectual function; and one patient died of respiratory failure.
Conclusion:
Agenesis of the corpus callosum is found in a significant portion of patients with SONTD. When associated with hydrocephalus, its presence affects neuro-developmental outcome.
Agenesis of the corpus callosum (ACC) is a congenital abnormality in which there is partial or complete absence of the corpus callosum (CC). It was first recognized and documented in 1887 by John Langdon Down, a British physician best known for his description of the common genetic disorder that is now called Down syndrome.1 With ultrasound (U/S), and MRI screening, agenesis and hypoplasia of the CC are now being detected more easily. To date, the exact cause of ACC is unknown, and the incidence of ACC associated with spinal open neural tube defect (SONTD) is not determined.1 Agenesis of the CC may be complete or partial. It can result from disruption of callosal development, such as cellular proliferation and migration, axonal growth, or glial patterning at the midline. In partial ACC, the rostrum may be preferentially affected, with preservation of the other parts. Since each part develops at a slightly different time, in general following a rostrocaudal gradient, this selectivity suggests different timing of the insult.2 The clinical manifestations of partial ACC are similar to those of total ACC but often less severe, because some connections are still preserved. Most cases of sporadic, non-syndromic ACC are asymptomatic, and are detected purely incidentally. Patients don’t present with seizures or intellectual disability. When ACC is seen with SONTD, patients are symptomatic. They are either mentally retarded and/or have seizures. The connection between ACC and SONTD should be taken into account for the purpose of counseling. In the current study, we reviewed the incidence of ACC in association with SONTD, and studied its effect on neurodevelopmental outcome.3
Methods
We retrospectively reviewed 83 patients diagnosed with SONTD out of 155 children registered in the Spina Bifida Clinic at King Khalid University Hospital, Riyadh, Saudi Arabia, between 1995 and 2010. An MRI was performed on all subjects using a GE 1.5-Tesla scanner (General Electric, Medical Systems, Milwaukee, WI, USA). The following sequences were acquired: sagittal, T2-weighted, fast spin-echo, 27 4-mm contiguous sections (repetition time, 2500 milliseconds; and effective echo time, 85 milliseconds); axial, T2-weighted, double-echo, fast spin-echo, 28 5-mm contiguous sections (repetition time, 2900 milliseconds; and effective echo time, 19 and 95 milliseconds); and a 3-dimensional, T1-weighted, gradient-echo sequence that allowed reconstruction in any plane of 124 1.5-mm sections (repetition time, 35 milliseconds; echo time, 5 milliseconds; and flip angle, 35°).
The CC was measured from the mid-sagittal section, defined as the most mid-sagittal view of the genu and splenium (Figure 1). Images were measured blind to group membership. The CC was divided into 4 components (rostrum, genu, body, and splenium), following the method of Woodruff et al4 and Nosarti et al.5 This is used as a frame of reference for dividing the CC into quarters. The outline of the CC is traced manually. We defined ACC as either total absence (complete ACC) or absence of at least one, but not all, of the 4 components of the CC, while in hypoplasia, the CC is thinner than usual, but has a normal anterior-posterior extent.6 Two neuroradiologists experienced in the interpretation of brain images obtained with these sequences evaluated the CC. Both neuroradiologists were blinded to the patients’ neurodevelopmental histories. They independently evaluated the CC with each sequence in isolation twice, and at different times to control for intra-observer variation. Imaging studies revealed 38 patients with agenesis or hypoplasia of the CC. Each scan was reviewed with particular attention to the degree of callosal abnormality (agenesis versus hypoplasia, severity of agenesis), and the range of abnormalities associated with the characteristic hydrocephalus and Chiari malformation. Other specific abnormalities were also recorded and analyzed.
Figure 1.
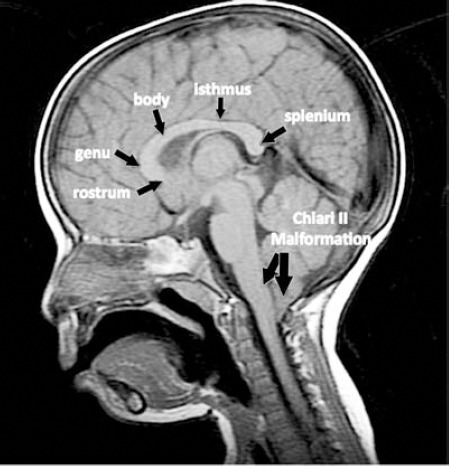
Mid-sagittal, T1-weighted image showing the 4 components of a normal corpus callosum (rostrum, genu, body including the isthmus, and splenium) in a patient with spinal open neural tube defect. Note the presence of tonsillar herniation.
Clinical information on neurologic and developmental status, in particular, pragmatic language, problem-solving skills, and social communication were reviewed. Presence or absence of seizures was recorded. Neurodevelopmental outcome was classified as poor in children with seizures, severe neurodevelopmental impairment, or death. Most visits to the spina bifida clinic included evaluation by a pediatric neurosurgeon, pediatric neurologist, pediatric orthopedic surgeon, pediatric urologist, physical therapist, and a psychologist. Arabic speaking examiners administered the Bayley Scales of Infant Development,7 the Bayley Scales of Infant Development (BSID 2nd ed) and the Preschool Language Scales-III (PLS) for cognitive development. The BSID is a well-standardized assessment measure for children, which yield scores on 2 indexes: the Psychomotor Development Index (PDI), assesses the child’s fine and gross motor skills, and the Mental Development Index (MDI), assesses the child’s level of cognitive, language, personal-social skills. The PLS is a standardized assessment of speech and language, and consists of PLS-EXP (expressive) that evaluates use of expressive language and PLS-REC (receptive) that assesses comprehension of language. The BSID and PLS yield scores with a mean of 100 and a standard deviation of 15.8
Results
Thirty-eight patients (45.8%) with ACC were identified among 83 cases with SONTD. There were 18 males and 20 females, and patients’ age ranged between one and 16 years; median age 4.3 years. All patients were born with open lumbar or lumbosacral myelomeningocele. Total ACC was found in 10 patients, partial ACC in 25, and in 3 patients the CC was hypoplastic (Figures 2, 3, & 4). Ten patients had atypical callosal dysgenesis, with the posterior corpus (isthmus, splenium, or both) being formed in the absence of the anterior portion. Eighteen patients had the body of the CC well formed with absence of the splenium or genu, and in 14 of them both splenium and rostrum were absent (Figure 4). In these 18 patients, the interhemispheric fissure was present in the occipital regions, and the CC appeared partially formed at the site of an almost completely formed interhemispheric fissure. In patients with complete ACC, almost complete hemispheric fusion (lack of normal interhemispheric fissure) was seen in 4 of them (40%). Active hydrocephalus was an associated finding in 34 patients (89.5%), and tonsillar herniation was observed in all patients with ACC associated SONTD (Figure 1).
Figure 2.
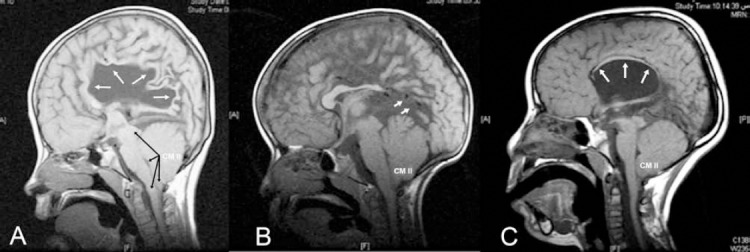
Mid-sagittal, T1-weighted image in patients with spinal open neural tube defect and Chiari II malformation (CM II), showing A) complete agenesis of the corpus callosum (ACC, white arrows) and CM II (black arrows), B) partial ACC where splenium is absent (white arrows), and C) hypoplasia of the CC (white arrows pointing at CM II very thin hypoplastic corpus callosum).
Figure 3.
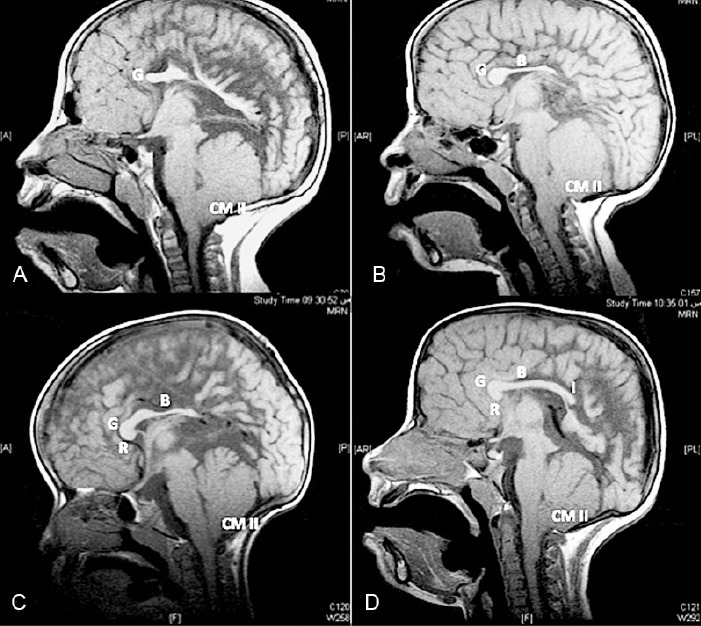
Mid-sagittal, T1-weighted images of patients with spinal open neural tube defect (SONTD) – associated partial agenesis of the corpus callosum (CC) A) Only genu of the CC is formed, B) genu and body are present, C) rostrum, genu, body, and isthmus of the CC, are formed, and D) shows CC missing splenium. Note all patients have Chiari II malformation (CM II). G - genu, B - body, R - rostrum, I - isthmus
Figure 4.
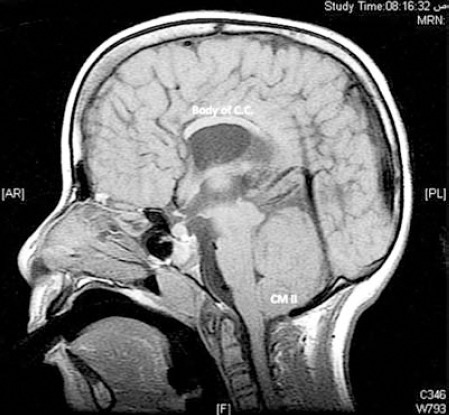
Mid-sagittal, T1-weighted image showing that the body of corpus callosum (CC) is formed with absence of both rostrum and splenium in a patient with partial agenesis of CC, spinal open neural tube defect, and Chiari II malformation (CM II).
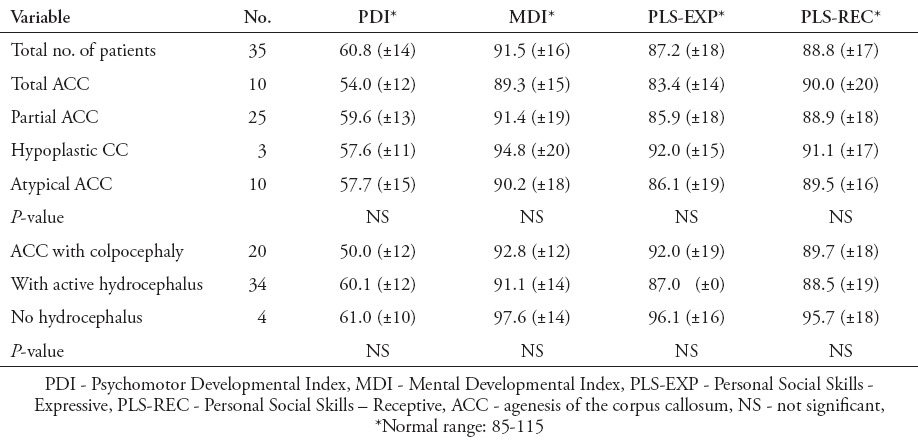
In 13 patients (34.2%), intellectual function was normal, whereas 25 (65.8%) patients were found to have learning disability, seizures, or poor intellectual function; one of them died of respiratory failure. The mean PDI was 60.8 (±14), which is moderately delayed, and the mean MDI was in the normal average range of normal 91.5 (±16). Cognitive scores in 6 (15.8%) of these children were significantly delayed, 19 (50%) were borderline, and 13 (34.2%) were in the average range for their age. Speech and language function were commensurate with mental function, with a mean receptive score of 88.8 (±17) and expressive score of 87.2 (±18), both in the low-normal range (Table 1).
Table 1.
Results of neurocognitive and developmental testing in different types of corpus callosum agenesis in spinal open neural tube defect.
Discussion
Agenesis of the CC is among the most common brain malformations observed in humans.9 In the general population, its estimated prevalence is 3-7 per 1000 birth, while in children with developmental disabilities it is 2-3 per 100.4 It can occur as an isolated finding on MRI, but is often associated with anomalies and disorders of neural migration.6 In our series, SONTD-associated ACC was found in almost half of the sample studied 38/83 (48.5%).10-12
The CC is a densely packed white matter structure, on MRI it appears as high signal on T1 weighted image (T1WI) and low signal in T2WI. Prenatal diagnosis of complete callosal agenesis is feasible from the mid-trimester onwards by expert sonographers.5 Fetal MRI is worthy of recommendation in order to reinforce a difficult sonographic diagnosis and to exclude possible additional cerebral anomalies overlooked at U/S.10,11,13
In complete agenesis, the CC is not visualized, whereas in a partial ACC, the later-forming structures are usually absent. Therefore, the CC may show a posterior genu; a posterior genu and anterior body; a genu and an entire body; or an entire genu, body, and splenium with the exception of the rostrum. In our series, 10 out of 83 patients with SONTD (12%) were found with complete ACC, and 25 patients (30%) with partial ACC, the posterior component was absent in most of them (splenium alone in 17 patients, or splenium and isthmus in 22 patients, representing 68% of partial ACC (Figure 3). This supports the theory that CC normally grows in a ventral to dorsal direction with the genu appearing first followed by posterior growth to form the body and splenium.10,14 On the other hand in 12 patients, the body of CC was formed with absence of both rostrum and splenium, this could be due to the disturbed or random order of CC formation in patients with SONTD (Figure 4). Tovar-Moll et al15 described the CC to be topographically organized, such that fibers connecting a given cortical area are adjacent.
Although the basis of CC abnormalities is not always clear in children with SONTD, who may have a combination of congenital and acquired effects on the cerebral white matter, there are 2 possible processes responsible for CC anomalies in children with SONTD; the first is a disorder involving prolonged disruption of neuroembryogenesis that results in partial agenesis and hypoplasia of the CC, primarily in the rostrum and splenium.16 In some forms of callosal agenesis, termed partial segmental agenesis of the CC, a 2-stage mechanism has been identified that produces an absent body, but present splenium. The second is a destructive process, involving secondary hydrocephalus that tends to produce hypoplasia, but not absence, of the genu and body.17,18 The lateral ventricles may have a colpocephalic appearance with a localized dilatation of the atria and occipital horns. They also appear widely separated and are medially concave, with the upper corners pointed up.15 Marked colpocephaly with enlargement of the occipital horn of the ventricles due to underdevelopment of the white matter in the posterior cerebrum was seen in 51.2% of patients with SONTD.19,20 Our series represent a group of patients referred to the spina bifida clinic from different regions. Because of this, selection and referral bias an estimation of the prevalence of ACC in SONTD is not possible.
Children with SONTD are at increased risk for cognitive impairments including nonverbal skills, arithmetic achievement, and visual-motor integration that become more evident with increasing age.21 Both shunted hydrocephalus and CC anomalies have been shown to affect cognitive functioning.22 The relationship between the degree of cognitive impairment and ACC have been variable suggesting that the presence of hydrocephalus may also have a profound effect on cognitive function.23 In our cohort group of patients, hydrocephalus was present in almost 90% of the patients and we found no significant differences between neurocognitive and developmental test scores among different types of SONTD-associated ACC in children with or without active hydrocephalus (Table 1).24
Our observation of neurocognitive outcome of ACC in SONTD is different from the systematic review published data25 on the neurodevelopment of children that were diagnosed prenatally with isolated ACC, where the rates of normal outcome were 71.2%, borderline or moderate disability 13.6%, and severe disability 15.2%. In this series, in 13 patients (34.2%) intellectual function was normal, whereas 19 (50%) were borderline, and 6 (15.8%) were significantly delayed.25
The cognitive and neurological manifestations of ACC vary considerably from mild behavioral problems to severe neurological deficits.6 The clinical manifestations of partial ACC are similar to those of total ACC, but often less severe because some connections across this commissure are preserved. Difficulties with interhemispheric transfer of stereognosis and naming information is seen more in patients with focal agenesis of the splenium than of rostrum, because the somatosensory reception is in the parietal lobe, and Wernicke’s area for speech is in the posterior temporal lobe, though articulation must be transferred to Broca’s area of the frontal lobe.26
Fletcher et al27 proposed that CC abnormalities lead to deficits in spatial cognition that are commonly observed in children with shunted hydrocephalus. Although 57% of our children had deficiencies of the CC, no significant differences were found between test scores when compared with children with a normal CC, suggesting that development of ventriculomegaly early in brain development may be the important factor here. However, deficits may appear as the children become older and neuropsychologic testing becomes more specific and focused on associative, processing, and interpretation skills.
To date, it is not clear which changes in the brains of children with ACC are related to compensatory adaptation and which are associated with functional deficits. The concept of compensatory mechanisms has been invoked to explain why children with ACC associated with SONTD differ so dramatically from children and adults with acquired CC damage or section. In SONTD, ACC does not appear to have a direct or dramatic impact on general cognitive ability, and does not appear to have a significant negative impact on general language skills.16,28
In conclusion, complete or partial ACC is common in children with SONTD and are often associated with functional problems. One should take account of the mode of ascertainment, to make a connection between ACC and NTD for counseling purposes. There are no known treatments to cure the anomaly; the only treatment available for individuals with ACC is for the management of its symptoms and control of seizures.
Footnotes
Disclosure.
Related Articles.
Mansouri HA. Central nervous system anomalies diagnosed antenatally and post-delivery management. Saudi Med J 2010; 31: 257-261.
Al-Mendalawi MD. Folic acid awareness among female college students. Neural tube defect prevention. Saudi Med J 2009; 30: 723; author reply 723-724.
Kari JA, Bardisi ES, Baitalmal RM, Ageely GA. Folic acid awareness among female college students: neural tube defects prevention. Saudi Med J 2008; 29: 1749-1751.
Chacko A, Koul R, Sankhla DK. Corpus callosum agenesis. Saudi Med J 2001; 22: 22-25.
References
- 1.Alexiou GA, Zarifi MK, Georgoulis G, Mpouza E, Prodromou C, Moutafi A, et al. Cerebral abnormalities in infants with myelomeningocele. Neurol Neurochir Pol. 2011;45:18–23. doi: 10.1016/s0028-3843(14)60055-4. [DOI] [PubMed] [Google Scholar]
- 2.Juranek J, Salman MS. Anomalous development of brain structure and function in spina bifida myelomeningocele. Dev Disabil Res Rev. 2010;16:23–30. doi: 10.1002/ddrr.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glass HC, Shaw GM, Ma C, Sherr EH. Agenesis of the corpus callosum in California 1983-2003: a population-based study. Am J Med Genet A. 2008;146A:2495–2500. doi: 10.1002/ajmg.a.32418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woodruff PW, Pearlson GD, Geer MJ, Barta PE, Chilcoat HD. A computerized magnetic resonance imaging study of corpus callosum morphology in schizophrenia. Psychol Med. 1993;23:45–56. doi: 10.1017/s0033291700038836. [DOI] [PubMed] [Google Scholar]
- 5.Nosarti C, Rushe TM, Woodruff PW, Stewart AL, Rifkin L, Murray RM. Corpus callosum size and very preterm birth: relationship to neuropsychological outcome. Brain. 2004;127:2080–2089. doi: 10.1093/brain/awh230. [DOI] [PubMed] [Google Scholar]
- 6.Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain. 2014;137:1579–1613. doi: 10.1093/brain/awt358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedrich WN, Lovejoy MC, Shaffer J, Shurtleff DB, Beilke RL. Cognitive abilities and achievement status of children with myelomeningocele: a contemporary sample. J Pediatr Psychol. 1991;16:423–428. doi: 10.1093/jpepsy/16.4.423. [DOI] [PubMed] [Google Scholar]
- 8.Tew B, Laurence KM. The effects of hydrocephalus on intelligence, visual perception and school attainment. Dev Med Child Neurol Suppl. 1975;35:129–134. doi: 10.1111/j.1469-8749.1975.tb03592.x. [DOI] [PubMed] [Google Scholar]
- 9.Dobyns WB. Absence makes the search grow longer. Am J Hum Genet. 1996;58:7–16. [PMC free article] [PubMed] [Google Scholar]
- 10.Hetts SW, Sherr EH, Chao S, Gobuty S, Barkovich AJ. Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. AJR Am J Roentgenol. 2006;187:1343–1348. doi: 10.2214/AJR.05.0146. [DOI] [PubMed] [Google Scholar]
- 11.Hannay HJ, Dennis M, Kramer L, Blaser S, Fletcher JM. Partial agenesis of the corpus callosum in spina bifida meningomyelocele and potential compensatory mechanisms. J Clin Exp Neuropsychol. 2009;31:180–194. doi: 10.1080/13803390802209954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawamura T, Nishio S, Morioka T, Fukui K. Callosal anomalies in patients with spinal dysraphism: correlation of clinical and neuroimaging features with hemispheric abnormalities. Neurol Res. 2002;24:463–467. doi: 10.1179/016164102101200348. [DOI] [PubMed] [Google Scholar]
- 13.Ren T, Anderson A, Shen WB, Huang H, Plachez C, Zhang J, et al. Imaging, anatomical, and molecular analysis of callosal formation in the developing human fetal brain. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:191–204. doi: 10.1002/ar.a.20282. [DOI] [PubMed] [Google Scholar]
- 14.Rados M, Judas M, Kostović I. In vitro MRI of brain development. Eur J Radiol. 2006;57:187–198. doi: 10.1016/j.ejrad.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Tovar-Moll F, Moll J, de Oliveira-Souza R, Bramati I, Andreiuolo PA, Lent R. Neuroplasticity in human callosal dysgenesis: a diffusion tensor imaging study. Cereb Cortex. 2007;17:531–541. doi: 10.1093/cercor/bhj178. [DOI] [PubMed] [Google Scholar]
- 16.Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, et al. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- 17.Dennis M, Edelstein K, Frederick J, Copeland K, Francis D, Blaser SE, et al. Peripersonal spatial attention in children with spina bifida: associations between horizontal and vertical line bisection and congenital malformations of the corpus callosum, midbrain, and posterior cortex. Neuropsychologia. 2005;43:2000–2010. doi: 10.1016/j.neuropsychologia.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 18.Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elgamal EA. Natural history of hydrocephalus in children with spinal open neural tube defect. Surg Neurol Int. 2012;3:112. doi: 10.4103/2152-7806.101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller E, Widjaja E, Blaser S, Dennis M, Raybaud C. The old and the new: supratentorial MR findings in Chiari II malformation. Childs Nerv Syst. 2008;24:563–575. doi: 10.1007/s00381-007-0528-x. [DOI] [PubMed] [Google Scholar]
- 21.Chiappedi M, Bejor M. Corpus callosum agenesis and rehabilitative treatment. Ital J Pediatr. 2010;36:64. doi: 10.1186/1824-7288-36-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wills KE, Holmbeck GN, Dillon K, McLone DG. Intelligence and achievement in children with myelomeningocele. J Pediatr Psychol. 1990;15:161–176. doi: 10.1093/jpepsy/15.2.161. [DOI] [PubMed] [Google Scholar]
- 23.Friedrich WN, Lovejoy MC, Shaffer J, Shurtleff DB, Beilke RL. Cognitive abilities and achievement status of children with myelomeningocele: a contemporary sample. J Pediatr Psychol. 1991;16:423–428. doi: 10.1093/jpepsy/16.4.423. [DOI] [PubMed] [Google Scholar]
- 24.Mazur JM, Aylward GP, Colliver J, Stacey J, Menelaus M. Impaired mental capabilities and hand function in myelomeningocele patients. Z Kinderchir. 1988;43(Suppl 2):24–27. doi: 10.1055/s-2008-1044150. [DOI] [PubMed] [Google Scholar]
- 25.Sotiriadis A, Makrydimas G. Neurodevelopment after prenatal diagnosis of isolated agenesis of the corpus callosum: an integrative review. Am J Obstet Gynecol. 2012;206:337.e1–5. doi: 10.1016/j.ajog.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 26.Johnson MP, Gerdes M, Rintoul N, Pasquariello P, Melchionni J, Sutton LN, et al. Maternal-fetal surgery for myelomeningocele: neurodevelopmental outcomes at 2 years of age. Am J Obstet Gynecol. 2006;194:1145–1150. doi: 10.1016/j.ajog.2006.01.072. [DOI] [PubMed] [Google Scholar]
- 27.Fletcher JM, Bohan TP, Brandt ME, Kramer LA, Brookshire BL, Thorstad K, et al. Morphometric evaluation of the hydrocephalic brain: relationships with cognitive development. Childs Nerv Syst. 1996;12:192–199. doi: 10.1007/BF00301250. [DOI] [PubMed] [Google Scholar]
- 28.Tyszka JM, Kennedy DP, Adolphs R, Paul LK. Intact bilateral resting-state networks in the absence of the corpus callosum. J Neurosci. 2011;31:15154–15162. doi: 10.1523/JNEUROSCI.1453-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]