

Abstract
Objective:
To ascertain the incidence, and describe the various forms of neural tube defects (NTDs) due to genetic, chromosomal, and syndromic causes.
Methods:
We carried out a retrospective analysis of data retrieved from the medical records of newborn infants admitted to the Neonatal Intensive Care Unit with NTDs and their mothers spanning 14 years (1996-2009) at the Security Forces Hospital, Riyadh, Saudi Arabia. The cases were ascertained by a perinatologist, neonatologist, geneticist, radiologist, and neurologist. The literature was reviewed via a MEDLINE search. Only liveborn babies were included. Permission from the Educational Committee at the Security Forces Hospital was obtained prior to the collection of data.
Results:
Out of 103 infants with NTDs admitted during this period, 20 (19.4%) were found to have an underlying genetic syndromic, chromosomal and/or other anomalies. There were 5 cases of Meckel-Gruber syndrome, 2 Joubert syndrome, one Waardenburg syndrome, one Walker-Warburg syndrome, 2 chromosomal disorders, 2 caudal regression, one amniotic band disruption sequence, one associated with omphalocele, one with diaphragmatic hernia, and 4 with multiple congenital anomalies.
Conclusions:
There is a high rate of underlying genetic syndromic and/or chromosomal causes of NTDs in the Saudi Arabian population due to the high consanguinity rate. Identification of such association can lead to more accurate provisions of genetic counseling to the family including preimplantation genetic diagnosis or early termination of pregnancies associated with lethal conditions.
Neural tube defects (NTDs) result from multifactorial disturbance in embryonic neurulation during the first 4 weeks of gestation. The majority of NTDs (approximately 70%) occur in isolation, and are said to show multifactorial inheritance.1-2 Between 2-16% of isolated NTDs will have a cytogenetic abnormality.3-5 This figure increases up to 24% if the NTD occurs in association with other congenital abnormalities.4 Up to 53% of spontaneous abortions with NTDs will have a karyotypic abnormality,6 and they can occur in association with other congenital abnormalities and show a normal karyotype (syndromic NTDs). Many are due to single gene mutations and inherited in an autosomal recessive, autosomal dominant; or X-linked recessive manner.7 Some NTDs are sporadic and can result from a teratogenic insult affecting the mother in critical periods. Antenatal diagnosis of NTDs utilizing modern ultrasound scans and karyotype analysis and thorough clinical examination of the child, or fetus are essential parts of the diagnostic process.7 Molecular characterization and identification of gene mutations of the syndromic and genetic NTDs will lead to more accurate provision of genetic counseling to the family including preimplantation genetic diagnosis (PGD), or early termination of pregnancy. The objective of this study is to ascertain the incidence, and describe the various forms of NTDs due to genetic, chromosomal, and syndromic causes.
Methods
We carried out a retrospective analysis of data retrieved from the medical records of newborn infants admitted to the Neonatal Intensive Care at the Security Forces Hospital, Riyadh, Saudi Arabia, diagnosed to have NTDs during the period from 1996 to 2009 (14 years). Infants with NTDs associated with genetic, chromosomal, syndromic, and multiple congenital anomalies were identified utilizing clinical data, physical examination, laboratory tests, genetic tests, and radiologic work up. They were assessed by a neonatologist, pediatric neurologist, neurosurgeon, and geneticist.
Results
The total number of liveborn infants during this period was 85,672. The number of NTDs was 103 (1.2 per 1000 liveborn). Out of these, 20 (19.4%) were associated with genetic, chromosomal, syndromic and/or multiple congenital anomalies. We identified 5 patients who fulfilled the diagnostic criteria of Meckel-Gruber Syndrome (MKS):(Figures 1A, 1B, & 1C). The clinical characteristics of the patients are summarized in Table 1. Two patients, a boy and his sister, were diagnosed as Joubert syndrome (JS). They presented in the neonatal period with occipital encephalocele, respiratory problems, with periods of apnea and tachypnea. Later on, both developed visual problems (ocular motor apraxia) and spastic quadriplegia, and had severe global neurodevelopmental retardation. Brain MRI (Figures 2A & B) showed the typical features of JS including the “molar tooth” sign. Genetic studies using autozygome-guided candidate gene mutation analysis and exome sequencing revealed truncating mutation in TCTN1 and C5ORF42 gene. One patient was diagnosed as Walker-Warburg syndrome (WWS), and had 3 similarly affected siblings. He was a product of termination of pregnancy at 30 weeks based on antenatal ultrasound, which showed severe hydrocephalus and polyhydramnios. When delivered, the patient showed severe macrocephaly (Figures 3A & 3B), lumbar myelomeningocele microphthalmia, cataract, bilateral hip dislocation, and bilateral talipes equinovarus. He had generalized hypotonia with absent reflexes and flaccid paraplegia. Cranial CT scan showed (Figure 3C) severe hydrocephalus similar to that reported in the brain MRI of a previously affected sibling (Figure 3D), which also revealed Cobblestone lissencephaly, absent corpus callosum, and pontocerebellar hypoplasia. Creatinine kinase (CK) was elevated at 3763 U/l (N=38-174).
Figure 1.
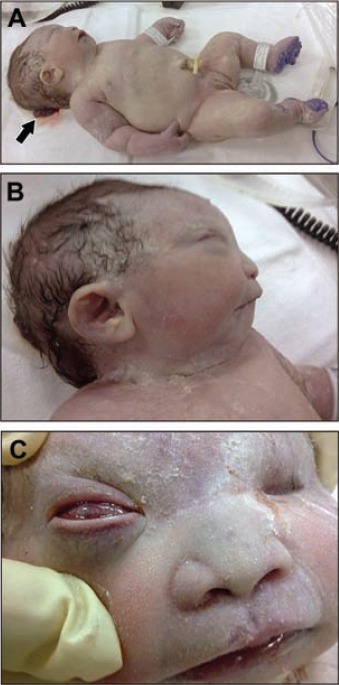
Whole body showing the triad of Meckel-Gruber syndrome. A) Occipital encephalocele (arrow), postaxial polydactyly of hands and feet, and large kidneys. B) Face showing sloping forehead, micrognathia, and low set ears. C) Bilateral anophthalmia.
Table 1.
Summary of clinical features of Meckel-Gruber syndrome.
Figure 2.
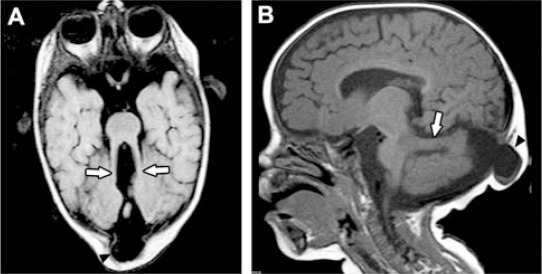
Joubert syndrome. A) Axial MRI brain showing molar tooth sign (arrows). B) Sagittal MRI showing horizontal cerebellar peduncle (arrow) and occipital encephalocele (arrowhead).
Figure 3.
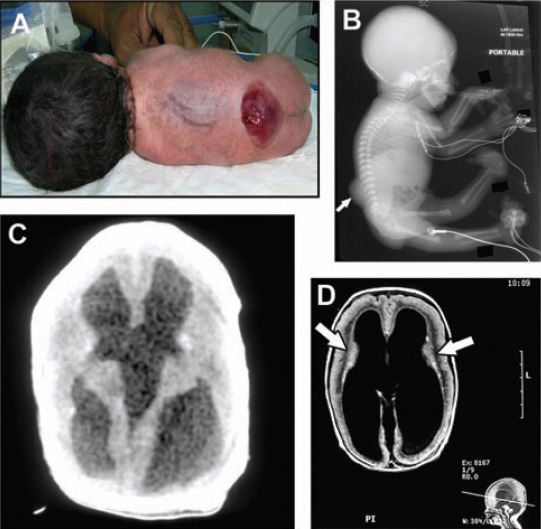
Walker-Warburg Syndrome. A) Macrocephaly and lumbosacral myelomeningocele in the proband (also shown on X-ray [arrow in B]). C) Cranial CT scan of the proband showing hydrocephalus. D) Axial brain MRI of an affected sibling showing hydrocephalus, lissencephaly, and layering of the cortex (arrows). Note the pontocerebellar hypoplasia in the sagittal image below.
One patient was diagnosed as Waardenburg syndrome (Figures 4A & 4B). She presented with dysmorphic features consisting of wide nasal bridge, lateral displacement of the inner canthi, low set ears, white forelock, white eyebrows, and white eyelashes of the left eye, heterochromia iridis, polydactyly, lumbosacral myelomeningocele, flaccid paraplegia, and severe sensorineural hearing loss. She developed intestinal obstruction due to duodenal atresia and malrotation of the bowel. Cranial CT brain showed absent corpus callosum.
Figure 4.
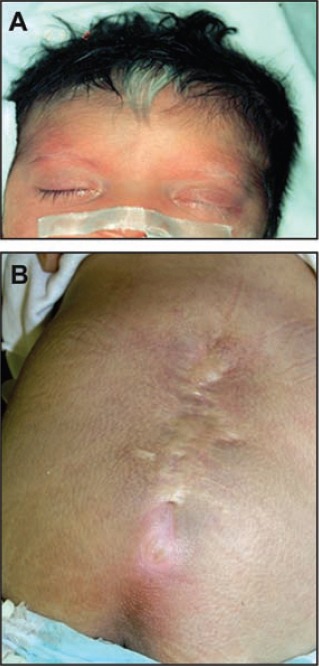
Waardenburg syndrome. A) Face showing white forelock and white left eyebrow. B) Surgically repaired myelomeningocele.
Two patients were found to have chromosomal abnormality associated with NTDs. A boy with dorsolumbar myelomeningocele (Figure 5) had flaccid paraplegia with incontinence of urine and stool. Cranial CT brain showed hydrocephalus and partial agenesis of the corpus callosum. Chromosome analysis revealed pericentric inversion of the Y chromosome with break and fusion points at Y p11.2 and Yp12: inv (Y) (P11.2q12). Additional study by fluorescence in situ hybridization [inv (Y) (Ypter+, DYZ3, Yqter+)] showed that the subtelomeric Ypter and Yqter regions were correctly situated. Only one centromere was present on chromosome Y. His father carried the same karyotype and had normal phenotype. Family history revealed the presence of cleft palate in the family of our patient, one sister, and 2 cousins. The other patient with chromosomal abnormality was a preterm male with a huge occipital encephalocele who died at the age of 2 months. He was found to have an apparently balanced Robertsonian translocation between chromosome 13 and 14 [45, XY, der (13,14) (q10,q10)]. His mother had 8 abortions for which she was investigated and found to have Robertsonian balanced translocation between Chromosome 13 and 14 [45,XX, t (13q 14q)]; the father had normal karyotype. The family has 2 normal living daughters. They also had an anencephalic term baby.
Figure 5.
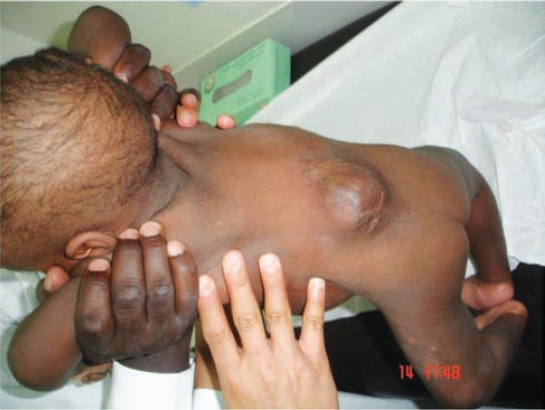
Patient with inversion of Y chromosome dorsolumbar myelomeningocele (repaired).
There were 2 patients with caudal regression syndrome. One boy was delivered at term by cesarean section due to fetal distress. The mother was gravida 6 para 5 and had insulin dependent diabetes mellitus (IDDM). Antenatal ultrasound scan revealed a defect in the occipital region including no brain tissue. Birth weight was 2.7 kg. Examination showed an occipital mass and bilateral clubbed feet (talipes equinovarus and vertical talus). Skeletal survey revealed abnormal cervical spine, hemivertebrae, and sacral agenesis S2 to S5. Brain MRI (Figures 6A & 6B) showed a heterogenous extra-axial cystic mass on the midline of the posterior fossa at the cisterna magna causing midline separation of the cerebellar hemispheres. There was an extracranial cyst at the site of posterior fontanelles (dermoid cyst). A spinal MRI (Figure 6C) showed only 2 sacral segments associated with short spinal cord ending in the lower dorsal spine, thickening of filum terminale, and cord tethering. The patient had craniotomy for removal of the posterior fossa dermoid cyst and ventriculoperitoneal shunt insertion. Histopathology confirmed cerebellar dermoid cyst. He also suffered from repeated sepsis, constipation, and enuresis. Chromosome analysis showed normal male 46, XY karyotype.
Figure 6.
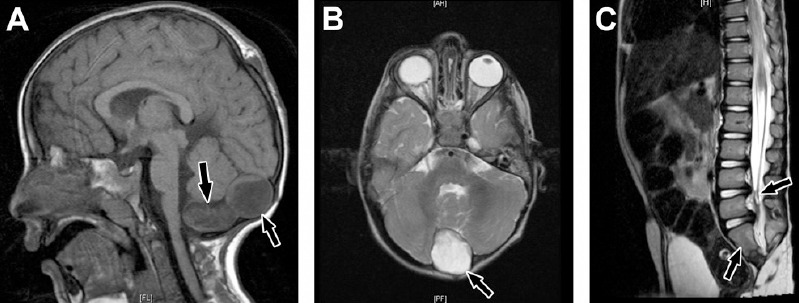
Caudal regression syndrome. A) Brain MRI showing 2 cystic structures at the region of cisterna magna demonstrating high signal intensity on T1-weighted image (arrows). B) A cyst is also shown on axial T2-weighted image (arrow). C) Sagittal MRI dorsolumbar spine showing abrupt cutoff (truncation) and bulbar configuration of conus medullaris at the level of L1 vertebral body and thickening of filum terminale (right arrow). There is also sacral agenesis (only 2 sacral segments are visualized, left arrow).
The other patient, one of twins born to a mother with insulin dependent diabetes mellitus (IDDM), was a case of sirenomelia who died within one hour of birth.
Discussion
Neural Tube Defects (NTDs) have an incidence of 1-2 per 1000 births and are considered to be a heterogeneous condition resulting from failure of normal neural tube closure between the third and fourth weeks of embryonic development. The incidence of NTDs varies with race, geographic variation, socioeconomic status, and multiple predisposing factors such as gene disorder, chromosomal abnormalities, and maternal risk factors, like diabetes mellitus, hyperthermia, and exposure to teratogens.
At the Security Forces Hospital, Riyadh, Saudi Arabia, and over a 14 year period (1996-2009), we identified approximately 20% of 103 NTDs cases to be associated with genetic, chromosomal, syndromic, and other anomalies. This incidence is from liveborn infants. Five of the affected patients had MKS (OMIM 249000), a severe pleiotropic autosomal recessive developmental disorder caused by dysfunction of primary cilia during early embryogenesis. The classical triad of MKS comprises 1) cystic renal disease, 2) a CNS malformation most commonly occipital encephalocele, and 3) polydactyly most often postaxial. Besides this triad, the phenotype also includes liver fibrosis, microphthalmia, cleft palate, and a wide array of associated brain anomalies. Our patients fulfilled all the phenotypic criteria of MKS (Table 1, Figure 1). In the Arab world, because of the high rate of consanguinity, the prevalence of MKS is 1:35008 compared with most populations (<1:20,000). Our prevalence was 1:17,134; but the true prevalence will be much higher than this as most cases of MKS are still born or follow termination of pregnancy,9 and not included in our study.
Ten genes have been identified so far to be mutated in MKS, encoding ciliary proteins.10-18 Shaheen et al19-20 utilizing homozygosity mapping and mutation analysis identified a novel pathogenic mutation: c.1506_2A>G in TCTN2 (NM_024809.3) in one of our patients (Patient 5, Table 1). Tectonic was recently identified in a murine model of NTD, Reiter and Skarnes, 2006,21 TCTN2 is causally linked to MKS, thus expanding the NTD phenotype associated with mutation in this family of proteins. The parents were offered preimplantation genetic diagnosis (PGD). We identified 2 patients with Joubert syndrome (JS, OMIM #610188), an autosomal recessive disorder presenting with developmental delay, hypotonia, ataxia, ocular motor apraxia, and neonatal breathing abnormalities.22-27 Neuroradiologically, JS is characterized by a peculiar malformation of the midbrain - hindbrain junction known as the “molar tooth sign,” consisting of cerebellar vermis hypoplasia or aplasia, thick and mal oriented superior cerebellar peduncles, and abnormally deep interpeduncular fossa.28-30 Joubert syndrome is a ciliopathy with genetic heterogeneity with 16 genes identified to date. Recently, Alazami et al22 utilizing exome sequencing identified truncating mutation in C5ORF42 in our patients. The parents were offered PGD.
Walker-Warburg syndrome (OMIM #236670) is a genetically heterogenous autosomal recessive disease characterized by congenital muscular dystrophy (CMD), hydrocephalus, cobblestone lissencephaly, and ocular malformation.31-35 Mutations in 6 genes involved in the glycosylation of a-dystroglycan, POMT1, POMT2, POMGNT1, FCDM, FKRP, and LARGE had been identified.36-43 These genes account for only a portion of WWS cases.43 LARGE gene was identified in one of our WWS families as a novel causative gene of WWS.42 The parents were first cousins.
Bedri et al44 described a patient demonstrating the typical clinical features of WWS in addition to other features including hydrocephalus, occipital encephalocele, agenesis of the corpus callosum, microphthalmia, ventricular septal defect, and rocker bottom feet. Several of these features were seen in our patient apart from a lumbar myelomeningocele instead of occipital encephalocele. Whitley et al45 reviewed 10 cases of WWS and occipital encephalocele was present in 4.
Waardenburg syndrome type 1 (OMIM #193500), is an autosomal dominant disorder characterized by pigmentary abnormalities of the hair (including a white forelock), skin and eyes (heterochromia irides and brilliant blue eyes), congenital sensorineural hearing loss, and lateral displacement of the ocular inner canthi (dystopia canthorum).46-48 Our patient with WS was found to have lumbosacral myelomeningocele. This association has been reported by Carezani-Gavin49 in a patient with WS type 1 who also had a myelomeningocele (MMC) at the L3-S1 level. In this family, Hoth el al50 identified a heterozygous mutation in the PAX3 gene. Chatkupt et al51 stated that spina bifida had been noted in at least 4 patients with WS, and reported cases of brothers with both WS and lumbosacral MMC. Nye et al52 reported lumbosacral MMC and WS type III in a patient with an interstitial deletion of 2q35 and the PAX3 gene. Hol and associates53 described a girl with spina bifida with mild signs of WS and a frameshift mutation in the PAX3 gene. Of note is that our patient was also found to have intestinal obstruction due to duodenal atresia, a feature which is rarely reported. Hirschsprung disease associated with WS was reported by Shah et al54 (Waardenburg-Shah syndrome or WS type 4), Read and Newton,46 and Tamayo et al.47 One of our patients with WS was found to have severe long segment Hirschsprung disease and died at the age of 3 months (unreported).
We had 2 patients with chromosome abnormality associated with NTDs, one of whom had balanced pericentric inversion of chromosome Y associated with dorsolumbar myelomeningocele. This chromosome rearrangement had been observed in individuals with normal phenotype. The pericentric inverted Y is inherited from generation to generation and has apparently no clinical significance.55 The prevalence of males with pericentric Y inversion in the general population is approximately one per 1000.56,57 The father of this patient is normal male with chromosome Y inversion, so the occurrence of MMC in his son might be just a coincidence. Jacobs et al58,59 reported the first male pericentric Y inversion, who had left sided cleft palate. Of note, is the presence of cleft palate in the family of our patient in one sister and two cousins. Whether these are related to the pericentric inversion of Y chromosome or coincidence, needs further investigations and chromosome analysis of this family.
The other patient had occipital encephalocele and balanced Robertsonian translocation between chromosomes 13 and 14 [45,XY, der (13,14) (q10,q10)]. The mother is a carrier of the same balanced translocation with normal phenotype. Fusion of chromosomes 13 and 14 leads to an increased risk of miscarriage and risk of liveborn trisomy 13 (Patau syndrome), but normal pregnancies are also possible, and they have 2 normal living daughters, but also had an anencephalic term baby. The father’s karyotype was normal. This finding is interesting as our patient, although he carried a balanced translocation as reported by cytogenetic lab, was found to have brain malformation (encephalocele). Although a carrier of a balanced Robertsonia translocation is phenotypically normal, there is a risk of unbalanced gametes and therefore of unbalanced offspring.60 The family with balanced Robertsonian translocation should be thoroughly counseled to avoid unnecessary reproductive wastage, by offering preimplantation genetic diagnosis (PGD), chorionic villous sampling, or prenatal amniocentesis to determine the karyotype and offer early termination of pregnancy if the fetus is found to be unbalanced.
Two patients with caudal regression syndrome were identified. One of them had sacral agenesis from S2-S5 together with cerebellar dermoid cyst and tethered cord, chronic constipation and enuresis. Although he had chronic constipation, he had no anorectal abnormalities. The other patient had an extreme form of caudal regression syndrome, sirenomelia. The mothers of both patients had IDDM, which is known to be associated with caudal regression syndrome. Multiple genetic and environmental factors also play role in the pathogenesis of caudal regression syndrome.61-62
The limitations of this study are its retrospective design, including only liveborn infants and thus excluding stillbirths, termination of pregnancies, and abortions due to lethal genetic disorders like MKS. Chromosomal analysis was not carried out on all cases of NTDs, so some cases might have been missed. We recommend performing chromosomal studies for all cases of NTDs and genetic work up for syndromic causes, to offer sound genetic counselling for the family.
In conclusion, genetic, syndromic, chromosomal, and associated multiple congenital anomalies constituted nearly 20% of the total number of NTDs identified at the Security Forces Hospital, Riyadh, Saudi Arabia. The percentage of these conditions will he higher than this if still births and termination of pregnancies due to them were included. The recurrence risk for these conditions is higher than those for non-syndromic multifactorial NTDs. Most of these conditions like MKS, JS, and WWS syndrome are inherited as autosomal recessive, with a recurrence rate of 25%. Waardenburg syndrome is autosomal dominant with a recurrence rate of 50%. Thanks to the advances in molecular genetics and whole exome sequencing, several causative genes involved in syndromic NTDs have been identified, making pre-implantation genetic diagnosis or early termination of pregnancy feasible measures to prevent recurrence of such cases like our families with MKS and JS. Identification of a chromosome anomaly or syndrome in a child with a NTD should lead to more accurate provision of genetic counseling to family members. The role of folic acid in preventing such cases is not as gratifying as for multifactorial NTDs. Strict control of diabetes is important both preconceptional and during pregnancy to avoid NTDs like sirenomelia and caudal regression syndrome which are strongly related to poor diabetic control.
Acknowledgments
We thank Maria Josefa A. Borromeo of the Department of Pediatrics for manuscript preparation, and Mandy Sarmiento from Medical Photography, Academic and Health Education Department at Security Forces Hospital, Riyadh, Kingdom of Saudi Arabia.
Footnotes
Disclosure.
References
- 1.Hall G, Solehdin F. Genetics of neural tube defects. Mental retardation and developmental disabilities. Res Rev. 1998;4:269–281. [Google Scholar]
- 2.Aguiar MJ, Campos AS, Aguiar RA, Lana AM, Magalhães RL, Babeto LT. [Neural tube defects and associated factors in liveborn and stillborn infants] J Pediatr (Rio J) 2003;79:129–134. [PubMed] [Google Scholar]
- 3.Harmon JP, Hiett AK, Palmer CG, Golichowski AM. Prenatal ultrasound detection of isolated neural tube defects: is cytogenetic evaluation warranted? Obstet Gynecol. 1995;86:595–599. doi: 10.1016/0029-7844(95)00206-7. [DOI] [PubMed] [Google Scholar]
- 4.Hume RF, Jr, Drugan A, Reichler A, Lampinen J, Martin LS, Johnson MP, et al. Aneuploidy among prenatally detected neural tube defects. Am J Med Genet. 1996;61:171–173. doi: 10.1002/(SICI)1096-8628(19960111)61:2<171::AID-AJMG14>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 5.Coerdt W, Miller K, Holzgreve W, Rauskolb R, Schwinger E, Rehder H. Neural tube defects in chromosomally normal and abnormal human embryos. Ultrasound Obstet Gynecol. 1997;10:410–415. doi: 10.1046/j.1469-0705.1997.10060410.x. [DOI] [PubMed] [Google Scholar]
- 6.Creasy MR, Alberman ED. Congenital malformations of the central nervous system in spontaneous abortions. J Med Genet. 1976;13:9–16. doi: 10.1136/jmg.13.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch SA. Non-multifactorial neural tube defects. Am J Med Genet C Semin Med Genet. 2005;135C:69–76. doi: 10.1002/ajmg.c.30055. [DOI] [PubMed] [Google Scholar]
- 8.Teebi AS, Teebi SA. Genetic diversity among the Arabs. Community Genet. 2005;8:21–26. doi: 10.1159/000083333. [DOI] [PubMed] [Google Scholar]
- 9.Kheir AEM, Imam A, Omer IM, Hassan IMA, Elamin SA, Awadalla EA, et al. Meckel-Gruber Syndrome: A rare and lethal anomaly. Sudan J Paediatr. 2012;12:93–96. [PMC free article] [PubMed] [Google Scholar]
- 10.Kyttälä M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, et al. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 11.Consugar MB, Kubly VJ, Lager DJ, Hommerding CJ, Wong WC, Bakker E, et al. Molecular diagnostics of Meckel-Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum Genet. 2007;121:591–599. doi: 10.1007/s00439-007-0341-3. [DOI] [PubMed] [Google Scholar]
- 12.Dawe HR, Smith UM, Cullinane AR, Gerrelli D, Cox P, Badano JL, et al. The Meckel-Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 13.Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 15.Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank V, den Hollander AI, Brüchle NO, Zonneveld MN, Nürnberg G, Becker C, et al. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 17.Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 18.Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, AlQahtani K, et al. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat. 2011;32:573–578. doi: 10.1002/humu.21507. [DOI] [PubMed] [Google Scholar]
- 20.Shaheen R, Faqeih E, Alshammari MJ, Swaid A, Al-Gazali L, Mardawi E, et al. Genomic analysis of Meckel-Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. Eur J Hum Genet. 2013;21:762–768. doi: 10.1038/ejhg.2012.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiter JF, Skarnes WC. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev. 2006;20:22–27. doi: 10.1101/gad.1363606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alazami AM, Alshammari MJ, Salih MA, Alzahrani F, Hijazi H, Seidahmed MZ, et al. Molecular characterization of Joubert syndrome in Saudi Arabia. Hum Mutat. 2012;33:1423–1428. doi: 10.1002/humu.22134. [DOI] [PubMed] [Google Scholar]
- 23.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 24.Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropediatrics. 1977;8:57–66. doi: 10.1055/s-0028-1091505. [DOI] [PubMed] [Google Scholar]
- 25.Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726–731. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 26.Khan AO, Oystreck DT, Seidahmed MZ, AlDrees A, Elmalik SA, Alorainy IA, et al. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008;115:2286–2289. doi: 10.1016/j.ophtha.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 28.Alorainy IA, Sabir S, Seidahmed MZ, Farooqu HA, Salih MA. Brain stem and cerebellar findings in Joubert syndrome. J Comput Assist Tomogr. 2006;30:116–121. doi: 10.1097/01.rct.0000191681.05473.13. [DOI] [PubMed] [Google Scholar]
- 29.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905. doi: 10.1016/S1474-4422(13)70136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 2011;32:1459–1463. doi: 10.3174/ajnr.A2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobyns WB, Pagon RA, Armstrong D, Curry CJ, Greenberg F, Grix A, et al. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32:195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 32.Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis. 2006;1:29. doi: 10.1186/1750-1172-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathews KD. Muscular dystrophy overview: genetics and diagnosis. Neurol Clin. 2003;21:795–816. doi: 10.1016/s0733-8619(03)00065-3. [DOI] [PubMed] [Google Scholar]
- 34.Salih MAM. Muscular Dystrophies and Myopathies in Arab Populations. In: Teebi AS, editor. Genetic Disorders among Arab Populations. 2nd ed. Berlin Heidelberg (DE): Springer-Verlag; 2010. pp. 158–179. [Google Scholar]
- 35.Salih MAM. Hereditary and acquired myopathies. In: Elzouki AY, Stapleton FB, Whitley RJ, Oh W, Nazer H, Harfi HA, editors. Textbook of Clinical Pediatrics. 2nd ed. New York (NY): Springer-Verlag; 2012. pp. 3503–3541. [Google Scholar]
- 36.Beltrán-Valero de Bernabé D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Currier SC, Lee CK, Chang BS, Bodell AL, Pai GS, Job L, et al. Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am J Med Genet A. 2005;133A:53–57. doi: 10.1002/ajmg.a.30487. [DOI] [PubMed] [Google Scholar]
- 38.van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabé D, Sabatelli P, Merlini L, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 41.Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Reeuwijk J, Grewal PK, Salih MA, Beltrán-Valero de Bernabé D, McLaughlan JM, Michielse CB, et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet. 2007;121:685–690. doi: 10.1007/s00439-007-0362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manzini MC, Gleason D, Chang BS, Hill RS, Barry BJ, Partlow JN, et al. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mutat. 2008;29:E231–E241. doi: 10.1002/humu.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bedri H, Mustafa B, Jadallah Y. Walker-Warburg syndrome: A case with multiple uncommon features. Sudan J Paediatr. 2011;11:59–63. [PMC free article] [PubMed] [Google Scholar]
- 45.Whitley CB, Thompson TR, Mastri AR, Gorlin RJ. Warburg syndrome: lethal neurodysplasia with autosomal recessive inheritance. J Pediatr. 1983;102:547–551. doi: 10.1016/s0022-3476(83)80182-6. [DOI] [PubMed] [Google Scholar]
- 46.Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34:656–665. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamayo ML, Gelvez N, Rodriguez M, Florez S, Varon C, Medina D, et al. Screening program for Waardenburg syndrome in Colombia: clinical definition and phenotypic variability. Am J Med Genet A. 2008;146A:1026–1031. doi: 10.1002/ajmg.a.32189. [DOI] [PubMed] [Google Scholar]
- 48.Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 49.Carezani-Gavin M, Clarren SK, Steege T. Waardenburg syndrome associated with meningomyelocele. Am J Med Genet. 1992;42:135–136. doi: 10.1002/ajmg.1320420127. [DOI] [PubMed] [Google Scholar]
- 50.Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I) Am J Hum Genet. 1993;52:455–462. [PMC free article] [PubMed] [Google Scholar]
- 51.Chatkupt S, Chatkupt S, Johnson WG. Waardenburg syndrome and myelomeningocele in a family. J Med Genet. 1993;30:83–84. doi: 10.1136/jmg.30.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nye JS, Balkin N, Lucas H, Knepper PA, McLone DG, Charrow J. Myelomeningocele and Waardenburg syndrome (type 3) in patients with interstitial deletions of 2q35 and the PAX3 gene: possible digenic inheritance of a neural tube defect. Am J Med Genet. 1998;75:401–408. [PubMed] [Google Scholar]
- 53.Hol FA, Geurds MP, Jensson O, Hamel BC, Moore GE, Newton R, et al. Exclusion mapping of the gene for X-linked neural tube defects in an Icelandic family. Hum Genet. 1994;93:452–456. doi: 10.1007/BF00201674. [DOI] [PubMed] [Google Scholar]
- 54.Shah KN, Dalal SJ, Desai MP, Sheth PN, Joshi NC, Ambani LM. White forelock, pigmentary disorder of irides, and long segment Hirschsprung disease: possible variant of Waardenburg syndrome. J Pediatr. 1981;99:432–435. doi: 10.1016/s0022-3476(81)80339-3. [DOI] [PubMed] [Google Scholar]
- 55.Zeuthen E, Nielsen J. Pericentric Y inversion in the general population. Humangenetik. 1973;19:265–270. doi: 10.1007/BF00278400. [DOI] [PubMed] [Google Scholar]
- 56.Walzer S, Breau G, Gerald PS. A chromosome survey of 2,400 normal newborn infants. J Pediatr. 1969;74:438–448. doi: 10.1016/s0022-3476(69)80202-7. [DOI] [PubMed] [Google Scholar]
- 57.Gerald PS, Walzer S. Chromosome studies of normal newborn infants. In: Jacobs PA, editor. Human population cytogenetics. Edinburgh (UK): University Press; 1970. pp. 143–151. [Google Scholar]
- 58.Jacobs PA, Brunton M, Brown WM. Cytogenetic studies in leucocytes on the general population: subjects of ages 65 years and more. Ann Hum Genet. 1964;27:353–365. doi: 10.1111/j.1469-1809.1963.tb01532.x. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs PA, Ross A. Structural abnormalities of the Y chromosome in man. Nature. 1966;210:352–354. doi: 10.1038/210352a0. [DOI] [PubMed] [Google Scholar]
- 60.Jiang J, Fu M, Wang D. Cytogenetic analysis in 61 couples with spontaneous abortions. Chin Med J (Engl) 2001;114:200–201. [PubMed] [Google Scholar]
- 61.Duesterhoeft SM, Ernst LM, Siebert JR, Kapur RP. Five cases of caudal regression with an aberrant abdominal umbilical artery: Further support for a caudal regression-sirenomelia spectrum. Am J Med Genet A. 2007;143A:3175–3184. doi: 10.1002/ajmg.a.32028. [DOI] [PubMed] [Google Scholar]
- 62.Orioli IM, Amar E, Arteaga-Vazquez J, Bakker MK, Bianca S, Botto LD, et al. Sirenomelia: an epidemiologic study in a large dataset from the International Clearinghouse of Birth Defects Surveillance and Research, and literature review. Am J Med Genet C Semin Med Genet. 2011;157C:358–373. doi: 10.1002/ajmg.c.30324. [DOI] [PMC free article] [PubMed] [Google Scholar]