

Abstract
Objective:
To illustrate the clinical and radiological findings of split cord malformation (SCM) in patients with spinal open neural tube defect (SONTD), and report the outcome of their treatment.
Methods:
A retrospective study of the clinical and radiological findings of 11 patients diagnosed with SCM, identified among 83 patients with SONTD at King Khalid University Hospital, in Riyadh, Saudi Arabia between 1995 and 2010.
Results:
There were 6 girls and 5 boys; their age ranged from less than a year to 9 years (mean 4.2 years). Six patients had type I SCM, and 5 patients type II SCM. The CT and MRI imaging showed characteristic bony, cartilaginous, or fibrous septum, and other SONTD-associated anomalies. Seven patients were graded A & B according to the Frankel grading score, and none of them required surgery, while worsening neurology led to surgical intervention in 3 patients, with clinical improvement after surgery, and one patient that underwent cord untethering remained stable.
Conclusion:
Split cord malformation is not uncommon among patients with SONTD. It tends to involve mainly the lumbar spine, and female predominance is more remarkable in type I. Neurological manifestations of SCM may be superimposed with SONTD. Surgery is effective for symptomatic patients, and not indicated in the severely disabled.
Split cord malformation (SCM), also known as diastematomyelia, is a congenital spinal dysraphism in which a segment of the spinal cord is divided into 2 hemicords. It can occur at any part of the spinal column, but is mainly found in the lumbar region. Split cord malformation has been classified into 2 types, type I consists of 2 hemicords, which are separated by a bony spur, each contained within its own dural sac. In type II the 2 hemicords are separated by a fibrous septum and are contained in a single dural sac.1-3 Children born with spinal open neural tube defect (SONTD) are usually investigated by MRI of the entire neuroaxis, which in many cases shows abnormal findings such as: Chiari II malformation, low lying conus, thickened filum terminale, intraspinal lipomas, dermal sinus tracts, and SCM.4-6 A CT scan is usually considered complimentary to MRI and is helpful in evaluating bony anomalies.5-8 Between 10-13% of cases with SCM have an associated SONTD, because of the close association between the development of the spinal cord and vertebral column.4-8 Clinical manifestations related to SCM in patients with SONTD either appear do novo or superimposed by symptoms related to NTD. This may include a combination of upper and lower motor neuron signs in the lower extremities associated with wasting of the leg muscles and feet deformity. It is difficult to diagnose in the presence of disabling paraplegia and double sphincter incontinence, which makes a surgical decision also difficult.2 Neurosurgeons decide surgical treatment based on neurological assessment and in correlation with radiological findings associated with SCM. Surgical treatment in both types of SCM aims at removing the median septum and all other associated bands, untethering of the spinal cord, and reconstitution of a single dural sac.9-12 Surgery is more effective for patients with type 1 SCM; patients without surgery show no improvement. In this study, we aimed to illustrate the clinical and radiological findings of SCM in patients with SONTD, and report on the outcome of their treatment.
Methods
A retrospective study of the clinical and radiological findings, and the outcome of surgical treatment of 11 patients with SCM associated with SONTD, was carried out at King Khalid University Hospital, in Riyadh, Saudi Arabia between 1995 and 2010. During this period, 83 cases were treated for SONTD. All patients diagnosed with SCM were included in the study. Exclusion criteria were cranial neural tube defects, spina bifida occulta, closed meningocele, or meningomyelocele. The data was obtained from case files, and radiology images. Frankel grading system was used for assessment of the motor and sensory functions of the patients.13 Spinal MRI was performed in all cases, where the entire spine was screened (Figures 1A & 1B), and thoraco-lumbar CT scan was carried out when SCM was diagnosed on the MRI. Radiological findings including the level of spur, lower level of the conus, presence of syrinx, and other spine anomalies, if any, were all recorded. The SCM was classified into type I, and type II depending on the presence of bony spur or fibrous septum.14 All patients that underwent surgery had SCM type I. Laminectomy was performed around the attachment of the rigid median septum, the septum was dissected subperiosteally from its dural sleeve within the dural cleft, and the septum was removed using a rongeur. The dura was opened on both sides and the sleeve was resected, followed by closing the posterior dura only.
Figure 1.
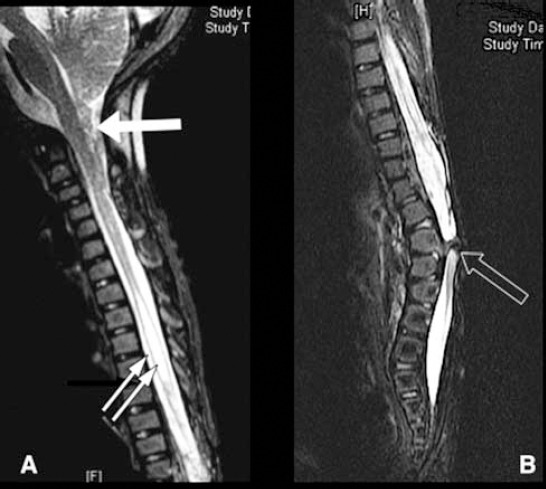
Whole spine MRI T2WI, sagittal view showing tonsillar herniation (white arrow), syringomyelia (2 white arrows), and type I - split cord malformation (black arrow).
Results
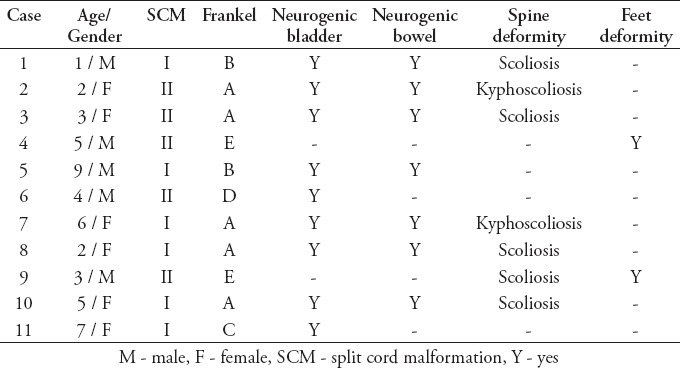
All patients with SONTD (83 patients) underwent screening MRI of the entire neuroaxis to rule out possible concomitant anomaly. Split cord malformation was diagnosed in 11 patients. They constituted 7% of all cases with spinal dysraphism (11/156) registered in the spina bifida clinic, and 13% of patients with SONTD (11/83). There were 6 girls and 5 boys ranging in age from less than one year to 9 years with a mean age 4.2 years, and female predominance was remarkable in type I - SCM (2:1). A total of 6 patients had type I - SCM (Figures 2A, 2B, & 2C), and 5 patients were diagnosed with type II - SCM (Figures 3A & 3B). Radiological findings of the spine are summarized in Table 1. All patients were born with lumbar or lumbosacral SONTD (meningomyelocele, meningocele, or myeloschisis) and underwent closure soon after birth. The level of SCM was found to be in the lumbar region in all patients; most were at the L1-L2 level (6 patients), and at the L3-L4 (5 patients). In 2 patients, the conus ended above the level of the split, which involved the cauda (Figure 2C). An MRI revealed syringomyelia in 5 patients (Figures 4A & 4B), and Chiari malformation type II in 7 patients. There was no associated dermoid or lipoma. The clinical findings in all patients with SCM are shown in Table 2, and the clinical manifestations related to SCM in the 3 patients (patients 4, 9, and 10) who underwent surgery were; scoliosis (one patient), foot deformity (2 patients), gait disturbance (one patient), back pain (one patient), and abnormal urodynamic function in (one patient). All patients underwent surgery improved after surgery, while the remaining 8 patients that were treated conservatively remained stable.
Figure 2.
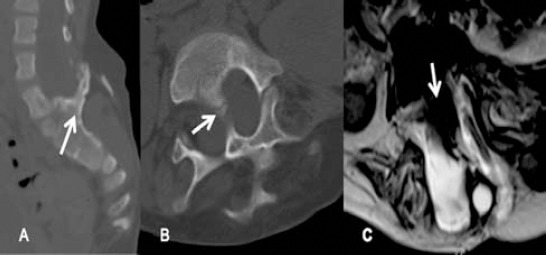
Lumbosacral CT scan of type I split cord malformation; A) sagittal reconstruction, and B) axial view showing bony spur dividing spinal canal (arrow). C) An MRI, T2WI, axial view showing a large spur (arrows) splitting the spinal canal into 2, each contains part of the cauda equina.
Figure 3.
Sagittal MRI scan A), and B) axial T2WI view of a child with a lumbar meningocele and type II split cord malformation showing the meningocele (wide arrow) and fibrous band (narrow arrow) dividing the conus into 2 hemicords, both contained in a single dural sheath.
Table 1.
Radiological findings of 11 cases with split cord malformation.
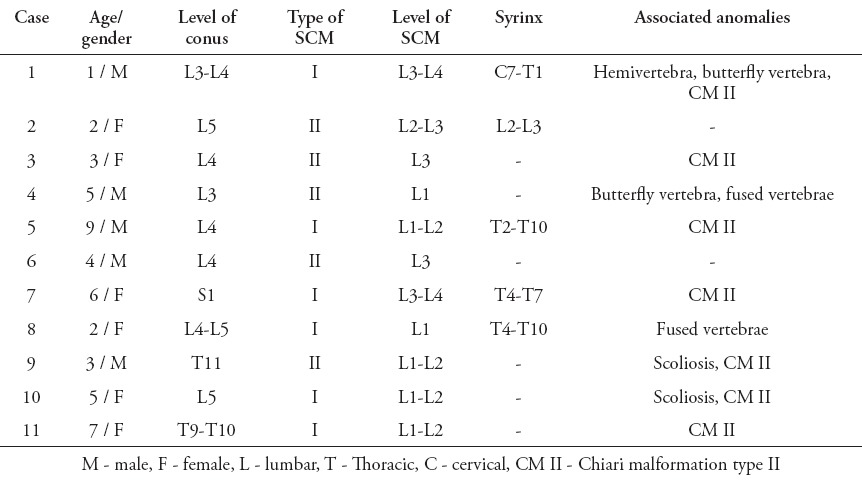
Figure 4.
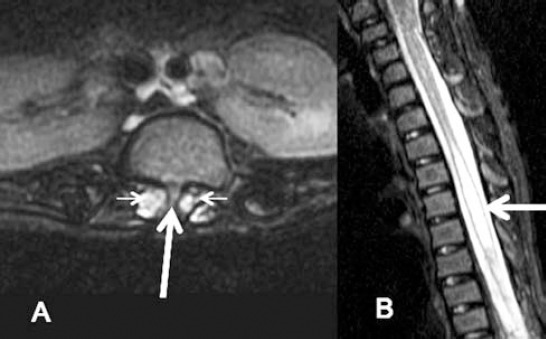
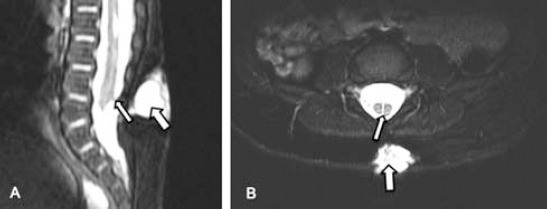
Lumbar spine axial T2WI MRI A) showing small spur (arrow) dividing the spinal canal at the L1/L2 level with 2 hemicords (small arrows). B) Sagittal T2WI for cervical and upper thoracic spine showing focal thoracic spinal cord syrinx (arrow).
Table 2.
Clinical and neurological presentation of 11 patients with split cord malformation.
Discussion
Split cord malformation is a rare form of NTD, classified by Pang et al2,3 into 2 types. In type I, the 2 hemicords are always invested in 2 separate dural sheaths, and separated by a bony spur. In type II, the 2 halves of the spinal cord are separated by a fibrous septum but contained in a single dural sheath.2,3 Pang and colleagues3 also proposed a unified theory that explained the embryogenesis of SCMs, in which they described the origin of all SCMs as an error occurring approximately at the time of closure of the neuroenteric canal, and an accessory canal is formed between the yolk sac and the amnion, which subsequently invested with the mesenchymal tract, which splits the notochord and the neural plate.2,3
An SCM could be found alone or in combination with a NTD, and might affect any level of the spinal column. This study included 11 cases of SONTD-associated SCM, representing 13% of all patients with SONTD, and 7% of all patients with NTD registered in the spina bifida clinic. The incidence in this series is similar to a study from Iran, where 33 patients (10%) had SCM among 330 cases with meningomyelocele,4 and is lower than its incidence in India when 300 cases (20%) with SCM were found out of approximately 1500 patients of NTD between 1991 and 2006.12
Females are usually affected more commonly than males, and female predominance is noticed to be more remarkable in type I - SCM than in type II.1,5,14 In our series, females predominate in a ratio of 1.2:1.0, and in type I - SCM the female to male ratio was 2:1 (p>0.05). However, the sample in our study is small.
Screening of the whole spine with MRI in patients with SONTD is essential to rule out associated anomalies. It displays the fine anatomy of the associated myelomeningocele, and soft tissue bands of type II SCM. Imaging of the whole spine is a routine practice in spina bifida patients not only for patients with SCM. Most SCM patients are diagnosed in early life after MRI, and not when they present with symptoms suggestive of spinal cord pathology other than MMC. In the presence of severe scoliosis, or kyphoscoliosis, coronal images should always be obtained. A CT scan is important to show the bony anatomy such as the neural arches, and the bony spur in type I SCM, and it provides precise information on the exact localization of the bony septum, its origin, size, and direction (Figures 5A, 5B, & 5C).8,15 The abnormal findings found on MRI or CT of our patients included; syringomyelia, butterfly vertebra, hemivertebra, fused vertebrae, and scoliosis. All of which has also been previously reported.
Figure 5.

Lumbar spine CT type I SCM; sagittal reconstruction A) showing large bony spur in the spinal canal (black arrow). B) Coronal reconstruction view showing wide interpedicular space and spur in the center of the canal. C) Axial CT showing the bony spur arising from the vertebral body dividing spinal canal into 2.
The spinal cord was low lying below its normal level in 10 patients, and in all cases the SCM was located in the lumbar region, the majority was at L1-L2 (6 patients), followed by L3-L4 (5 patients), and the cauda equina was split in 2 patients. Mahapatra12 also reported that SCM was most common in the lumbar and dorsolumbar area (78%), and in the lumbosacral region (8%).
Spinal cord tethering caused by SCM is difficult to diagnose in patients with SONTD who are paraplegic and have double sphincter incontinence, which makes surgical decision also difficult. Persistent back pain could be an important symptom indicating symptomatic SCM requiring attention.
We could observe neurological deterioration due to SCM in 3 patients with good Frankel grade. They underwent timely surgical intervention, after which they showed marked recovery. In both types of SCM, surgical treatment aims at removing the median septum and all other associated bands, untethering of the spinal cord, and reconstitution of a single dural sac, which is better performed through laminectomy.9,16,17
The presence of syringomyelia did not interfere with the surgical decision for SCM in 5 patients who were treated conservatively. This was supported earlier by Gan et al18 who observed no change in syringomyelia after untethering of the spinal cord in (8/15) patients with type I - SCM, and rarely contributes to their neurological symptoms.18
There is no evidence to support prophylactic surgery in asymptomatic or severely disabled children, and there is no available data on the natural history of tethered cord syndrome in asymptomatic adults as well, but it is known that neurological deterioration can be precipitated after a fall or strenuous exercise. We therefore did not recommend operating on asymptomatic children and those who are otherwise severely disabled and managed them conservatively.
In conclusion, children with SONTD should be screened for SCM by MRI. In SONTD, (1) there is a female predominance in type I, compared with type II - SCM, (2) the SCM tends to involve mainly the lumbar spine, (3) it is difficult to detect neurological deterioration of SCM in paraplegic patients with sphincter incontinence, (4) the presence of syringomyelia is not a sole indication of surgery for SCM. (5) Surgery is effective for symptomatic patients, while asymptomatic, and severely disabled patients can be closely observed. The limitation of this study is its small sample size. Multicenter collaboration is needed to help improve our understanding of future cases.
Footnotes
Disclosure.
References
- 1.Dias MS, Pang D. Split cord malformations. Neurosurg Clin N Am. 1995;6:339–358. [PubMed] [Google Scholar]
- 2.Pang D. Split cord malformation: Part II: Clinical syndrome. Neurosurgery. 1992;31:481–500. doi: 10.1227/00006123-199209000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451–480. doi: 10.1227/00006123-199209000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Ansari S, Nejat F, Yazdani S, Dadmehr M. Split cord malformation associated with myelomeningocele. J Neurosurg. 2007;107(4 Suppl):281–285. doi: 10.3171/PED-07/10/281. [DOI] [PubMed] [Google Scholar]
- 5.Iskandar BJ, McLaughlin C, Oakes WJ. Split cord malformations in myelomeningocele patients. Br J Neurosurg. 2000;14:200–203. doi: 10.1080/026886900408360. [DOI] [PubMed] [Google Scholar]
- 6.Kumar R, Bansal KK, Chhabra DK. Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg. 2002;36:119–127. doi: 10.1159/000048366. [DOI] [PubMed] [Google Scholar]
- 7.Erşahin Y. Split cord malformation types I and II: a personal series of 131 patients. Childs Nerv Syst. 2013;29:1515–1526. doi: 10.1007/s00381-013-2115-7. [DOI] [PubMed] [Google Scholar]
- 8.Borkar SA, Mahapatra AK. Split cord malformations: A two years experience at AIIMS. Asian J Neurosurg. 2012;7:56–60. doi: 10.4103/1793-5482.98643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Proctor MR, Scott RM. Long-term outcome for patients with split cord malformation. Neurosurg Focus. 2001;10:e5. doi: 10.3171/foc.2001.10.1.6. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Prakash M. Unusual split cord with neurenteric cyst and cerebellar heterotopia over spinal cord. Childs Nerv Syst. 2007;23:243–247. doi: 10.1007/s00381-006-0211-7. [DOI] [PubMed] [Google Scholar]
- 11.Rowley VB, Johnson AJ. Lumbar split cord malformation with lateral hemimyelomeningocele and associated Chiari II malformation and other visceral and osseous anomalies: a case report. J Comput Assist Tomogr. 2009;33:923–926. doi: 10.1097/RCT.0b013e318198947d. [DOI] [PubMed] [Google Scholar]
- 12.Mahapatra AK. Split cord malformation - A study of 300 cases at AIIMS 1990-2006. J Pediatr Neurosci. 2011;6(Suppl 1):S41–S45. doi: 10.4103/1817-1745.85708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankel HL, Hancock DO, Hyslop G, Melzak J, Michaelis LS, Ungar GH, et al. The value of postural reduction in the initial management of closed injuries of the spine with paraplegia and tetraplegia. I. Paraplegia. 1969;7:179–192. doi: 10.1038/sc.1969.30. [DOI] [PubMed] [Google Scholar]
- 14.Kumar R, Singh SN, Bansal KK, Singh V. Comparative study of complex spina bifida and split cord malformation. Indian J Pediatr. 2005;72:109–115. doi: 10.1007/BF02760692. [DOI] [PubMed] [Google Scholar]
- 15.Ozturk E, Sonmez G, Mutlu H, Sildiroglu HO, Velioglu M, Basekim CC, et al. Split-cord malformation and accompanying anomalies. J Neuroradiol. 2008;35:150–156. doi: 10.1016/j.neurad.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Higashida T, Sasano M, Sato H, Sekido K, Ito S. Myelomeningocele associated with split cord malformation type I -three case reports. Neurol Med Chir (Tokyo) 2010;50:426–430. doi: 10.2176/nmc.50.426. [DOI] [PubMed] [Google Scholar]
- 17.Erşahin Y. An unusual split cord malformation. Pediatr Neurosurg. 2000;32:109. doi: 10.1159/000028909. [DOI] [PubMed] [Google Scholar]
- 18.Gan YC, Sgouros S, Walsh AR, Hockley AD. Diastematomyelia in children: treatment outcome and natural history of associated syringomyelia. Childs Nerv Syst. 2007;23:515–519. doi: 10.1007/s00381-006-0205-5. [DOI] [PubMed] [Google Scholar]