

Abstract
Role of the funding source: Funding from the NIH was used for support of the participating clinical centers and the coordinating center. The funding source did not participate in the collection or the analysis of the data.
BACKGROUND. The β cell killing that characterizes type 1 diabetes (T1D) is thought to begin years before patients present clinically with metabolic decompensation; however, this primary pathologic process of the disease has not been measured.
METHODS. Here, we measured β cell death with an assay that detects β cell–derived unmethylated insulin (INS) DNA. Using this assay, we performed an observational study of 50 participants from 2 cohorts at risk for developing T1D from the TrialNet Pathway to Prevention study and of 4 subjects who received islet autotransplants.
RESULTS. In at-risk subjects, those who progressed to T1D had average levels of unmethylated INS DNA that were elevated modestly compared with those of healthy control subjects. In at-risk individuals that progressed to T1D, the observed increases in unmethylated INS DNA were associated with decreases in insulin secretion, indicating that the changes in unmethylated INS DNA are indicative of β cell killing. Subjects at high risk for T1D had levels of unmethylated INS DNA that were higher than those of healthy controls and higher than the levels of unmethylated INS DNA in the at-risk progressor and at-risk nonprogressor groups followed for 4 years. Evaluation of insulin secretory kinetics also distinguished high-risk subjects who progressed to overt disease from those who did not.
CONCLUSION. We conclude that a blood test that measures unmethylated INS DNA serves as a marker of active β cell killing as the result of T1D-associated autoimmunity. Together, the data support the concept that β cell killing occurs sporadically during the years prior to diagnosis of T1D and is more intense in the peridiagnosis period.
TRIAL REGISTRATION. Clinicaltrials.gov NCT00097292.
FUNDING. Funding was from the NIH, the Juvenile Diabetes Research Foundation, and the American Diabetes Association.
Introduction
Type 1 diabetes (T1D) begins years before clinical presentation with hyperglycemia (1, 2). Following the initial breech of immune tolerance, the autoimmune response against β cells is thought to expand and diversify. Over time, there are increasing numbers of antigenic epitopes that are recognized by autoantibodies and by autoreactive T cells. Some investigators have postulated that there is a waxing and waning progression of disease over time that explains its chronicity and possibly its diversity (3). Nonetheless, based largely on the model proposed by Eisenbarth in 1986, it has become generally accepted that there is a steady progression of β cell killing by pathologic T cells that eventually results in the loss of 80% to 90% of β cell function at the time of diagnosis (4). However, the data that led to the development of this model were indirect and involved serial measurement of autoantibodies and the insulin secretory responses to intravenous glucose. Autoantibodies are not the primary mediators of β cell killing and an intravenous glucose tolerance test does not provide dynamic or insulin secretory responses to a physiologic stimulus.
Immune therapies, such as anti-CD3 mAbs, CTLA4-Ig, LFA3-Ig, and anti-CD20 mAb have been able to modify the loss of β cell function in the short term in patients with disease, but none have reversed or even completely preserved metabolic function (5–11). Because the effects of these drugs have waned with time, investigators have focused on disease prevention, with the goal of arresting the course of disease when β cell mass is sufficient to maintain normal glucose levels. Identifying those individuals who are progressing to T1D during the asymptomatic period with normoglycemia is therefore an important goal. Autoantibodies have largely been used to identify individuals at risk, but their appearance may be transient, and they do not directly measure the pathologic process and cannot provide information about the pace of disease progression on an individual basis (12, 13).
Our understanding of the kinetics of disease progression is limited because we do not have methods that directly measure β cell death, the primary pathologic process. We recently developed a method for assessing β cell death in vivo by measuring the relative amount of β cell–derived INS DNA in the circulation (14–16). The analysis involved quantitative PCR studies of INS DNA, with epigenetic marks that identify the DNA source as cells that actively transcribe insulin. The only significant source of unmethylated INS DNA is β cells, and therefore, the level of unmethylated INS DNA in the serum is proportional to the rate of β cell death. We previously validated this approach in mice with diabetes induced by streptozotocin or in NOD mice with autoimmune diabetes (15). Using the same approach adapted to measurement of human INS DNA in serum, we found increased rates of β cell death in patients with recent-onset T1D compared with those in healthy control subjects (14). Patients with recent-onset T1D who were treated with anti-CD3 mAb showed improvement in C-peptide responses and also showed reduced levels of unmethylated INS DNA in the circulation. We then improved the specificity and sensitivity of the method by using droplet digital PCR (ddPCR) to analyze differentially methylated CpG sites in circulating INS (16).
A number of reports have described changes in C-peptide and glucose responses to oral glucose during progression to T1D in at-risk individuals (17–19). These studies have shown statistically significant but modest quantitative changes in C-peptide release very close to the time of clinical presentation (20). The small changes that are observed during the prediabetic period may reflect the destruction of the majority of β cells before insulin secretion declines, but previously, there has not been a way to determine when exactly β cell killing is occurring. Furthermore, insulin secretion may be modulated by a number of factors in addition to the absolute mass, including insulin sensitivity and ambient glucose, as well as including immunologic mediators, such as IL-1β, and other inflammatory mediators (21–24). The effects of these factors may involve cytotoxicity or cellular stress as well as impaired or delayed function without cell killing. Indeed, a number of studies have shown that the early insulin secretory defect in type 2 diabetes arises from delays in the timing of the β cell response to glucose (25, 26). Similar changes in insulin secretion may occur in T1D, since we previously reported that patients with T1D, nondiabetic at-risk individuals who progress to overt disease, and those with long-standing disease show a delayed secretory response compared with nondiabetic individuals (27, 28).
Identifying individuals who are progressing to T1D and when β cell killing occurs will be important for designing prevention strategies. In this study, we determined the levels of β cell killing in 2 cohorts of individuals at risk for T1D using the ddPCR assay. One cohort was followed for as long as 4.4 years (or 4.1 years prior to onset of T1D). In the other high-risk cohort, we analyzed β cell death and function to determine the timing of β cell killing and the factors that lead to disease presentation. In addition, we measured the disappearance of β cell–derived, unmethylated INS DNA in patients with chronic pancreatitis who received autologous islet transplants following total pancreatectomy in order to understand the kinetics of disappearance of unmethylated INS DNA from dying β cells. Our studies indicate that β cell killing can be detected sporadically in at-risk individuals at a time when glucose tolerance and insulin secretion are normal. In the time period near presentation of clinical disease, β cell killing was consistently increased. In addition to β cell death, the progression to disease was indicated by a reversible delay in the kinetics of insulin secretion. Our findings suggest a new model of disease progression in which β cell destruction and metabolic dysfunction are events closely associated with disease onset.
Results
Demographic and metabolic features of high-risk participants in the TrialNet Natural History study.
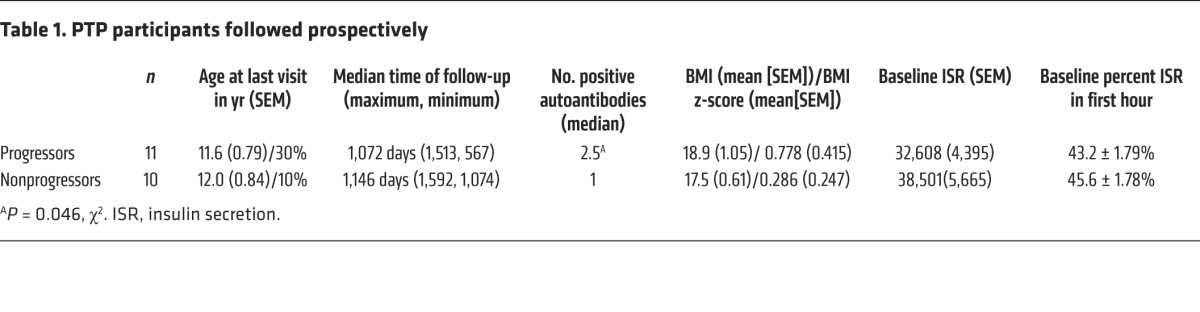
We studied 50 relatives of patients with T1D who were at risk for the disease, from 2 cohorts in the TrialNet Pathway to Prevention (PTP) study (Figure 1). All of the individuals had normal HbA1c levels. We identified 10 at-risk participants who developed T1D over a 3- to 4-year follow-up period (progressors, n = 10) and a group of at-risk participants of similar age who were followed over a similar time period but did not develop T1D (nonprogressors, n = 10). The demographic and metabolic features of these two groups were comparable (Table 1), but the progressors had a higher frequency of autoantibodies (P < 0.05), consistent with previous findings that individuals with a greater number of autoantibodies are more likely to progress to T1D (13). Day 0 was designated the date of diagnosis of T1D in the progressors and the last visit of the nonprogressors. These participants were prospectively studied at approximately 6-month intervals for up to 1,513 days, prior to diagnosis of diabetes or before the last visit. Each subject had at least 4 serial samples drawn during the observation period.
Figure 1. CONSORT diagram showing allocation of the study subjects.

The TrialNet Natural History study commenced in 2004. The number of subjects screened reflects the total number of subjects since enrollment. The first at-risk subject was enrolled in 2005 and the last was enrolled in 2008. The high-risk subjects were identified between 2010 and 2013. The diagram shows the allocation of subjects to the “at-risk” group that was prospectively followed and the “high-risk” group that was studied on 2 occasions.
Table 1. PTP participants followed prospectively.
β Cell death in at-risk progressors and nonprogressors to T1D.
We measured the level of β cell death with a ddPCR method that compares the levels of unmethylated and methylated INS DNA in bisulfite-treated DNA that is isolated from the serum (referred to herein as the ratio). When the average of all of the values taken from each individual was compared with the averages of the measures of healthy control subjects there was a significant increase in the ratios from the progressors (P = 0.048, ANOVA; P < 0.05, Bonferroni multiple comparison) (Figure 2A). However, the differences in the average values were modest, which may have been due to sporadic increases in the ratios in some but not all individuals. Indeed, of the measurements from these individuals (Figure 2B), 24.5% (12 of 49) from at-risk progressors and 14.2% (8 of 56) from the nonprogressors were more than 2 SD above the mean of the healthy control subjects (P = NS); 7 of the 10 individual progressors and 4 of the 10 nonprogressors had at least 1 positive value. The time of the increased ratios in the progressors and nonprogressors was similar (data not shown). In addition, we did not find differences in the progressors and nonprogressors at specific time points by repeated-measures ANOVA.
Figure 2. β Cell death in progressors and nonprogressors to T1D in the PTP study followed for long periods of time.

(A) Comparison of the average levels (mean ± SEM) of unmethylated INS DNA (i.e., ratio of unmethylated/methylated INS DNA) in the at-risk progressors, nonprogressors, and healthy control subjects (HC) (P = 0.048, ANOVA; *P < 0.05, Bonferroni multiple comparison test). (B) All of the individual ratio measurements are shown for the at-risk progressors (n = 10, 49 measurements) and nonprogressors (n = 10, 56 measurements) and were compared with the average levels (n = 32, 62 measurements). Six measurements (of a total of 105) were unobtainable for technical reasons. The dashed line represents the mean + 2 SD of the nondiabetic control subjects. (C) The ISR AUC for the progressors and nonprogressors at the study visits over the observation period prior to the diagnosis of T1D in the progressors or the last study visit in the nonprogressors. The least-square mean ± SEM from the repeated-measures ANOVA is shown. Neither the 2 curves nor the individual time points differ significantly. (D) The relationship between the change in the ratio and the change in the ISR AUC (from the first study visit) is shown for the progressors and nonprogressors (progressors: Pearson correlation coefficient, r = –0.475, P = 0.0026; nonprogressors: P = NS). All of the data points are shown.
We also analyzed the insulin secretion during the oral glucose tolerance tests (OGTTs) that were performed approximately every 6 months during the follow-up period by deconvoluting the C-peptide data (29) and determining the total insulin secreted during the 2-hour test (ISR AUC) (Figure 2C). Insulin secretion was not significantly different between the progressors and nonprogressors, similar to previous reports that have shown that β cell secretory responses are preserved until shortly before diagnosis of T1D (20, 28). However, it is possible that there were modest changes in insulin secretion during disease progression that might not have been identified with the overall analysis by ANOVA. Therefore, to determine whether the increase in the levels of unmethylated INS DNA corresponds to pathologic changes, we studied the relationship between changes in the ratio and insulin secretion at the study visits (Figure 2D). There was a strong negative correlation between increases in the ratio (compared with the earliest study visit) and decreases in ISR AUC in the progressors (r = –0.475, P = 0.0026) but not the nonprogressors when data from all study visits were used. A significant inverse relationship was also found in the progressors (P = 0.043; nonprogressors, P = 0.457) when the values for each subject were considered together. This indicates that increases in the ratio are associated with β cell killing during the prediabetes period.
The duration of the elevated levels of unmethylated INS DNA reflects both the disease activity and the half-life of the DNA in the serum, which is not known. To address this issue, we measured the appearance and disappearance of unmethylated INS DNA in the sera of 4 patients with chronic pancreatitis who underwent autologous islet transplants following a total pancreatectomy for their primary disease (Figure 3). It has previously been reported that there is a wave of β cell death that occurs immediately after heterotopic allo-islet or auto-islet transplants into the liver (30–32). Within 60 minutes of the transplant, we were able to detect an increase in the ratio, which reached a maximum by 360 minutes in all subjects. On the basis of the decline in the ratio, the average (± SEM) half-life of the unmethylated INS DNA was determined to be 117 ± 37 minutes.
Figure 3. Levels of unmethylated INS DNA in recipients of autologous islet transplants.

The levels of unmethylated INS DNA were measured in 4 individuals with pancreatitis who underwent a pancreatectomy, followed by intraportal transplantation of the autologous islets. The number of islet equivalents (IEQ) that were transplanted is shown.
Because of the short half-life of the unmethylated INS DNA, our studies of at-risk subjects, sampled approximately every 6 months, may have missed episodes of increased β cell death. We therefore also studied PTP participants who were at high risk for progression of T1D, in whom, we postulated, there may be active β cell killing (Table 2). Their designation as “high risk” was based on the presence of at least 2 autoantibodies and “dysglycemia” (i.e., glucose at 15, 30, 60, or 90 minutes >200 mg/dl during an OGTT) or impaired glucose tolerance (i.e., fasting glucose, 110–126 mg/dl, or 2-hour glucose at the end of an OGTT, 140–199 mg/dl). The rate of progression of this group was significantly different from that of the at-risk subjects in the prospective study and was approximately 90% by 7 years (ref. 33 and P < 0.001, Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI78142DS1). These high-risk individuals were selected based on the outcomes of the OGTTs performed on 2 occasions at relatively close intervals. At the second visit, 10 subjects repeated their finding of dysglycemia or IGT (impaired glucose tolerance), 10 subjects had an OGTT result that was diagnostic of diabetes, and 10 had a normal OGTT result. The time intervals between the visits were not significantly different in these 3 subgroups (impaired glucose tolerance: 70 ± 6.96 mg/dl, range: 6–175 days; diabetes: 99 ± 33.8 mg/dl, range: 7–291 days; normal: 108 ± 20.2 mg/dl, range: 22–204 days). The progression to diabetes in other members of these groups of PTP participants is shown in Supplemental Figure 1. The changes in glucose AUC at the 2 OGTTs in these 30 participants are shown in Figure 4C (P < 0.0001). We hypothesized that the level of β cell death at the first visit would predict progression to diabetes or reversion to normal glucose tolerance at the second visit.
Table 2. High-risk PTP participantsA.
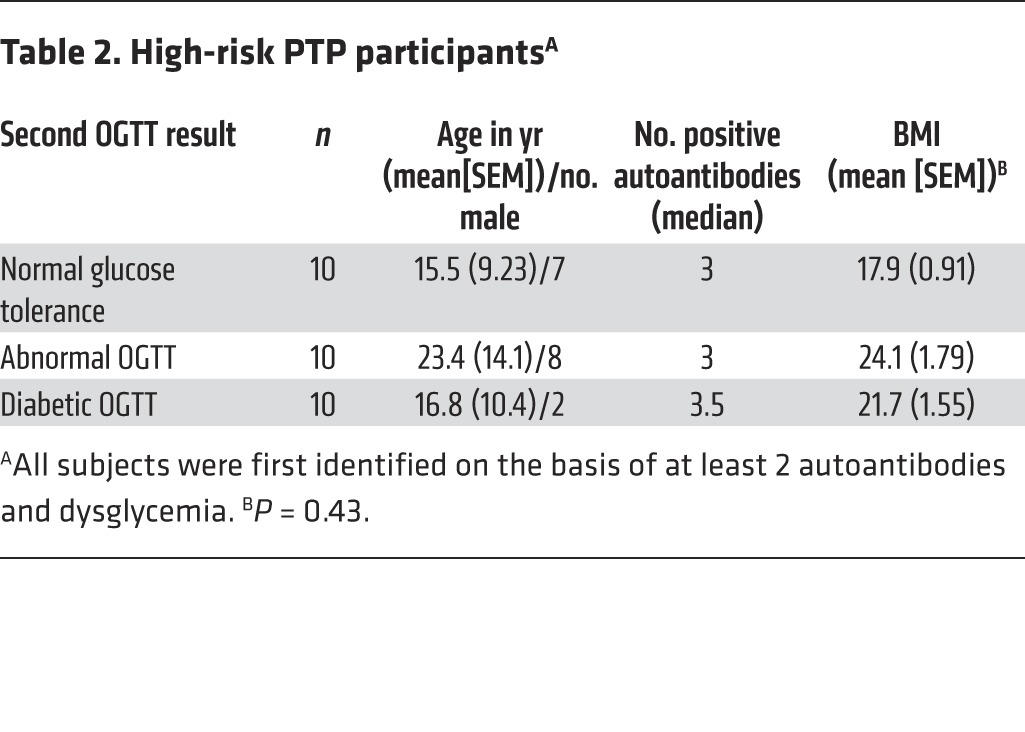
Figure 4. β Cell death and glucose tolerance in high-risk individuals.

(A) The level of unmethylated INS DNA to methylated INS DNA measured by ddPCR in serum samples from the first visit (n = 27; results from 3 samples were unavailable for technical reasons) was compared with age-matched nondiabetic control subjects (HC) (****P < 0.0001, Student’s t test). The dashed line represents the mean + 2 SD of the nondiabetic control subjects (n = 32) is shown. The dashed line represents the mean + 2 SD of the nondiabetic control subjects. (B) Comparison of ratios in high-risk subjects, progressors, and nonprogressors from the PTP study and healthy control subjects. Data shown are the least squares (mean ± SEM) from the mixed model (using ID as a class variable). Ratios were higher in the high-risk group compared with the others (P < 0.0001, ANOVA; ****P < 0.0001). (C) Change in glucose AUC during OGTTs performed on 2 visits in high-risk participants. The second test was performed 94 ± 15 days after the first. Groups were designated by the outcome of the OGTT at the second visit. One-third of subjects had a normal glucose tolerance test when they returned (Nl), one-third repeated the finding of dysglycemia (AbnGT), and one-third showed a diabetic glucose tolerance test (DM). Change in glucose AUC during the 2-hour OGTT is shown (n = 10 each, P = 0.0002, ANOVA; **P < 0.01, ****P < 0.0001, Bonferroni multiple comparison test). (D) Ratios at the time of the first visit are shown for those who returned with a normal (n = 10), an abnormal (n = 8), and a diabetic (n = 9) OGTT and compared with the ratios in controls (mean + SEM; **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. controls, Bonferroni multiple comparison test and ANOVA). Ratios were elevated in each subgroup compared with controls but were not significantly different between groups.
The levels of unmethylated INS DNA were higher in these high-risk subjects compared with those in healthy control subjects or the progressors and nonprogressors that were prospectively followed. At the first visit, 22 of 27 subjects had ratio levels greater than mean + 2 SD of the healthy control subjects (Figure 4A), which is significantly greater than that of the progressors and nonprogressors in the PTP study (P < 0.0001, χ2). Furthermore, in a mixed model, there were significantly higher ratios in the high-risk group compared with those in control subjects or the progressors and nonprogressors followed for longer periods from the PTP study (Figure 4B, P < 0.0001).
While the level of β cell death was higher in each of the 3 subgroups of high-risk subjects than in nondiabetic control subjects, the levels in those who returned with a normal, abnormal, or diabetic OGTT did not differ from each other at the first visit (Figure 4D). Moreover, we did not find a significant difference in the change in the measurement of β cell death when the levels of unmethylated INS DNA were compared between the 2 visits (data not shown).
Analysis of insulin secretory function in at-risk individuals.
These findings indicated that there were increased levels of β cell killing in this high-risk cohort compared with that in subjects at earlier stages of the disease, but the levels of β cell death did not differentiate individuals who rapidly progressed to disease from those whose course was less aggressive. We compared other measurements that may have accounted for changes in insulin sensitivity and deterioration in glucose tolerance, including BMI and the homeostasis model assessment-estimated insulin resistance, but these did not differ between the subgroups (Table 2 and P = 0.74). The interval between the 2 study visits was also similar between the groups. To further understand the basis for progression, we analyzed insulin secretion during the OGTTs.
The total amount of insulin secreted (AUC) was similar among the 3 groups at the first study visit (Figure 5A) and did not change significantly between the 2 visits, even though those individuals who returned with a normal and a diabetic OGTT showed a decrease and increase in the glucose AUC between the 2 visits (Figure 5, A and B, and Supplemental Figure 2).
Figure 5. Insulin secretion during the repeat OGTTs in individuals at high risk for T1D.

(A) The insulin secretory AUC (n = 10 each, mean ± SEM) and (B) change in the AUC (second visit to first visit) are shown for each subgroup. There was not a significant change in the ISR AUC between the 2 visits. (C) The time of the peak rate of insulin secretion (ISR) during the 2-hour OGTT is shown for the first and second visits for the 3 subgroups of high-risk subjects. The time of peak insulin secretion was not significantly different at the first visit, at a time when all participants had an abnormal OGTT. Those who returned with normal glucose tolerance had an earlier time of peak insulin secretion when they returned (the lines represent median values, P = 0.0004, Kruskal-Wallis, overall group comparison; *P < 0.05, ***P < 0.001, Dunn’s multiple comparison test). (D) The percentage of total insulin secreted within the first hour of the second OGTT is shown. The high-risk subjects who returned with normal OGTT secreted a significantly greater proportion of insulin in the first hour compared with those who returned with an abnormal OGTT or diabetic OGTT (P = 0.0005, ANOVA; **P < 0.01, Bonferroni multiple comparisons test) (mean ± SEM).
We previously showed that some patients with T1D have a delayed secretion of insulin in response to a glucose load (27). We therefore compared the kinetics of insulin secretion in the subgroups of subjects. At the time that the high-risk subjects were first studied, the median time of peak insulin secretion during the OGTT was 105 minutes and was similar among the 3 subgroups (Figure 5C). At the first visit, the proportion of insulin secreted within the first hour of the test also did not differ among the 3 subgroups (data not shown). The time of peak secretion was relatively late but consistent with our previous findings (28). At the second visit, subjects who returned with a normal OGTT had peak insulin secretion at an earlier time than those who returned with dysglycemia or a diabetic OGTT (Figure 5C; P = 0.0004, Kruskal-Wallis), and the proportion of the total insulin secreted during the first hour was significantly greater than that in those who returned with an abnormal or diabetic OGTT (Figure 5D, ANOVA P = 0.0005). These results suggest that improvement of metabolic dysfunction in the high-risk individuals is associated with recovery of an early insulin secretory response.
To understand the development of the secretory dysfunction during disease progression, we analyzed, in a similar manner, insulin secretion from the OGTTs in the progressors and nonprogressors shown in Figure 2. Although the ISR AUC did not change during the 4-year follow-up period (Figure 2B), the proportion of insulin secreted during the first hour of the OGTT was decreased significantly (–3.4%, P = 0.04) in the progressors compared with that in the nonprogressors (by repeated-measures ANOVA) (Figure 6). The greatest decline in the percentage of insulin secreted during the first hour (–12.6%) was seen 250 days prior to diagnosis in the progressors (P = 0.03).
Figure 6. Changes in insulin secretory pattern during progression to T1D in PTP participants followed for up to 4 years.

The proportion of insulin secreted in the first hour of the OGTTs shown in Figure 1C is shown. Day 0 was designated the date of diagnosis of T1D in the progressors and the last visit of the nonprogressors. There was a significant difference in the dynamics of insulin secretion in the progressors versus nonprogressors (n = 10 each group, mean ± SEM from the mixed model are shown; P = 0.04, repeated-measures ANOVA). *P = 0.03.
Discussion
It has been known for more than 3 decades that relatives of patients with T1D who progress to T1D may display autoantibodies prior to disease onset. However, the presence of autoantibodies alone does not predict when the disease will occur, and these serologic markers do not directly reflect the pathologic process that leads to T1D, most likely because autoantibodies are not the primary mediators of β cell killing. We measured the levels of unmethylated INS DNA in 50 participants from 2 cohorts in the TrialNet PTP study, using a method involving a quantitative analysis that is specific for epigenetically modified forms of INS DNA. This strategy is based on the notion that unmethylated INS DNA in the serum is derived primarily from β cells that have died and released their cellular contents. Our results show that, over a 4-year observation period, the average levels of unmethylated INS DNA are greater in at-risk relatives who progress to T1D compared with those in healthy control subjects. Twenty-five percent of the measurements in the at-risk progressors are greater than levels found in healthy control subjects, whereas 14% of the levels are elevated in the nonprogressors. Moreover, increases in the levels of unmethylated INS DNA (ratio) were associated with decreased insulin secretion during OGTTs, suggesting that there was a pathologic correlate of the findings from the molecular assay. The increased ratios were sporadic over the 4-year observation period, which likely explains why we did not find a change in the ISR AUC in the progressors over the same time. However, in individuals identified as high risk, β cell killing is significantly increased and persistent. This observation suggests that there is a marked increase in β cell killing late in the progression of disease and close to the time of diagnosis.
Our analysis of the appearance and disappearance of unmethylated INS DNA in islet autotransplant recipients provided a unique opportunity to determine the half-life of the unmethylated INS DNA. We found that the average half-life of unmethylated INS DNA was relatively short: <2 hours. Intrahepatic islet transplantation is known to trigger an instant blood-mediated inflammatory reaction that is initiated by tissue factor and cytokines and chemokines expressed by pancreatic islets in the portal vein environment (30–32). In one of the patients, samples were available before (ratio = 0.14) and after (ratio = 0.125) pancreatectomy, suggesting that the ratios that were found in the healthy control subjects reflected the detection of INS DNA with unmethylated CpG sites from nonpancreatic tissues. The relatively short half-life that we found is consistent with the published information on the survival of human DNA in the circulation based on studies of Y chromosomal DNA from male offspring in their mothers (34). However, this finding also suggests that there may have been episodes of β cell killing in the at-risk cohorts that were followed prospectively that were missed between the sampling every 6 months. Therefore, future studies in at-risk subjects and use of this assay to follow individuals with the disease may require sampling more frequently than every 6 months to track more precisely the progression of β cell killing.
The sporadic findings of elevated ratios in the PTP subjects followed prospectively may not only reflect an infrequent occurrence of β cell killing but also missed episodes because of the sampling schedule in the study. We therefore focused our attention on relatives at high risk for progression. The levels of unmethylated INS DNA were significantly greater in these individuals compared with those in healthy control subjects and with the subjects followed for 4 years. At the time that dysglycemia was found, they had been referred for screening for a prevention trial, which created an opportunity to repeat the analysis of these subjects within about 100 days. The levels were again elevated when the subjects were studied, indicating that, unlike the subjects who had been studied at earlier time points in disease progression, there are persistent high levels of β cell killing within this time interval. (Unfortunately, we were unable to obtain samples over a similar time period in the at-risk subjects followed for 4 years.) Interestingly, a similar pattern was seen in NOD mice during progression to diabetes: the greatest rates of β cell killing were found just prior to diagnosis (15). After hyperglycemia developed, the levels declined.
However, the level of unmethylated INS DNA was not the only factor that led to disease presentation, and the levels were unable to distinguish the high-risk subjects who were progressing quickly to diabetes from those in whom the disease had a more protracted course. Instead, differences in the pattern of insulin secretion were associated with progression to overt disease or transient remission. A similar delay in insulin secretion was found in the PTP participants followed for 4 years, suggesting that there may be ongoing inflammation that leads to function impairment in insulin secretion in the progressors but that β cell killing is sporadic until the peridiagnosis period when it is consistent and at high levels.
In addition to β cell killing, the changes in insulin secretory dynamics suggest that there are revisable chronic cellular stressors during the disease progression. Cytokines that are produced by β cells or infiltrating immune cells, such as IL-1β, TNF, IFN-γ, and IFN family members as well as high glucose, have been associated with cell dysfunction (35–39). Our studies of the PTP subjects followed for long periods of time suggest that these factors may be present for years before disease onset. Of note, the individuals we studied all had normal HbA1c levels, and therefore, glucose alone would not likely be the primary cause. However, it is also known that very modest elevations in blood glucose, levels that could be classified as normal, can cause dysfunction of early insulin release insulin release in response to glucose (40). Clearly, the subjects with dysglycemia have transient elevations in glucose levels, despite normal HbA1c levels. In addition, despite the “remission” of glucose responses during an OGTT, those high-risk individuals who returned with a normal OGTT after dysglycemia still have elevated ratios and retain a >75% risk of developing diabetes in 5 years. Our prospective data from the PTP participants showed a significant delay in insulin secretion, occurring about 250 days prior to diagnosis of T1D in the progressors. The time of the first visit in the high-risk group may be similar to the visit represented as 250 days before diagnosis in those studied for longer periods. The ISR AUC at the time of diagnosis of diabetes in the high-risk subjects who had a diabetic OGTT was greater than that at the time of diagnosis in the prospectively followed progressors (51,733 ± 13,194 pmol vs. 40,464 ± 8,077 pmol, P = NS). It is also possible that the ratio may be affected by the β cell mass itself, since the ratio would be expected to be higher with more cells available for killing.
This is the first study to our knowledge that has measured β cell death during progression of T1D. There are a number of potential limitations of these measurements to consider. We do not know the absolute number of β cells that must die to detect a change in the levels of unmethylated INS DNA. We found that the ddPCR reactions can detect unmethylated INS DNA at concentrations of 1 pg/μl and 1 copy per 2 μl, but the corresponding number of β cells that need to die to achieve these concentrations in the peripheral blood is not clear. Our analysis of transplanted islets suggested a relationship between the number of islets transplanted and the ratio but suggested that a positive signal in the assays is likely to require significant levels of killing. We don’t know the number of β cells that acutely died after transplantation, but we were able to detect an increased ratio when as few as 100,000 islets were transplanted. It is possible, therefore, that a more smoldering process, involving fewer islets, is ongoing in the at-risk subjects but not detectable with our methods in the prediabetic period. Closer sampling intervals might also identify episodes that are missed with sampling every 6 months. In addition, if there was any degradation of DNA with time in storage, it might affect the ratio, since the concentration of unmethylated INS DNA is considerably lower than that of methylated INS DNA.
In addition, the significance of the background levels that are detected in healthy control subjects needs interpretation. We have performed high-throughput epigenetic analyses of the Ins1 and Ins2 genes in non–β cells in mice and found that the rate of methylation across 28 sites was between 70% and 84%, consistent with findings reported by Husseiny et al. (41). In one of the islet autotransplant recipients, we measured the ratio before and after pancreatectomy, and these levels were similar (0.14 and 0.125, respectively). The increased levels following transplantation, and the metabolic outcomes that were associated with increases in the ratio in the at-risk progressors, indicates that the unmethylated INS DNA detected is coming from β cells and is associated with their death. The ratios that are found in healthy individuals most likely represent the “background” levels of unmethylated CpG sites in the INS gene at non–β cells.
There are a number of factors in our study design that warrant caution in the broader application of our findings. We analyzed a selected group of a limited number of subjects, all of whom were relatives of patients with T1D. The high-risk individuals were older than the subjects that were followed in the prospective study. Therefore, the inclusion of individuals aged >20 years in which the disease progression is slower may have affected the studies of metabolic function as well as their insulin secretory capacity (42). The designation of individuals as nonprogressors can only be applied to the duration of this analysis — some of the nonprogressors may eventually develop diabetes.
In summary, our studies of β cell death and dysfunction indicate that both can be found before the onset of T1D. There are detectable episodes of β cell killing associated with a decline in insulin secretion in the prediabetes period, but there is a dramatic increase in killing in the peridiagnosis period. Metabolic dysfunction, which may be due to factors that do not directly cause killing, also is found prior to diagnosis. When both consistent cytotoxicity and metabolic dysfunction occur, metabolic decompensation ensues.
Methods
Study subjects.
Serum samples were obtained from 2 cohorts of participants in the TrialNet Natural History PTP study (Clinical Trials.gov NCT00097292). These subjects were designated as “at risk” (Figure 1). Relatives of patients with T1D were enrolled in the PTP study if they had a positive autoantibody (anti-GAD65, anti-insulin, anti-ICA512, anti-ZnT8, or ICA). They were studied approximately every 6 months. Ten subjects who developed T1D were identified (progressors), and 10 who did not develop T1D were identified (nonprogressors). Of the 10 progressors, 2 were followed for up to 550 days, 3 were followed for up to 900 days, 3 were followed for up to 1,000 days, and 2 were followed for up to 1,300 days. Of the nonprogressors, 1 was followed for up to 550 days, 2 were followed for up to 900 days, 5 were followed for up to 1,000 days, and 2 were followed for up to 1,300 days (P = NS). All of the subjects had a normal HgA1c level and did not have symptoms of diabetes. The samples were obtained between 6/22/2005 and 3/7/2013.
We also studied 30 PTP participants who were identified as at “high risk” for development of T1D. These participants had at least 2 biochemical autoantibodies and dysglycemia during a 2-hour OGTT (defined as either a fasting glucose between 100 and 125 mg/dl, a 2-hour glucose between 140 and 200 mg/dl, or an intermediate glucose >200 mg/dl). This high-risk cohort was studied on 2 occasions approximately 100 days apart. At the time of repeat testing, 10 of the subjects had a normal OGTT, an OGTT that fulfilled the diagnosis of T1D (i.e., fasting glucose, >125 mg/dl, or 2-hour glucose, >200 mg/dl), or repeat dysglycemia. These subjects also had a normal HbA1c level and were asymptomatic. The samples were obtained between 12/27/2010 and 11/15/2013. All samples were acquired and selected by the TrialNet Coordinating Center. The investigative staff were blinded to the group assignments in each analysis.
Finally, we obtained serum samples from 4 individuals who underwent islet autotransplantation at the University of Minnesota following pancreatectomy for chronic pancreatitis. The primary diseases for these subjects were cystic fibrosis (Pt 1), PRSS1 mutation (Pt 4), and idiopathic (Pts 2 and 3). Sera from age-matched nondiabetic control subjects were obtained from the clinical laboratory at the Yale New Haven Hospital.
Analysis of β cell death.
DNA was purified from 200 μl serum using QIAamp DNA Blood Kits, as suggested by the manufacturer (Qiagen), with a modified incubation period of 20 minutes at 45°C in the final step. DNA was bisulfite treated using the EZ DNA Methylation Kit (Zymo Research).
We measured the levels of unmethylated INS DNA by ddPCR (16). Each 25-μl volume consisted of Droplet PCR Supermix (Bio-Rad), 900 nM of primer, 250 nM of probe, 5 μl of sample. The mixture and droplet generation oil were loaded onto a droplet generator (Bio-Rad), and the generated droplets were transferred to a 96 well PCR plate. The PCR reaction was run on a thermal cycler with 10 minutes activation at 95°C, 40 repetitions of a 2-step amplification protocol (30 seconds at 94°C denaturation and 60 seconds at 58°C), and a 10-minute inactivation step at 98°C. The DNA content of the droplets were analyzed with a QX100 Droplet Reader (Bio-Rad) and QuantaSoft (Bio-Rad) Analysis software. Discrimination between droplets that contained the target (positives) and those which did not (negatives) was achieved by applying a fluorescence amplitude threshold based on the amplitude read from the negative template control. For each sample, the ratio of unmethylated INS DNA to methylated INS was calculated.
Calculation of insulin secretion.
Plasma C-peptide levels were measured at Northwest Lipid Metabolism and Diabetes Research Laboratories using the Tosoh AIA 1800 assay. The lower limit of detection was 0.017 nmol/l, with intra- and interassay coefficients of variation of 1.71% and 4.68%, respectively. The C-peptide levels obtained during oral glucose tolerance tests were deconvoluted using a 2-compartment model for hormone clearance with Chronobiological Series Analyzer software (27, 43–45). Standard kinetic parameters for C-peptide were used based on the findings of Van Cauter et al., who estimated rate constants based on extrapolations from C-peptide decay curves of 200 subjects (45). The parameters that were used accounted for the patient’s age, sex, height, and weight.
To calculate the AUC of insulin secreted, a trapezoidal rule was applied. The percentage of insulin secreted in the first hour of the OGTT was calculated by dividing the insulin secreted within the first hour of the test by the total insulin secreted (×100).
Statistics.
The sample sizes were based on measurements of differences of unmethylated INS DNA in subjects with new-onset T1D that we have reported previously (14–16). Unless indicated otherwise, the data are presented as mean ± SEM. To align comparisons, studies performed on the Natural History Study cohort were grouped according to intervals of 250 days (one-half interval = 125 days) prior to the development of T1D for the progressors or prior to the last study visit for the nonprogressors. The time of entry into the follow-up study varied for subjects, but all subjects were followed for at least 567 days. All subjects reached the last study visit. Comparisons between 2 groups were made with Student’s t test. For comparisons between more than 2 groups, an ANOVA with Bonferroni corrections for multiple comparisons or a Kruskal-Wallis test for nonparametric data was used. Analyses over time were performed by repeated-measures ANOVA with mixed models. No adjustments were made for missing data. There were 5–19 values for each time point used in the model, depending on the subjects’ time of entry into the study and the day prior to the final visit. In the analysis of the ratio between PTP progressor and nonprogressors, however, basic statistical assumptions required for repeated-measures analysis were not met, and so the means from each individual were compared using ANOVA. Subsequent bootstrapping analysis confirmed these findings. Some analyses of the change in measurements from the baseline values (i.e., the first study visit) for each individual were performed. The times of peak insulin secretion were identified from the calculations of the insulin secretion rates during the OGTT, and times of peak insulin were compared between groups at time points by χ2 test. All statistical comparisons were 2 sided, and a P value of less than 0.05 was considered to be statistically significant. Statistical analyses were performed with GraphPad/Prism 5 or SAS 9.4.
Study approval.
Institutional review board approval was obtained for these studies at each study site (see Supplemental Methods for a full list of the study sites). All subjects gave written consent, with guardians providing consent for subjects under age 18 years. The use of study samples from TrialNet and the University of Minnesota was considered nonhuman research and exempt from study approval.
Supplementary Material
Acknowledgments
This study was supported by NIH grants DP3DK101122, R01DK057846, R41DK095639, U01DK085466, and R03DK102469 and JDRF grant 2012-546. The Type 1 Diabetes TrialNet Study Group is a clinical trials network funded by the NIH through the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development through the cooperative agreements U01 DK061010, U01 DK061016, U01 DK061034, U01 DK061036, U01 DK061040, U01 DK061041, U01 DK061042, U01 DK061055, U01 DK061058, U01 DK084565, U01 DK085453, U01 DK085461, U01 DK085463, U01 DK085499, U01 DK085505, and U01 DK085509 as well as contract HHSN267200800019C; National Center for Research Resources awards UL1 RR024131, UL1 RR024139, UL1 RR024153, UL1 RR024975, UL1 RR024982, UL1 RR025744, UL1 RR025761, UL1 RR025780, UL1 RR029890, UL1 RR031986, and P30 DK017047; General Clinical Research Center award M01 RR00400; and JDRF and American Diabetes Association. Kevan C. Herold represents the Type I Diabetes TrialNet Study Group.
Footnotes
Conflict of interest: Kevan C. Herold and Sahar Usmani-Brown have patent applications for assays to measure β cell death.
Reference information:J Clin Invest. 2015;125(3):1163–1173. doi:10.1172/JCI78142.
References
- 1.Herold K, Vignali DA, Cooke A, Bluestone J. Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol. 2013;13(4):243–256. doi: 10.1038/nri3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464(7293):1293–1300. doi: 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol. 2007;7(12):988–994. doi: 10.1038/nri2192. [DOI] [PubMed] [Google Scholar]
- 4.Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314(21):1360–1368. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- 5.Herold KC, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes. 2013;62(11):3766–3774. doi: 10.2337/db13-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold KC, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54(6):1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herold KC, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 8.Keymeulen B, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 9.Orban T, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–419. doi: 10.1016/S0140-6736(11)60886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pescovitz MD, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rigby MR, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Endocrinology Metabolism. 2013;1(4):284–294. doi: 10.1016/S2213-8587(13)70111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orban T, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32(12):2269–2274. doi: 10.2337/dc09-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vehik K, et al. Development of autoantibodies in the TrialNet Natural History Study. Diabetes Care. 2011;34(9):1897–1901. doi: 10.2337/dc11-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebastchi J, et al. Immune therapy and β-cell death in type 1 diabetes. Diabetes. 2013;62(5):1676–1680. doi: 10.2337/db12-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akirav EM, et al. Detection of beta cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci U S A. 2011;108(47):19018–19023. doi: 10.1073/pnas.1111008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Usmani-Brown S, Lebastchi J, Steck AK, Beam C, Herold KC, Ledizet M. Analysis of β-cell death in type 1 diabetes by droplet digital PCR. Endocrinology. 2014;155(9):3694–3698. doi: 10.1210/en.2014-1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sosenko JM, et al. Increasing the accuracy of oral glucose tolerance testing and extending its application to individuals with normal glucose tolerance for the prediction of type 1 diabetes: the Diabetes Prevention Trial-Type 1. Diabetes Care. 2007;30(1):38–42. doi: 10.2337/dc06-1615. [DOI] [PubMed] [Google Scholar]
- 18.Sosenko JM, et al. Glucose and C-peptide changes in the perionset period of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2008;31(11):2188–2192. doi: 10.2337/dc08-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu P, et al. Prognostic performance of metabolic indexes in predicting onset of type 1 diabetes. Diabetes Care. 2010;33(12):2508–2513. doi: 10.2337/dc10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sosenko JM, et al. Patterns of metabolic progression to type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2006;29(3):643–649. doi: 10.2337/diacare.29.03.06.dc05-1006. [DOI] [PubMed] [Google Scholar]
- 21.Donath MY, Storling J, Maedler K, Mandrup-Poulsen T. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med. 2003;81(8):455–470. doi: 10.1007/s00109-003-0450-y. [DOI] [PubMed] [Google Scholar]
- 22.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39(9):1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 23.Mandrup-Poulsen T, Bendtzen K, Nerup J, Dinarello CA, Svenson M, Nielsen JH. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia. 1986;29(1):63–67. doi: 10.1007/BF02427283. [DOI] [PubMed] [Google Scholar]
- 24.Spinas GA, et al. Interleukin 1 dose-dependently affects the biosynthesis of (pro)insulin in isolated rat islets of Langerhans. Diabetologia. 1987;30(7):474–480. doi: 10.1007/BF00279615. [DOI] [PubMed] [Google Scholar]
- 25.Breda E, Toffolo G, Polonsky KS, Cobelli C. Insulin release in impaired glucose tolerance: oral minimal model predicts normal sensitivity to glucose but defective response times. Diabetes. 2002;51(suppl 1):S227–S233. doi: 10.2337/diabetes.51.2007.s227. [DOI] [PubMed] [Google Scholar]
- 26.Del Prato S, Marchetti P, Bonadonna RC. Phasic insulin release and metabolic regulation in type 2 diabetes. Diabetes. 2002;51(suppl 1):S109–S116. doi: 10.2337/diabetes.51.2007.s109. [DOI] [PubMed] [Google Scholar]
- 27.Steele C, et al. Insulin secretion in type 1 diabetes. Diabetes. 2004;53(2):426–433. doi: 10.2337/diabetes.53.2.426. [DOI] [PubMed] [Google Scholar]
- 28.Sosenko JM, Skyler JS, Herold KC, Palmer JP. The metabolic progression to type 1 diabetes as indicated by serial oral glucose tolerance testing in the Diabetes Prevention Trial-type 1. Diabetes. 2012;61(6):1331–1337. doi: 10.2337/db11-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polonsky KS, et al. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest. 1988;81(2):435–441. doi: 10.1172/JCI113338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennet W, et al. Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation? Diabetes. 1999;48(10):1907–1914. doi: 10.2337/diabetes.48.10.1907. [DOI] [PubMed] [Google Scholar]
- 31.Moberg L, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. 2002;360(9350):2039–2045. doi: 10.1016/S0140-6736(02)12020-4. [DOI] [PubMed] [Google Scholar]
- 32.Carlsson PO. Influence of microenvironment on engraftment of transplanted β-cells. Ups J Med Sci. 2011;116(1):1–7. doi: 10.3109/03009734.2010.548609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab. 2008;4(6):334–343. doi: 10.1038/ncpendmet0832. [DOI] [PubMed] [Google Scholar]
- 34.Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64(1):218–224. doi: 10.1086/302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eizirik DL, Darville MI. β-Cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes. 2001;50(suppl 1):S64–S69. doi: 10.2337/diabetes.50.2007.s64. [DOI] [PubMed] [Google Scholar]
- 36.Strandell E, Eizirik DL, Sandler S. Reversal of β-cell suppression in vitro in pancreatic islets isolated from nonobese diabetic mice during the phase preceding insulin-dependent diabetes mellitus. J Clin Invest. 1990;85(6):1944–1950. doi: 10.1172/JCI114657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabinovitch A, Baquerizo H, Sumoski W. Cytotoxic effects of cytokines on islet beta-cells: evidence for involvement of eicosanoids. Endocrinology. 1990;126(1):67–71. doi: 10.1210/endo-126-1-67. [DOI] [PubMed] [Google Scholar]
- 38.Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J Clin Endocrinol Metab. 1990;71(1):152–156. doi: 10.1210/jcem-71-1-152. [DOI] [PubMed] [Google Scholar]
- 39.Diana J, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. 2013;19(1):65–73. doi: 10.1038/nm.3042. [DOI] [PubMed] [Google Scholar]
- 40.Brunzell JD, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42(2):222–229. doi: 10.1210/jcem-42-2-222. [DOI] [PubMed] [Google Scholar]
- 41.Husseiny MI, Kaye A, Zebadua E, Kandeel F, Ferreri K. Tissue-specific methylation of human insulin gene and PCR assay for monitoring beta cell death. PLoS One. 2014;9(4):e94591. doi: 10.1371/journal.pone.0094591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenbaum CJ, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes. 2012;61(8):2066–2073. doi: 10.2337/db11-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polonsky KS, et al. Use of biosynthetic human C-peptide in the measurement of insulin secretion rates in normal volunteers and type I diabetic patients. J Clin Invest. 1986;77(1):98–105. doi: 10.1172/JCI112308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherry NA, Tsai EB, Herold KC. Natural history of β-cell function in type 1 diabetes. Diabetes. 2005;54(suppl 2):S32–S39. doi: 10.2337/diabetes.54.suppl_2.s32. [DOI] [PubMed] [Google Scholar]
- 45.Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes. 1992;41(3):368–377. doi: 10.2337/diab.41.3.368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.