

Abstract
Effective colonic motility involves an intricate pattern of excitatory and inhibitory neuromuscular signals that arise from the enteric neural circuitry of the colon. Recent investigations have demonstrated that inflammation leads to a variety of changes in the physiological properties of the neurons in this circuitry, including hyperexcitability of neurons at the afferent end of the peristaltic reflex, synaptic facilitation, and attenuated inhibitory neuromuscular transmission. Furthermore, links have been established between these changes and disrupted motor activity in the colon, and we now know that some of these changes persist long after recovery from inflammation. It is highly likely that inflammation-induced neuroplasticity, which is not detectable by clinical diagnostics, contributes to disrupted motility in active and quiescent inflammatory bowel disease and in functional gastrointestinal disorders.
Introduction
The nervous and immune systems represent two of the final frontiers in our attempts to understand how complex organisms function, and it is becoming clear that these two systems interact extensively in health and disease. For example, neurons have the ability to upregulate or downregulate the extent of inflammatory responses, and pro-inflammatory mediators can act on nearby nerves to mediate short- and long-term changes in neuronal function.
Intestinal functions are regulated by the most complex system of nerves outside of the CNS. During the past decade, significant research efforts have been directed toward unraveling the relationship between the colonic inflammatory response and neuronal functions in the gut. Inflammation-induced changes have been reported in neurons in dorsal root ganglia, sympathetic prevertebral ganglia, and enteric ganglia. This Review concentrates on those components of the enteric nervous system (ENS) that regulate propulsive motility and explores how propulsive motility is altered through inflammation-induced neuroplastic changes in the colon secondary to intestinal inflammatory disorders such as inflammatory bowel disease (IBD) and enterocolitis.
Neuronal circuitry that underlies propulsive motility
Over a century ago, Bayliss and Starling demonstrated that the innervation of the intestines is unique compared with other organ systems (1). They showed that stimulation of the mucosal surface in one location results in contraction proximal and relaxation distal to the site of stimulation. They called this phenomenon the “law of the intestine” and attributed this complicated reflex response to the actions of the “local nervous mechanism.” Based on this knowledge that intrinsic reflex circuits can regulate the functions of the intestine, Langley subsequently divided the autonomic nervous system into three parts — parasympathetic, sympathetic, and enteric, with the enteric division comprising the nerves housed within the wall of the gastrointestinal tract (2).
During the last two decades of the twentieth century, significant effort was directed toward identifying the neuronal elements of the reflex circuits that are responsible for peristaltic propulsion of luminal contents and active secretion of water in the intestines (3–5). Compared with the peristaltic reflex circuit, the circuitry responsible for secretion is relatively simple. Active secretion is housed in the submucosal plexus and involves a two-neuron reflex that includes an afferent neuron, which can be activated by secretory products of enteroendocrine cells (e.g., serotonin secreted by enterochromaffin [EC] cells), and a secretomotor neuron that projects into the lamina propria.
The reflex circuitry that mediates propulsive motility includes ascending and descending limbs that lead to proximal contraction and distal relaxation, as described by Bayliss and Starling (Figure 1 and ref. 1). Like the secretory reflex, the motor reflex is initiated by the activation of an afferent neuron by enteroendocrine secretions and/or by stretch. Interneurons projecting upstream in the intestine selectively form synapses with excitatory motor neurons that cause contraction of the smooth muscle via the release of acetylcholine and tachykinins. Interneurons that project downstream in the gut cause relaxation of the smooth muscle via the release of nitric oxide and purines. Collectively, the upstream contraction and downstream relaxation results in a pressure gradient that propels the luminal contents in an aboral direction.
Figure 1. The basic elements of the peristaltic reflex circuit in the colon and changes detected at specific sites of this circuitry in colitis.

(i) A stimulus in the lumen, such as stretch or distension, results in the release of serotonin by the epithelial cells in the gut mucosa. (ii) The serotonin and mechanical stretch activate afferent neurons. These neurons, in turn, activate sensory afferent fibers that send projections into the submucosal and myenteric plexuses. This in turn activates two limbs of the peristaltic reflex. (iii) Upstream of the stimulus, interneurons synapse with and activate excitatory motor neurons that release acetylcholine and tachykinins to trigger smooth muscle contractions. (iv) Downstream of the stimulus, interneurons synapse with and activate inhibitory motor neurons that release NO and purines to trigger smooth muscle relaxation. The combination of upstream contraction and downstream relaxation creates a pressure gradient that drives the luminal contents from the proximal to the distal end of the gut. Colitis is characterized by increased serotonin availability, increased excitability of afferent neurons, facilitation of interneuronal synaptic activity, decreased purinergic neuromuscular transmission, and loss of myenteric neurons, which together lead to a mixture of overlapping excitatory and inhibitory motor outputs in the inflamed region (see Figure 2). EEC, enteroendocrine cell.
The neurons that serve at the afferent end of intrinsic reflex circuits in the gut are called AH neurons because one of their characteristic electrical properties is a prolonged afterhyperpolarization (AHP). AH neurons are sensitive to stretch, and they project to the lamina propria, where they can receive the chemical signals released from enteroendocrine cells. As a result of their functions, these neurons have been called intrinsic primary afferent neurons (6); however, some researchers feel that they do not fit the formal definition of primary afferent/sensory neurons and prefer to call them interneurons, as they form an AH neuron synaptic network (7). Here they are referred to as AH neurons. The other neurons in the propulsive motility circuitry are classified as S neurons electrophysiologically, but they can be distinguished by their projection patterns and chemical coding properties (3, 5). The ability to identify the individual components of the peristaltic reflex circuit, particularly in the guinea pig, has allowed for a systematic evaluation of inflammation-induced changes occurring along this reflex pathway, the mechanisms responsible for these changes, and the impact of these changes on propulsive motility.
Inflammation-induced changes in colonic motor circuitry
AH neurons.
Because AH neurons project to the lamina propria of the intestine, where inflammatory responses are centered, they are particularly susceptible to the effects of pro-inflammatory signaling molecules in the inflamed gut. Indeed, to date, all studies of inflammation-induced changes to enteric neurons have reported alterations in the electrical properties of AH neurons. The locations and forms of inflammation that have been studied include myenteric (8) and submucosal (9) neurons in the colon inflamed by trinitrobenzene sulfonic acid (TNBS), myenteric neurons in the TNBS-inflamed ileum (10), and ileal myenteric neurons in animals with Trichinella spiralis–induced enteritis (11, 12). Furthermore, submucosal AH neuron excitability is enhanced when tissue from β-lactoglobulin–sensitized guinea pigs is exposed to the milk protein (13, 14).
The mechanisms responsible for inflammation-induced AH neuron hyperexcitability have not been completely resolved, but they appear to vary depending on the location and type of inflammatory response. A consistent feature of inflammation-induced neuroplasticity in AH neurons is a reduction in the magnitude of the AHP, which is important in limiting the excitability of these neurons. In TNBS colitis, a major contributor to the hyperexcitability in AH neurons is an increase in a hyperpolarization-activated cation conductance (8). In this model, the AH neuron hyperexcitability involves activation of COX-2 because chronic exposure to prostaglandin E2 results in AH neuron hyperexcitability in normal preparations (15) and because inhibition of COX-2, but not COX-1, prevents AH neuron hyperexcitability in animals with TNBS colitis (16).
In T. spiralis enteritis, the AH neuron membrane potential is depolarized and the neurons exhibit a lower input resistance (11, 12). Both of these features, along with the decreased AH, indicate that a decrease in the activity of the intermediate conductance Ca2+-activated K+ channel (IKCa) in these cells is involved in the increase in excitability in this condition. In these neurons the cAMP/pCREB signaling pathway is facilitated and acute administration of histaminergic receptors, COX enzymes, and leukotriene inhibitors dampens hyperexcitability (11). Taken together, these reports indicate that hyperexcitability of AH neurons is a common feature of inflammatory conditions in the intestines.
Interneurons and motor neurons.
Neurons with the electrical properties classified as S neurons include neurons that function as interneurons and motor neurons, as well as S neurons that likely have a role as sensory neurons because they are sensitive to mechanical stimulation (3). The changes in electrical properties of myenteric S neurons in TNBS colitis are more subtle than those of AH neurons, and no significant changes are detected when considered as a single population (8). However, when the S neurons are divided into two subpopulations — those with ascending projections and those with descending projections — the S neurons with ascending projections exhibit a slight increase in excitability. While the electrical properties of colonic S neurons are not dramatically changed in response to colitis, the strength of interneuronal synaptic potentials is significantly increased in both the submucosal and myenteric plexuses of the TNBS-inflamed colon (8, 9, 17). In submucosal ganglia, synaptic facilitation appears to involve a pharmacological transformation from synaptic potentials mediated by nicotinic cholinergic transmission to a combination of nicotinic, purinergic, and in some cases serotonergic transmission (9). In the myenteric plexus, colitis-induced synaptic facilitation does not involve altered pharmacology, but rather an increase in the readily releasable pool of neurotransmitters available in the nerve terminals and a sustained activation of protein kinase A, which likely increases Ca2+ influx into the nerve terminals (17).
Neuromuscular transmission.
The status of neuromuscular transmission in the inflamed colon has been investigated in TNBS colitis. In an intracellular recording study of synaptic junction potentials from circular muscle cells, Strong and colleagues demonstrated that the ascending excitatory neuromuscular transmission was unchanged in the inflamed region, whereas the descending inhibitory neuromuscular transmission was dampened (18). Pharmacological isolation of the nitrergic and purinergic contributions to the inhibitory junction potential (IJP) demonstrated that nitrergic neuromuscular transmission is normal in colitis, whereas the purinergic component was significantly attenuated.
Since nitrergic and purinergic neuromuscular signals likely arise from the same nerve terminals in the intestines, the finding that purinergic IJPs are disrupted but nitrergic IJPs are normal was a bit perplexing. A recent investigation has shed light on this dilemma by demonstrating that the disruption of the purinergic IJP likely involves an interruption of purine production in the mitochondria due to oxidative stress in the inflamed region (19). IJPs in non-inflamed preparations are attenuated when the tissue is exposed to free radical donors or ATP synthase inhibitors, and neurally mediated purine release is decreased in inflamed preparations. In both guinea pig TNBS colitis and murine DSS colitis, treatment of animals with a free radical scavenger protects purinergic IJP. Addition of tempol to the drinking water during the development of colitis prevents oxidative stress in the tissue and maintains the purinergic IJPs at a normal amplitude even though the inflammatory scores of the colons are comparable to those not treated with the free radical scavenger. This disruption in the descending, inhibitor limb of the peristaltic reflex could affect propulsive motility by decreasing the extent of relaxation that occurs in the receiving segment of the gut during peristalsis.
To summarize, findings described above indicate that a number of changes are occurring in the intrinsic motor circuitry of the colon in response to inflammation (Figure 1). These include the following: (a) an increase in serotonin availability due to increased numbers of EC cells and decreased serotonin-selective reuptake transporter (SERT) levels; (b) an increase in the excitability of AH neurons that can result from a decrease in IKCa function and/or an increase in hyperpolarization-activated cation current (Ih); (c) a facilitation of interneuronal synaptic activity due to an increase in the readily releasable pool of neurotransmitters in myenteric nerve terminals; (d) a decrease in purinergic neuromuscular transmission due to an oxidative stress–induced decrease in purine release from the terminals of inhibitory motor neurons. In addition, colitis is associated with a loss of about 20% of the myenteric neurons (20, 21).
Linking inflammation-induced neuroplasticity to disrupted motility
The questions of whether and how these inflammation-induced changes in the neural circuitry affect motility has been studied using an ex vivo guinea pig colon propulsive motility assay that provides the investigator with an opportunity to run numerous trials and evaluate the effects of test compounds that interact with receptors and ion channels. Initial studies simply involved measuring the amount of time it took for a fecal pellet to progress along a segment of normal versus TNBS-inflamed distal colon and demonstrated that the velocity of propulsive motility was slower in the inflamed preparations (22). Subsequent studies that used a Gastrointestinal Motility Monitor (23) to evaluate the progress of the fecal pellet along the segment of colon demonstrated that propulsive motility was completely obstructed or temporarily halted in heavily inflamed regions (18, 24).
Attention deficit disorder in the ENS.
Given that major neurophysiological changes induced by colitis are associated with increased neuronal activity and enhanced synaptic transmission, one might intuit that these changes would be associated with enhanced reflex activity and accelerated motility. However, another possibility is that the increased neuronal activity in inflamed regions results in a form of attention deficit disorder in the ENS. Under normal, healthy conditions, the background activity in the ENS motor circuitry is thought to be rather quiescent due to the relative inexcitability of AH neurons. Thus, in response to a luminal stimulus or stretch at one site, there is an activation of AH neurons at that level of the gut, leading to unambiguous signals passing proximally and distally to contract and relax the bowel, respectively. As the bolus is propelled in the distal direction, this response repeats itself, resulting in a propagating wave motility pattern (Figure 2A).
Figure 2. Neuronal activity in a segment of colon under resting conditions and during peristalsis.
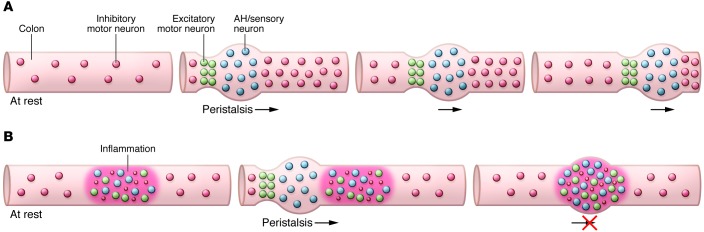
(A) Healthy tissue. In healthy tissue under resting conditions, spontaneous inhibitory neuromuscular transmission maintains the tissue in a slightly relaxed state. During peristalsis, chemical and or mechanical stimulation leads to activation of AH/sensory neurons at the site of the stimulation. These neurons, along with interneurons, transmit signals proximally to activate excitatory motor neurons and distally to activate inhibitory motor neurons. The result is a pressure gradient that propels the luminal contents distally, and as the sequence repeats itself, a wave of peristalsis is generated. (B) Inflamed tissue. In inflamed regions, AH/sensory neurons are spontaneously active and synaptic activity is augmented, leading to overlapping ascending excitatory and descending inhibitory signals throughout the inflamed region. Also, inhibitory neuromuscular transmission is attenuated, as represented by the smaller red dots. When a propulsive wave reaches the inflamed region, peristalsis is disrupted by the inability to overcome the mixed signals being received due to the spontaneous neuronal activity and the suppressed neuromuscular transmission in the inflamed region.
In inflamed regions of the colon, about 50% of the AH neurons exhibit spontaneous action potentials. This likely leads to the activation of ascending contractile and descending inhibitory signals arising from multiple regions along the inflamed segment of bowel. This aberrant neuronal behavior may then result in mixed excitatory and inhibitory messages converging on smooth muscle throughout the inflamed region — a form of intestinal fibrillation.
To test whether enhanced neuronal activity in the myenteric plexus would slow rather than accelerate propulsive motility, segments of guinea pig distal colon were treated with the opener of voltage-activated Na+ channels, veratridine, which at a concentration of 1 μM causes enteric neurons to generate action potentials repetitively (25). When veratridine was added to the bathing solution, forward progress of the pellet was obstructed even though muscle contractions were still clearly observed (24). Even though there was abundant neuronal and muscle activity in the tissue, it was not possible to generate an adequate pressure gradient to move the fecal pellet forward.
Links between neuroplasticity and disrupted motility in colitis.
Strong support for the concept that neuroplastic changes in colitis that are associated with increased neuronal activity contribute to disrupted propulsive motility comes from experiments in which similar neuroplastic changes were induced pharmacologically in normal colons or were reversed in TNBS-inflamed preparations (24). In normal colons, the velocity of propulsive motility is significantly decreased when IKCa channels are inhibited, resulting in an increase in AH neuron excitability. The velocity is further reduced when interneuronal synaptic activity is enhanced with a 5-hydroxytryptamine type 4 (5-HT4) receptor agonist (24, 26). Thus, by mimicking the AH neuronal hyperexcitability and synaptic facilitation features of colitis, motility is inhibited rather than accelerated.
Propulsive motility is restored in inflamed preparations when major features of neuroplasticity are reversed or prevented. As described above, in TNBS colitis, AH neuron hyperexcitability involves activation of COX-2 and an increase in the Ih current. The rate of colonic motility is significantly higher in TNBS-inflamed colons from animals that are treated daily with a COX-2 inhibitor (16). Consistent with this, propulsive motility is dramatically improved in TNBS-inflamed colons following application of hyperpolarization-activated cyclic nucleotide–gated (HCN) channel blockers to inhibit the Ih in AH neurons (24). In these experiments, obstructed or halted motility patterns were converted to relatively linear motility patterns, with the fecal pellets traversing the inflamed region without pausing. Motility is also improved in TNBS-inflamed animals that had been treated with a free radical scavenger to protect the purinergic IJP from the effects of oxidative stress, and when HCN channel blockers are applied to these colons, motility is further improved to a healthy control level.
Taken together, these findings underscore the delicate and complicated nature of the peristaltic reflex circuitry, and they demonstrate that overstimulation of the system can actually lead to disruptions in propulsive motility. These studies also demonstrate that even though mucosal erosion, muscularis hypertrophy, and inflammation are still present, propulsive motility can be protected or reestablished when neuronal activity is restored to its normal level.
Persistent postinflammatory changes in enteric neurons and motility
Understanding the changes in neuronal function during active inflammatory responses is important in the context of linking these changes with altered gut functions as described above. It is also important to consider which aspects of inflammation-induced neuroplasticity persist following recovery from the inflammatory response. This is a relevant question because alterations in gut function are often sustained during remission of IBD (27–31), and there is mounting evidence to suggest that irritable bowel syndrome (IBS) and other functional gastrointestinal disorders involve an inflammatory component (27, 32–35). In fact, in a large contingent (~35%) of diarrhea-predominant IBS patients, a careful history reveals that the symptoms began during a bout of infectious enteritis (36), and these individuals are described as having post-infectious IBS. It is important to note that the types of inflammation-induced neuronal changes that have been identified (i.e., changes in neuronal excitability, ion channel function, and synaptic strength) are invisible in any clinical test. Thus, inflammation-induced neuroplasticity that outlasts the inflammatory response offers a mechanism that could be responsible for hard-to-explain, persistent symptoms experienced during remission from inflammation in IBD or associated with IBS.
T. spiralis.
Myenteric neurons have been studied with electrophysiological techniques at “post-inflammatory” time points in T. spiralis- and TNBS-inflamed animals, and while AH neuronal hyperexcitability was detected in both of these models, the mechanisms appear to differ. Christofi and colleagues examined the excitability of jejunal AH neurons at a time point of 35 days post-infection, by which time the parasite would have been cleared from the gut (11). Strictly speaking, this model is probably best described as a post-infectious model rather than a post-inflammatory model because evidence of inflammation is still detectable in the wall of the gut, including elevated myeloperoxidase, mast cell tryptase, and prostaglandin levels. Regardless, AH neuron hyperexcitability in this model likely involves post-translational changes in ion channel function because it is reversed by acute exposure to blockers of adenylyl cyclase, histamine, cyclooxygenase, or leukotriene pathways. However, long-term transcriptional changes may also contribute to persistent neuroplasticity in these cells because elevations in adenylyl cyclase and CREB phosphorylation levels are still detectable 35 days after infection.
TNBS colitis.
When considering the TNBS colitis model, it is important to recall that six days after administration, during the active phase of the inflammatory response, AH neurons are rendered hyperexcitable through a COX-2–dependent mechanism, interneuronal synaptic activity is augmented, and purinergic transmission is blunted. In this model, macroscopic damage scores return to normal 14 days after TNBS administration, and all signs of inflammation, including elevated myeloperoxidase and prostaglandin levels, have disappeared by 28 days (37). At 56 days (four weeks) after recovery from inflammation, AH neurons are still hyperexcitable and synaptic facilitation is still augmented. As detailed above, AH neuron hyperexcitability contributes to dysmotility in TNBS colitis, but both excitability and motility are relatively normal in animals that have been treated with a COX-2 inhibitor for four days prior to euthanasia. To determine whether the post-inflammatory persistent changes in the colon involved a sustained low-level activation of COX-2 to maintain the neuroplasticity and dysmotility, TNBS-inflamed animals were treated with a COX-2 inhibitor on days 52 to 56. In these preparations, the colons still exhibited significant dysmotility, which suggest that the changes are somehow imprinted into the AH neurons during the inflammatory response when it can be prevented or reversed by COX-2 inhibitors. It remains to be determined what transcriptional or epigenetic factors are responsible for the changes in AH neurons, and what might be done to reverse them. Regardless of the mechanism, however, changes such as these may contribute to post-inflammatory and post-infectious changes in gut function and sensitivity.
While AH neuron hyperexcitability and synaptic facilitation persist in the post-inflamed colon, purinergic transmission recovers with the resolution of inflammation (18). This is not too surprising because its disruption is due to an effect of oxidative stress on mitochondria and the oxidative stress subsides with the recovery from inflammation.
Allergen sensitivity in the ENS.
While it is not necessarily a post-inflammatory condition, food sensitivity represents another circumstance in which memory in the system can lead to pathological responses involving immune cells and the ENS. To investigate this, guinea pigs have been sensitized to cow’s milk at weaning, and then responses to β-lactoglobulin are evaluated (13). When challenged with the allergen, mast cell degranulation leads to AH neuron depolarization and hyperexcitability and a suppression of sympathetic synaptic activity in enteric ganglia. In this paradigm, the memory is stored in mast cells, which in turn activate acute changes in local neuronal function. Together with increased secretion, these changes result in enhanced propulsive motility and a flushing of the allergens from the lumen.
Colonic motility in the inflamed human colon.
It is difficult to make direct comparisons between the experimental results of animal studies and the pathophysiological results of human procedures. Such comparisons are difficult due to differences in the chronicity of animal models versus human diseases, the lack of animal models that perfectly reflect the human disorders, the diversity in human populations versus the homogeneity of animal strains, the ability to tightly control experimental conditions in animal studies, and the wider variety of ex vivo and invasive in vivo approaches that can be used in animal studies. In the case of IBD, very few investigations of motor activity have been performed and most were conducted decades ago. That said, several reports indicate that the intuitive presumption that diarrhea, a common feature of IBD, is associated with increased motility in inflamed colons is not supported by clinical investigations of colonic motility. In fact, diarrhea in IBD is more closely linked to decreased contractile activity (38–43).
Rao and colleagues detected decreases in whole gut and small intestinal transit rates and found that subjects with active colitis exhibited proximal colon stasis (38). More distally, they reported that rectosigmoidal transit was increased and that stool frequencies were elevated in active colitis relative to quiescent disease. They concluded that diarrhea in colitis is due to rectosigmoid irritability and exudation of mucus and blood in the sigmoid and rectum, rather than rapid transit, and that caution should be used when treating active colitis with anti-diarrheal agents that could further inhibit proximal colon motility. Consistent with this, in an earlier study, Kern and colleagues reported that the most striking abnormality of the motility pattern in ulcerative colitis was a reduction or complete absence of phasic activity (39). They reported that the reduced phasic activity was closely correlated with severe diarrhea and a more normal motility pattern appeared when the diarrhea terminated. Interestingly, they noted, “although it might have been expected that the absence of phasic activity would be due to rigidity and fibrosis of the colon in advanced disease, this phenomenon is clearly unrelated to the degree of anatomical change” (39). The gastrocolic reflex response is also disrupted in ulcerative colitis. Snape et al., using simultaneous myoelectric and pressure recordings, reported that while colonic spike activity increased rapidly following a meal, the maximal response was smaller and of shorter duration than that observed in controls, and that no postprandial increase in contractility was observed in the inflamed patients (43). They concluded that the lack of contractility might contribute to the increase in diarrhea observed in these individuals after a meal.
While contractility appears to be dampened in IBD, the frequency of propagating contractions is increased. In ulcerative colitis, propagating contractions are always anterograde, resulting in rapid movement of contents to the sigmoid colon (41, 44). The lack of retrograde contractions could be another contributing factor in IBD-associated diarrhea.
Concluding remarks
Effective movement of luminal contents requires a coordinated orchestration of excitatory and inhibitory signals from enteric neurons to the muscularis in a given region of the intestine. Propulsive motility involves a series of overlapping reflex circuits along the intestine that are capable of first receiving and then propelling the luminal contents along. It is possible that alterations in enteric neural function contribute to the disrupted contractility that has been reported in the human colons of inflamed subjects. The data obtained from animal models indicate that inflammation-induced enteric neuroplasticity can, and likely does, contribute to disrupted motility in colitis. These findings indicate that the orchestrated pattern of neuronal activity in the peristaltic reflex circuitry is disrupted in colitis, largely through an increase in neuronal activity and interneuronal synaptic strength, as well as a decrease in inhibitory purinergic neuromuscular transmission. Furthermore, many of these changes persist long after the recovery of colitis. These changes may contribute to alterations in gut function that are maintained during remission from IBD, and to altered motility in IBS; however, these functional properties of gut neurons are currently undetectable in a clinical setting.
It is important to keep in mind that in addition to the neurophysiological changes that alter gut motility and that have been the focus of this Review, there are a number of other concurrent changes within and outside of the colon that also play a role. These include changes within the colon such as the loss of neurons (20, 21), muscle changes such as hypertrophy, altered ionic conductances and responsiveness to neurotransmitters (45, 46), changes in mucosal serotonin signaling that appear to result in increased serotonin availability in inflamed regions (47), and changes in the physical compliance of the bowel in inflamed regions due to fibrosis. Changes in the sensitivity of extrinsic afferent neurons and/or changes in sympathetic input could also contribute to altered colon function as well (33). In addition to alterations in sympathetic activity due to inflammation-induced changes in the electrical properties of prevertebral neurons, there is a loss of synaptic input to the prevertebral ganglia from the gut due to a loss of intestinofugal neurons in colitis (48).
To more fully understand the role of neuroplasticity in IBD, it will be necessary to undertake the difficult task of studying colonic neurons from human subjects. In an intracellular study of human myenteric neurons, Brookes and colleagues determined that neurons with electrical properties similar to those in guinea pig were observed and, as in guinea pig, about 4% of the colonic myenteric neurons exhibited the features of AH neurons (49). A more recent investigation has described the use of combined intracellular recording, dye filling, and immunohistochemistry in human myenteric neurons (50), demonstrating that it may be possible to ultimately determine whether the functional properties of myenteric neurons are altered by inflammation. Another approach that may be useful in investigating the pathophysiology of human enteric neurons involves the use of voltage-sensitive dyes (51). However, an imaging approach is less useful than electrical recordings for understanding ionic conductance, and this electrophysiological information has been critical for understanding, mimicking, and reversing inflammation-induced changes in animal models. As addressed in this Review, determining the role of neuroplasticity in human colonic function may prove to be crucial in providing more accurate information in the pathogenesis of IBD and ultimately lead to improved therapeutic techniques.
Acknowledgments
Research on this topic conducted in the Mawe lab was supported by NIH grant DK62267. The author thanks Rachel Scott and Conor O’Neill for comments on the manuscript as well as Keith Sharkey and the members of both of our labs for their friendship and for significant contributions to the studies that are summarized in this Review.
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(3):949–955. doi:10.1172/JCI76306.
References
- 1.Bayliss WM, Starling EH. The movements and innervation of the small intestine. J Physiol. 1899;24(2):99–143. doi: 10.1113/jphysiol.1899.sp000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Langley JN. The Autonomic Nervous System. Cambridge, United Kingdom: Heffer; 1921. [Google Scholar]
- 3.Brookes SJ. Classes of enteric nerve cells in the guinea-pig small intestine. Anat Rec. 2001;262(1):58–70. doi: 10.1002/1097-0185(20010101)262:1<58::AID-AR1011>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 4.Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9(5):286–294. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- 5. Furness J, Clerc N, Gola M, Kunze W, Fletcher E. Identification of component neurons and organisation of enteric circuits. In: Krammer H, Singer M, eds. Neurogastroenterology From the Basics to the Clinics. Dordrecht, the Netherlands: Kluwer Academic Publishers; 2000:137–150. [Google Scholar]
- 6.Furness JB, Jones C, Nurgali K, Clerc N. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog Neurobiol. 2004;72(2):143–164. doi: 10.1016/j.pneurobio.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Wood JD. Enteric nervous system: reflexes, pattern generators and motility. Curr Opin Gastroenterol. 2008;24(2):149–158. doi: 10.1097/MOG.0b013e3282f56125. [DOI] [PubMed] [Google Scholar]
- 8.Linden DR, Sharkey KA, Mawe GM. Enhanced excitability of myenteric AH neurones in the inflamed guinea-pig distal colon. J Physiol. 2003;547(pt 2):589–601. doi: 10.1113/jphysiol.2002.035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lomax AE, Mawe GM, Sharkey KA. Synaptic facilitation and enhanced neuronal excitability in the submucosal plexus during experimental colitis in guinea-pig. J Physiol. 2005;564(pt 3):863–875. doi: 10.1113/jphysiol.2005.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nurgali K, Nguyen TV, Matsuyama H, Thacker M, Robbins HL, Furness JB. Phenotypic changes of morphologically identified guinea-pig myenteric neurons following intestinal inflammation. J Physiol. 2007;583(pt 2):593–609. doi: 10.1113/jphysiol.2007.135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, et al. Cyclic AMP signaling contributes to neural plasticity and hyperexcitability in AH sensory neurons following intestinal Trichinella spiralis-induced inflammation. Int J Parasitol. 2007;37(7):743–761. doi: 10.1016/j.ijpara.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Palmer JM, Wong-Riley M, Sharkey KA. Functional alterations in jejunal myenteric neurons during inflammation in nematode-infected guinea pigs. Am J Physiol. 1998;275(5 pt 1):G922–G935. doi: 10.1152/ajpgi.1998.275.5.G922. [DOI] [PubMed] [Google Scholar]
- 13.Frieling T, Cooke HJ, Wood JD. Neuroimmune communication in the submucous plexus of guinea pig colon after sensitization to milk antigen. Am J Physiol. 1994;267(6 pt 1):G1087–G1093. doi: 10.1152/ajpgi.1994.267.6.G1087. [DOI] [PubMed] [Google Scholar]
- 14.Liu S, et al. Expression of type 1 corticotropin-releasing factor receptor in the guinea pig enteric nervous system. J Comp Neurol. 2005;481(3):284–298. doi: 10.1002/cne.20370. [DOI] [PubMed] [Google Scholar]
- 15.Manning BP, Sharkey KA, Mawe GM. Effects of PGE2 in guinea pig colonic myenteric ganglia. Am J Physiol Gastrointest Liver Physiol. 2002;283(6):G1388–G1397. doi: 10.1152/ajpgi.00141.2002. [DOI] [PubMed] [Google Scholar]
- 16.Linden DR, Sharkey KA, Ho W, Mawe GM. Cyclooxygenase-2 contributes to dysmotility and enhanced excitability of myenteric AH neurones in the inflamed guinea pig distal colon. J Physiol. 2004;557(pt 1):191–205. doi: 10.1113/jphysiol.2004.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krauter EM, Linden DR, Sharkey KA, Mawe GM. Synaptic plasticity in myenteric neurons of the guinea-pig distal colon: presynaptic mechanisms of inflammation-induced synaptic facilitation. J Physiol. 2007;581(pt 2):787–800. doi: 10.1113/jphysiol.2007.128082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strong DS, Cornbrooks CF, Roberts JA, Hoffman JM, Sharkey KA, Mawe GM. Purinergic neuromuscular transmission is selectively attenuated in ulcerated regions of inflamed guinea pig distal colon. J Physiol. 2010;588(pt 5):847–859. doi: 10.1113/jphysiol.2009.185082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts JA, Durnin L, Sharkey KA, Mutafova-Yambolieva VN, Mawe GM. Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J Physiol. 2013;591(pt 15):3725–3737. doi: 10.1113/jphysiol.2013.254136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulbransen BD, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18(4):600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linden DR, et al. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17(5):751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 22.Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;285(1):G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman JM, Brooks EM, Mawe GM. Gastrointestinal Motility Monitor (GIMM). J Vis Exp. 2010;(46):2435. doi: 10.3791/2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman JM, McKnight ND, Sharkey KA, Mawe GM. The relationship between inflammation-induced neuronal excitability and disrupted motor activity in the guinea pig distal colon. Neurogastroenterol Motil. 2011;23(7):673–e279. doi: 10.1111/j.1365-2982.2011.01702.x. [DOI] [PubMed] [Google Scholar]
- 25.Mawe GM, Gershon MD. Functional heterogeneity in the myenteric plexus: demonstration using cytochrome oxidase as a verified cytochemical probe of the activity of individual enteric neurons. J Comp Neurol. 1986;249(3):381–391. doi: 10.1002/cne.902490305. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman JM, et al. Activation of colonic mucosal 5-HT(4) receptors accelerates propulsive motility and inhibits visceral hypersensitivity. Gastroenterology. 2012;142(4):844–854.e4. doi: 10.1053/j.gastro.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bercik P, Verdu EF, Collins SM. Is irritable bowel syndrome a low-grade inflammatory bowel disease? Gastroenterol Clin North Am. 2005;34(2):235–245. doi: 10.1016/j.gtc.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Casellas F, et al. Mucosal healing restores normal health and quality of life in patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2012;24(7):762–769. doi: 10.1097/MEG.0b013e32835414b2. [DOI] [PubMed] [Google Scholar]
- 29.Higgins PD, Schwartz M, Mapili J, Krokos I, Leung J, Zimmermann EM. Patient defined dichotomous end points for remission and clinical improvement in ulcerative colitis. Gut. 2005;54(6):782–788. doi: 10.1136/gut.2004.056358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrokhyar F, Marshall JK, Easterbrook B, Irvine EJ. Functional gastrointestinal disorders and mood disorders in patients with inactive inflammatory bowel disease: prevalence and impact on health. Inflamm Bowel Dis. 2006;12(1):38–46. doi: 10.1097/01.MIB.0000195391.49762.89. [DOI] [PubMed] [Google Scholar]
- 31.Vivinus-Nebot M, et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut. 2014;63(5):744–752. doi: 10.1136/gutjnl-2012-304066. [DOI] [PubMed] [Google Scholar]
- 32.Bashashati M, et al. Cytokine imbalance in irritable bowel syndrome: a systematic review and meta-analysis. Neurogastroenterol Motil. 2014;26(7):1036–1048. doi: 10.1111/nmo.12358. [DOI] [PubMed] [Google Scholar]
- 33.Brierley SM, Linden DR. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat Rev Gastroenterol Hepatol. 2014;11(10):611–627. doi: 10.1038/nrgastro.2014.103. [DOI] [PubMed] [Google Scholar]
- 34.Nasser Y, Boeckxstaens GE, Wouters MM, Schemann M, Vanner S. Using human intestinal biopsies to study the pathogenesis of irritable bowel syndrome. Neurogastroenterol Motil. 2014;26(4):455–469. doi: 10.1111/nmo.12316. [DOI] [PubMed] [Google Scholar]
- 35.Piche T. Tight junctions and IBS — the link between epithelial permeability, low-grade inflammation, and symptom generation? Neurogastroenterol Motil. 2014;26(3):296–302. doi: 10.1111/nmo.12315. [DOI] [PubMed] [Google Scholar]
- 36.Spiller R, Garsed K. Postinfectious irritable bowel syndrome. Gastroenterology. 2009;136(6):1979–1988. doi: 10.1053/j.gastro.2009.02.074. [DOI] [PubMed] [Google Scholar]
- 37.Krauter EM, Strong DS, Brooks EM, Linden DR, Sharkey KA, Mawe GM. Changes in colonic motility and the electrophysiological properties of myenteric neurons persist following recovery from trinitrobenzene sulfonic acid colitis in the guinea pig. Neurogastroenterol Motil. 2007;19(12):990–1000. doi: 10.1111/j.1365-2982.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 38.Rao SS, Read NW, Brown C, Bruce C, Holdsworth CD. Studies on the mechanism of bowel disturbance in ulcerative colitis. Gastroenterology. 1987;93(5):934–940. doi: 10.1016/0016-5085(87)90554-3. [DOI] [PubMed] [Google Scholar]
- 39.Kern F, Jr, Almy TP, Abbot FK, Bogdonoff MD. The motility of the distal colon in nonspecific ulcerative colitis. Gastroenterology. 1951;19(3):492–503. [PubMed] [Google Scholar]
- 40.Bassotti G, Antonelli E, Villanacci V, Salemme M, Coppola M, Annese V. Gastrointestinal motility disorders in inflammatory bowel diseases. World J Gastroenterol. 2014;20(1):37–44. doi: 10.3748/wjg.v20.i1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddy SN, et al. Colonic motility and transit in health and ulcerative colitis. Gastroenterology. 1991;101(5):1289–1297. doi: 10.1016/0016-5085(91)90079-z. [DOI] [PubMed] [Google Scholar]
- 42.Connell AM. The motility of the pelvic colon. II. Paradoxical motility in diarrhoea and constipation. Gut. 1962;3:342–348. doi: 10.1136/gut.3.4.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snape WJ, Jr, Matarazzo SA, Cohen S. Abnormal gastrocolonic response in patients with ulcerative colitis. Gut. 1980;21(5):392–396. doi: 10.1136/gut.21.5.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bassotti G, de Roberto G, Chistolini F, Sietchiping-Nzepa F, Morelli O, Morelli A. Twenty-four-hour manometric study of colonic propulsive activity in patients with diarrhea due to inflammatory (ulcerative colitis) and non-inflammatory (irritable bowel syndrome) conditions. Int J Colorectal Dis. 2004;19(5):493–497. doi: 10.1007/s00384-004-0604-6. [DOI] [PubMed] [Google Scholar]
- 45.Akiho H, Ihara E, Motomura Y, Nakamura K. Cytokine-induced alterations of gastrointestinal motility in gastrointestinal disorders. World J Gastrointest Pathophysiol. 2011;2(5):72–81. doi: 10.4291/wjgp.v2.i5.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbarali HI, Hawkins EG, Ross GR, Kang M. Ion channel remodeling in gastrointestinal inflammation. Neurogastroenterol Motil. 2010;22(10):1045–1055. doi: 10.1111/j.1365-2982.2010.01560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mawe GM, Hoffman JM. Serotonin signalling in the gut—functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2013;10(8):473–486. doi: 10.1038/nrgastro.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linden DR. Colitis is associated with a loss of intestinofugal neurons. Am J Physiol Gastrointest Liver Physiol. 2012;303(10):G1096–G1104. doi: 10.1152/ajpgi.00176.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brookes SJ, Ewart WR, Wingate DL. Intracellular recordings from myenteric neurones in the human colon. J Physiol. 1987;390:305–318. doi: 10.1113/jphysiol.1987.sp016702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carbone SE, Jovanovska V, Nurgali K, Brookes SJ. Human enteric neurons: morphological, electrophysiological, and neurochemical identification. Neurogastroenterol Motil. 2014;26(12):1812–1816. doi: 10.1111/nmo.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vignali S, et al. Recordings from human myenteric neurons using voltage-sensitive dyes. J Neurosci Methods. 2010;192(2):240–248. doi: 10.1016/j.jneumeth.2010.07.038. [DOI] [PubMed] [Google Scholar]