

Abstract
Cholangiocarcinoma is a relatively rare cancer of the biliary ducts that is highly refractory to treatment. The factors that drive cholangiocarcinoma are poorly understood, though chronic liver fluke infection is a risk factor for disease. In this issue of the JCI, Boulter and colleagues demonstrate that the WNT/β-catenin signaling pathway is upregulated in patients with sporadic cholangiocarcinoma. The authors determined that macrophages generate WNT ligands in cholangiocarcinomas and depletion or inhibition of this cell population in animal models of cholangiocarcinoma reduced tumor burden and proliferation. Moreover, pharmacological inhibition of WNT secretion or β-catenin activity was efficacious in animal models. Together the results of this study suggest that targeting WNT has potential as a therapeutic strategy for cholangiocarcinoma.
WNT-mediated pathways in cancer
In 1982, Roel Nusse and Harold Varmus described how overexpression of a Wnt gene could cause mouse mammary tumors (1, 2). Thirty years later, WNT signaling has been implicated in a wide range of diseases, including cholangiocarcinoma (CC) (3, 4). It is also clear that unlike murine cancers, human cancers are not caused by chromosomal translocations or the result of insertional mutagenesis driving WNT overexpression; therefore, the investigational spotlight shifted to mutations in targets downstream of WNTs that activate the WNT/β-catenin pathway. This focus on intracellular signaling followed the discovery that the adenomatous polyposis coli (APC) protein, which is mutated in hereditary and many sporadic colorectal cancers, regulates β-catenin abundance. Furthermore, pathological increases in β-catenin drive expression of proliferation-associated genes (4). Collectively, these findings led to an intense focus on understanding and trying to therapeutically block the pathological activity of stabilized β-catenin. This approach has provided multiple insights into intracellular WNT-mediated signaling pathways and produced several small molecule inhibitors of β-catenin signaling that are being tested for efficacy against human cancers with perturbations of WNT/β-catenin signaling (5).
In the last few years, the focus has shifted to the cell surface and the regulation of WNT receptors. We now understand more about how sensitivity to WNTs is coordinated by a tightly controlled ubiquitination cycle at the cell surface that involves two closely related integral membrane ubiquitin ligases, RNF43 and ZNRF3 (Figure 1 and ref. 6). Teh and coworkers reported the presence of mutations in the gene encoding RNF43 in CC related to ingestion of the liver fluke Opisthorchis viverrini, which is endemic to northern Thailand (7). Moreover, RNF43 loss-of-function mutations have also been reported in multiple cancer types, including sporadic pancreatic adenocarcinomas, endometrial cancers, and colorectal cancers with wild-type APC (8). Cong and coworkers along with others found that RNF43 and ZNRF3 regulate frizzled abundance and thereby determine cellular sensitivity to WNTs (9, 10). Ubiquitination of frizzled and perhaps low-density lipoprotein receptor–related protein 6 (LRP6) leads to their downregulation by internalization and proteolysis. Cong and colleagues also made the key discovery that RNF43/ZNRF3 activity at the cell surface is in turn regulated by the secreted WNT cofactor R-spondin (RSPO), which binds to RNF43/ZNRF3 in a complex with a member of the LGR4/5/6 family. RNF43 loss-of-function mutations increase WNT receptor abundance, making cells sensitive to WNTs (11). Rounding out the story, recurrent gain-of-function translocations in the genes encoding RSPO-2 and RSPO-3 were recently identified, first in colorectal cancers and then in several other cancer types (12, 13). The discovery of human mutations that result in sensitization to WNTs translates what Nusse and Varmus first found in mice to humans: a genetic basis for increased WNT ligand signaling in cancer.
Figure 1. A mutation-prone ubiquitin ligase module regulates cellular sensitivity to WNTs.
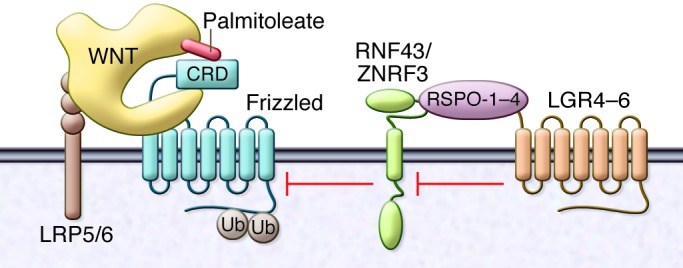
WNTs interact with LRP5/6 coreceptors, and the cysteine-rich domain (CRD) of frizzled in part via covalently bound palmitoleate (shown in red). The amount of LRP5/6 and frizzled present at the cell surface is regulated by the related ubiquitin ligases RNF43 and ZNRF3. The activity of RNF43 and ZNRF3 is in turn controlled by the WNT cofactor R-spondin family, RSPO-1–RSPO-4, and the coreceptor family encoded by the LGR4–LGR6 family. Loss of RNF43 function and gain of RSPO-2 or RSPO-3 function have recently been identified as drivers of a subset of cancers, including cholangiocarcinoma.
These advances toward understanding the role of WNT secretion in genetically defined cancers have been matched by the development of tools to pharmacologically block WNT secretion. All WNTs require post-translational modification by the addition of a monounsaturated palmitoleate group, which is catalyzed by an ER-resident palmitoyltransferase named porcupine for the Drosophila phenotype and referred to as PORCN in humans. WNTs require palmitoylation for transport from the ER to the cell surface (14, 15) and to interact with frizzled proteins (ref. 16 and Figure 1). PORCN inhibitors, which were first developed by Lum and coworkers (17), have proven to be useful tools in dissecting the role of WNT secretion in disease and have shown efficacy in WNT-dependent preclinical animal models of cancer (11, 18).
WNT signaling drives CC
Recently, WNT signaling was implicated in the development of a rare subset of CC (7, 19), and in this issue Boulter et al. report that WNT/β-catenin target genes as well as WNT7B and WNT10A are upregulated in human CC specimens collected from patients in Scotland (3), a region more associated with consumption of haggis and whisky than pla som (a Thai raw fish dish that carries CC-related liver flukes). The findings of Boulter and colleagues also extended to two rodent models of CC. In mice sensitized by p53 loss, thioacetamide-induced (TAA-induced) CC resulted in upregulation of WNT7B and WNT10A as well as WNT/β-catenin target genes. Similarly, rats given TAA exhibited increased WNT7B and WNT10A along with RSPO-1 and WNT/β-catenin target genes. These findings led to the question, What cell type is making the WNTs?
Macrophages are key regulators of inflammation, and in several systems they are known to produce WNTs (20). Boulter et al. determined that macrophages are a major source of WNT7B in all three CC models evaluated, human, mouse, and rat (3). As confirmation, rats with GFP-tagged (GFP+) hematopoietic cells were given TAA to induce CC. Evaluation of the GFP+ cells revealed that those expressing the macrophage marker CD68 were a major source of WNT7B in the resultant CCs. These macrophages were functionally important for CC development, as either macrophage depletion or small molecule inhibition of macrophage differentiation decreased tumor burden and proliferation in CC animal models.
Boulter and colleagues also made use of two small molecule inhibitors of the WNT pathway to evaluate whether the WNTs were indeed driving cancer progression (3). WNT-C59 is a PORCN inhibitor that prevents secretion of WNTs by blocking post-translational O-linked palmitoylation (18), while ICG-001 blocks the interaction of β-catenin with the co-activator CTBP1 (5). These drugs had a modest effect in vitro (3), a finding that is consistent with previous reports that autocrine WNT signaling is not essential for proliferation in standard tissue culture conditions (18). However, both of these WNT pathway inhibitors showed efficacy in mouse xenograft and rat carcinogen-induced CC without overt toxicity.
Together, the results from the study by Boulter et al. provide a strong case for the role of stroma-produced WNTs, especially macrophage-derived WNT7B, in the progression and maintenance of sporadic human CCs. Equally importantly, they demonstrate the efficacy of small molecule inhibitors of the WNT pathway in animal models of this disease (3). As related drugs are now in human trials, these findings may quickly translate into new therapies for this difficult disease.
Remaining questions and future directions
It is not clear why these CCs are particularly sensitive to WNTs. Macrophages can be induced to secrete WNT7B by inflammatory mediators; however, this does not regularly result in cancer. One possible explanation is that sporadic CCs have a mutagen-induced genetic or epigenetic lesion that results in WNT sensitization. At this point, the existence of such lesions is only speculative due to the absence of sequence data from these specific tumors. Mutations in RNF43 or ZNRF3, or translocations in RSPO2 and RSPO3 are candidates that should be examined further in this disease (13, 21). Epigenetic loss of WNT inhibitors such as DKK1 and secreted frizzled-related proteins (SFRPs) may also contribute to WNT sensitivity (22). Mutations in the genes encoding APC and β-catenin are unlikely to be causal in the PORCN inhibitor–sensitive tumors, as these mutations activate the WNT/β-catenin pathway downstream of the site of action of PORCN inhibitors. It is unlikely that physiologic WNT expression by macrophages alone is sufficient to drive carcinogenesis, but WNTs could be important cofactors required for sustaining tumor growth. One tantalizing possibility is that that other currently unknown regulators of WNT sensitivity are involved. For example, if loss of function of a ubiquitin ligase, such as RNF43, contributes to cancer, perhaps upregulation of a ubiquitin-specific protease that stabilizes frizzleds or LGR5 could have a similar effect.
There are still many lessons to be learned from the WNT pathway. The study of uncommon diseases such as fluke-associated CC can provide insights into the more common sporadic form of the disease. And as this study demonstrates, after thirty years of study, the WNT pathway still contains both surprises and important clinical opportunities. The recent advances in the development of WNT pathway inhibitors holds promise for patients with WNT-driven cancers. Boulter et al. now add sporadic CC to this list.
Acknowledgments
David Virshup is supported in part by the National Research Foundation Singapore through a Singapore Translational Research (STaR) Investigatorship award administered by the Singapore Ministry of Health’s National Medical Research Council.
Footnotes
Conflict of interest: David M. Virshup is a co-holder of a patent for a PORCN inhibitor.
Reference information:J Clin Invest. 2015;125(3):975–977. doi:10.1172/JCI80819.
See the related article beginning on page 1269.
References
- 1.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31(1):99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 2.Nusse R, Varmus H. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 2012;31(12):2670–2684. doi: 10.1038/emboj.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulter L, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125(3):1269–1285. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4(5):a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu J, Virshup DM. Updating the Wnt pathways. Biosci Rep. 2014;34(5):593–607. doi: 10.1042/BSR20140119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ong CK, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44(6):690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 8.Giannakis M, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46(12):1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koo BK, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488(7413):665–669. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 10.Hao HX, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. 2012;485(7397):195–200. doi: 10.1038/nature11019. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A. 2013;110(31):12649–12654. doi: 10.1073/pnas.1307218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seshagiri S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez Cardona G, et al. Identification of R-Spondin fusions in various types of human cancer. Cancer Res. 2014;74(19 suppl):2408. [Google Scholar]
- 14.Yu J, et al. WLS Retrograde transport to the endoplasmic reticulum during Wnt secretion. Dev Cell. 2014;29(3):277–291. doi: 10.1016/j.devcel.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Coombs GS, et al. WLS-dependent secretion of WNT3A requires Ser209 acylation and vacuolar acidification. J Cell Sci. 2010;123(19):3357–3367. doi: 10.1242/jcs.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural Basis of Wnt Recognition by Frizzled. Science. 2012;337(6090):59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proffitt KD, et al. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013;73(2):502–507. doi: 10.1158/0008-5472.CAN-12-2258. [DOI] [PubMed] [Google Scholar]
- 19.Loilome W, et al. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol. 2014;35(6):5357–5367. doi: 10.1007/s13277-014-1698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan-On W, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45(12):1474–1478. doi: 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki H, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]