

Abstract
We previously discovered one particular HLA-A*02:01 mutant that enhanced peptide-specific cytotoxic T lymphocyte (CTL) recognition in vitro compared to wild-type HLA-A*02:01. This mutant contains a single amino acid substitution from histidine to leucine at position 74 (H74L) that is located in the peptide-binding groove. To investigate the effect of the H74L mutation on the in vivo CTL priming, we took advantage of the technology of the HLA class I single-chain trimer (SCT) in which three components involving a peptide, β2 microglobulin and the HLA class I heavy chain are joined together via flexible linkers. We generated recombinant adenovirus expressing SCT comprised influenza A matrix protein (FMP)-derived peptide, β2 microglobulin and the H74L heavy chain. HLA-A*02:01 transgenic mice were immunized with the adenovirus, and the induction of peptide-specific CTLs and antitumor immunity was investigated. It was clearly shown that the H74L mutation enabled the HLA-A*02:01 SCT molecule to dramatically enhance both in vivo priming of FMP-specific CTLs and protection against a lethal challenge of tumor cells expressing FMP. These data present the first evidence that a simple point mutation in the HLA class I heavy chain of SCT is beneficial for improving CTL-based immunotherapy and prophylaxis to control tumors.
Introduction
Major histocompatibility complex (MHC) class I molecules are heterodimeric glycoproteins composed of a polymorphic heavy chain and an invariant β2-microglobulin (β2m), and play an essential role in the cytotoxic T lymphocyte (CTL)-mediated immunity. They bind naturally processed antigenic peptides, and present them to CD8+ CTLs for immune surveillance. Since CTLs play a major role in the specific clearance of neoplastic cells and virus-infected cells, the improvement of MHC class I presentation and peptide-specific CTL induction is a key issue for the development of prophylactic and therapeutic vaccines against tumors and intracellular pathogens.
The discovery of a peptide-binding groove in the MHC class I molecule1 was a major breakthrough in immunology. This groove is formed between two α helices of the α1 and α2 domains with an eight-stranded β-pleated sheet floor,1 and includes six peptide-binding pockets, designated pocket A-F.2 We previously constructed an extensive library of human leukocyte antigen (HLA)-A*0201 mutants with different single amino acid substitutions at almost all amino acid positions in the peptide-binding groove, and examined whether these changes affected HLA-A*0201-restricted, peptide-specific CTL responses.3–5 Most of the mutations had little or negative effects on the peptide-specific CTL killing activity against peptide-pulsed target cells expressing each of the mutant molecules. However, there was only one mutant that dramatically enhanced the presentation of exogenously loaded peptides to peptide-specific CTLs compared to the wild-type (WT) HLA-A*02:01 molecule.5 This HLA-A*02:01 mutant contains an amino acid substitution from histidine to leucine at position 74 (H74L) that is located in the α1 helix in the peptide-binding groove. We expected that the H74L mutant might be useful for the efficient induction of HLA-A*02:01-restricted, peptide-specific CTLs, leading to the development of a novel CTL-based vaccine. However, there was no appropriate method of verifying this hypothesis at that time.
Later on, the technology of the MHC class I single-chain trimer (SCT) has been developed.6 SCT is a polypeptide in which the three components, a peptide, β2m and the MHC class I heavy chain are joined together via flexible linkers.7,8 Since the peptide is covalently attached, SCTs are more stable at the cell surface than the native MHC class I/peptide complexes. It should be noted that SCTs could efficiently stimulate peptide-specific CTLs in vivo following DNA vaccination,6 and the SCT-based vaccination could provide protection against tumors9,10 and pathogens.11,12 These pre-assembled molecules bypass the normal antigen processing pathway, and hence, SCTs should be resistant to virus-encoded immune evasion proteins that prevent peptide processing, loading, and presentation.13 In addition, SCTs may not be greatly affected by aberrations of the antigen processing and presenting machinery that are frequently observed in tumors.14 Thus, this approach is expected to have applications as DNA vaccines against tumors and viral infection.
In this study, we took advantage of the SCT technology in evaluating the effect of the H74L mutation on the in vivo priming of peptide-specific CTLs. To examine in vivo CTL responses, the HLA-A*02:01-transgenic mice, designated as HHD mice,15 were employed. HHD mice express the transgenic HHD molecule, in which human β2m is covalently linked to a chimeric heavy chain composed of HLA-A*02:01 (α1 and α2 domains) and H-2Db (α3, transmembrane, and cytoplasmic domains).15 Because the innate H-2Db and mouse β2m genes have been disrupted by homologous recombination, HHD molecules are efficiently utilized for the HLA-A*02:01-restricted CTL responses in HHD mice. Therefore, HHD mice have been used as a versatile preclinical animal model for the study of HLA-A*02:01-restricted CTL responses.16,17 For in vivo priming experiments, we generated recombinant adenovirus expressing SCT composed of an influenza virus-derived peptide and either WT HHD or HHD with the H74L mutation. HHD mice were then immunized with these adenoviruses, and the efficiency of peptide-specific CTL induction was compared by various assays. Here, we clearly demonstrate for the first time that a simple point mutation in the MHC class I heavy chain of the SCT molecule induces enhancement of both in vivo CTL priming and antitumor protection.
Results
Enhanced presentation of exogenously loaded peptides to CTLs by the H74L mutant
Previous studies demonstrated that the mutant HLA-A*02:01 molecule with the H74L mutation possessed the unique ability to enhance the presentation of exogenously loaded peptides to peptide-specific CTLs.5,18 To confirm these results in the HHD system, we firstly constructed a mutant HHD gene that could generate this mutation in the HHD molecule (HHD-H74L) (Figure 1a,b) by the inverse polymerase chain reaction (PCR)-based site-directed mutagenesis. The HHD-H74L gene was then transfected and expressed in mouse lymphoma, RMA cells (RMA-H74L). The expression level of the H74L mutant molecules on RMA-H74L cells was almost equivalent to that of the HHD molecules on RMA cells expressing the WT HHD (RMA-HHD). To test the ability of the H74L mutant, HLA-A*02:01-restricted, peptide-specific CTL lines were generated from HHD mice, and examined for their killing activities against RMA-HHD and RMA-H74L in the presence of synthetic peptides at various concentrations in standard 51Cr-release assays. Consistent with the previous data,5 RMA-H74L cells were lysed by influenza A matrix peptide (FMP58-66)-specific CTLs at much lower peptide concentrations than RMA-HHD cells (Figure 1c). Similar results were obtained with different HLA-A*02:01-restricted peptides derived from human papillomavirus (HPV)-E6 (HPV-E629-38), human T-lymphotropic virus (HTLV)-tax (HTLV-tax11-19) and human immunodeficiency virus (HIV)-pol (HIV-pol476-484) (Figure 1c), indicating that this phenomenon was not peptide-specific as described in the previous studies.18
Figure 1.

HHD-H74L improves the presentation of exogenous peptides. (a) Structure of HHD-H74L. LS: leader sequence; huβ2m: human β2m; α1, α2, α3, Tm, Cyt: α1, α2, α3, transmembrane, cytoplasmic domains. (b) Diagram of HHD-H74L. (c) Enhanced cytotoxic T lymphocyte (CTL) recognition of HHD-H74L-expressing targets. CTL lines were examined for killing activities against RMA, RMA-HHD and RMA-H74L in the presence of a peptide (FMP58–66, HPV-E629–38, HTLV-tax11–19 or HIV-pol476–484) by 51Cr-release assays. Data are shown as the mean ± SD of triplicate wells. The experiments were repeated three times. *P < 0.05; **P < 0.01 compared to RMA-HHD, Student’s t-test. (d) Enhanced peptide-binding by HHD-H74L. RMA-S-HHD or RMA-S-H74L cells were cultured with a peptide, and examined for their surface expression of HHD or HHD-H74L by flow cytometry. The experiment was repeated twice with similar results. Data are standardized as the % relative binding, and shown as the mean ± SD of triplicate wells. *P < 0.05; **P < 0.01 compared to RMA-S-HHD, Student’s t-test. (e) CTL recognition of various HHD mutants. FMP-specific CTL lines were examined for killing activities against RMA expressing HHD mutants with various mutations at position 74 in the presence of 100 nmol/l FMP58-66 by 51Cr-release assays. The data are representative of one of three independent experiments. Data are standardized as the % relative lysis and shown as the mean ± SD of triplicate wells. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant, One-way analysis of variance.
The peptide-binding studies were conducted using the transporter associated antigen processing-2 (TAP-2) deficient cell line, RMA-S19 expressing HHD (RMA-S-HHD) or HHD-H74L (RMA-S-H74L). As shown in Figure 1d, the efficiency of peptide loading on the HHD molecule was significantly increased by the H74L mutation, suggesting that the improved peptide loading may result in the enhanced presentation of exogenous peptides to CTLs by the H74L mutation (Figure 1c). A series of amino acid substitutions at position 74 of the HHD molecule were also tested for their effects on the peptide presentation to FMP58-66-specific CTLs. In addition to H74L, three other mutations involving H74A, H74F, and H74T apparently enhanced the peptide presentation in the FMP58-66-specific CTL response (Figure 1e). In contrast, several mutations including H74E, H74K, H74N, and H74Y impaired the FMP58-66-specific CTL recognition. These data suggest that the effect of the H74 mutation depends on the amino acid to be substituted from histidine.
These data show that the functional phenotype of the H74L mutation in the HHD system is likely to be equal to that of the mutant HLA-A*02:01 with the H74L mutation as described previously.5,18
Construction of the SCT of HHD-H74L
It is well documented that the SCTs are potent stimulators of peptide-specific CTLs in vivo.6,9,20 The SCT polypeptide comprises three MHC class I components including an antigenic peptide, β2m and the MHC class I heavy chain attached sequentially with flexible linkers.6 Given the unique property of the H74L mutant, we hypothesized that the SCTs of HHD-H74L could prime peptide-specific CTLs in vivo more efficiently than the SCTs composed of WT HHD. To prove this hypothesis, we produced SCT constructs consisting of the following elements beginning with the amino terminus: the leader sequence of β2m, the FMP58–66 peptide, the first flexible linker of 15 residues, the mature portion of human β2m, the second flexible linker of 15 residues, and, finally, the heavy chain of the HHD (SCT-HHD) or HHD-H74L (SCT-H74L) molecule (Figure 2a,b). When the SCT-HHD or SCT-H74L construct was stably expressed in the TAP-2-deficient cell line, RMA-S, there was an apparent increase of the expression level of MHC class I molecules on the cell surface at 37°C in comparison with RMA-S-HHD in the absence of an exogenous peptide (Figure 1c). These results implied that SCT-HHD or SCT-H74L was expressed on the cell surface even in the absence of endogenous peptides, suggesting that the SCT molecules are likely to bypass MHC class I antigen processing due to their preassembled forms.
Figure 2.
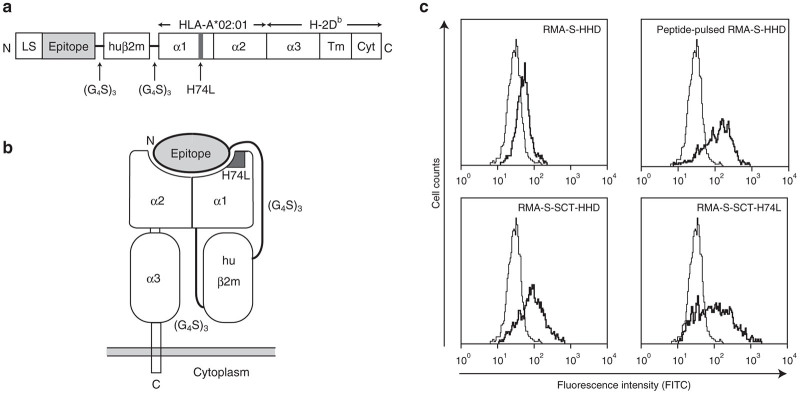
The SCT molecule comprises an epitope, human β2m and HHD with the H74L mutation. (a) Structure of the SCT molecule with the H74L mutation (SCT-H74L). LS: leader sequence; huβ2m: human β2m; α1, α2, α3: α1, α2, α3 domains; Tm: transmembrane domain; Cyt: cytoplasmic domain. (b) Diagram of SCT-H74L on the cell surface. (c) Expression of SCT-HHD or SCT-H74L on the surface of RMA-S cells. RMA-S and RMA-S transfectants were stained with the anti-HLA-A*02:01 mAb, followed by FITC-labeled goat antimouse IgG antibody. Thick lines: RMA-S-HHD, peptide-pulsed RMA-S-HHD (pulsed with 1 μmol/l FMP58–66), RMA-S-expressing SCT-HHD (RMA-S-SCT-HHD), and RMA-S-expressing SCT-H74L (RMA-S-SCT-H74L); Thin lines in all panels: RMA-S.
In the previous studies, DNA vaccinations were employed for stimulating peptide-specific CTLs with MHC class I SCTs in vivo.6,9,11 Although this approach has several advantages, the efficiency of CTL induction is relatively weak. Therefore, we generated a recombinant adenovirus expressing either SCT-HHD (Ad-SCT-HHD) or SCT-H74L (Ad-SCT-H74L) for in vivo priming in this study. As a control, a recombinant adenovirus expressing the full-length influenza A matrix protein (Ad-FMP) was used. Viral titers were accurately calculated by the 50% tissue culture infectious dose (TCID50).
SCT-H74L enhances in vivo CTL priming
To evaluate the effect of the H74L mutation, HHD mice were immunized once with either Ad-SCT-HHD, Ad-SCT-H74L, Ad-FMP or WT adenovirus (Ad-WT) at various doses ranging from 1.6 × 108 to 1.6 × 104 TCID50/mouse. After 2 weeks following immunization, spleen cells were prepared, stimulated in vitro with FMP58–66, and then examined for their FMP58–66-specific killing activities by the 51Cr-release assay (Figure 3). As shown in Figure 3a, FMP-specific CTLs were induced in all of the mice immunized with 1.6 × 108 TCID50 of either Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L. At an inoculation dose of 1.6 × 107 TCID50, however, percentages of CTL-positive mice were diminished to 60 and 80% in mice vaccinated with Ad-FMP and Ad-SCT-HHD, respectively (Figure 3a). In contrast, all of the mice generated FMP-specific CTLs after immunization with 1.6 × 107 TCID50 of Ad-SCT-H74L (Figure 3a). Furthermore, even at lower doses such as 1.6 × 106 and 1.6 × 105 TCID50, Ad-SCT-H74L primed FMP-specific CTLs in 60–80% of the immunized mice, whereas FMP-specific CTL responses were not detected in any of the mice injected with either Ad-FMP or Ad-SCT-HHD at the lower doses (Figure 3a,b). Ad-WT at any doses tested did not elicit FMP-specific CTLs in mice (Figure 3). These data indicate that Ad-SCT-H74L primes FMP-specific CTLs in mice more efficiently than Ad-SCT-HHD or Ad-FMP.
Figure 3.
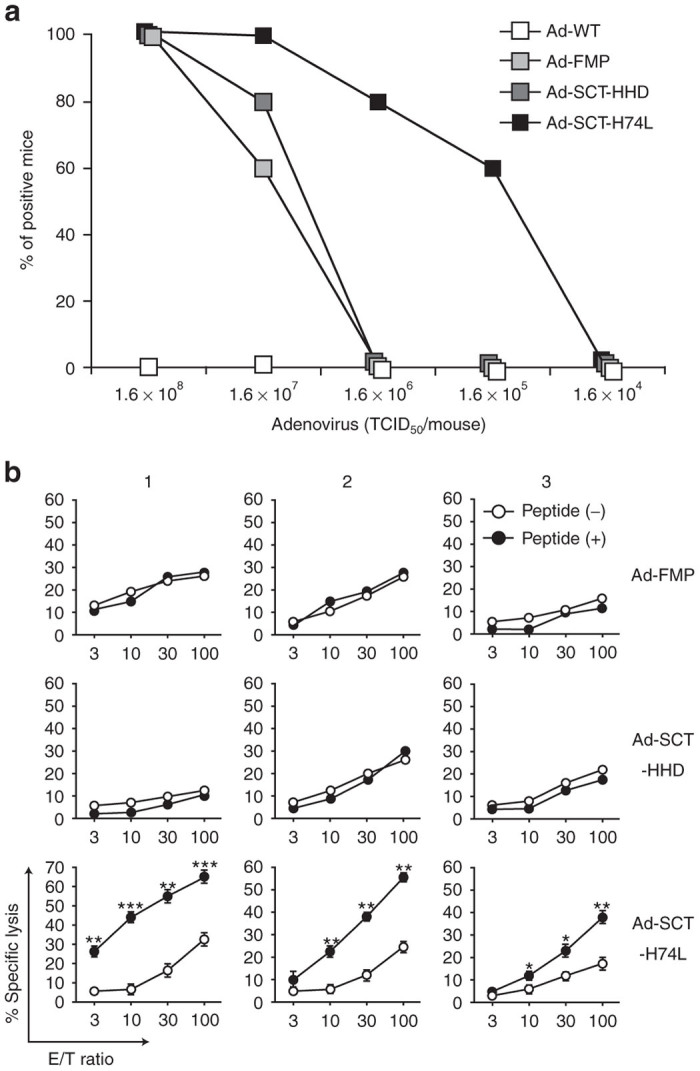
Vaccination with Ad-SCT-H74L enhances FMP-specific CTL induction. HHD mice (10 per group) were immunized with either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L at various doses. After 2 weeks following immunization, spleen cells were stimulated once with FMP58-66 and examined for their FMP-specific killing activities by 51Cr-release assays using RMA-HHD pulsed with or without FMP58-66 as targets. (a) Percents of positive mice in each group were shown. Mice that generated more than 10% relative lysis (% specific lysis of peptide-pulsed RMA-HHD − % specific lysis of unpulsed RMA-HHD) were defined to be positive. (b) Data are representative in mice immunized with each adenovirus at a dose of 1.6 × 106 TCID50. RMA-HHD cells pulsed with (+) or without (−) FMP58-66 were used as targets. The labels 1, 2, 3 indicate data from three different representative mice. *P < 0.05; **P < 0.01; ***P < 0.001 compared to RMA-HHD pulsed without a peptide, Student’s t test.
To further verify the effect of the H74L mutation, in vivo CTL assays were performed (Figure 4). At a high dose of 1.6 × 107 TCID50, mice that had been infected with either Ad-SCT-HHD or Ad-SCT-H74L gave the highest specific lysis of FMP58-66-pulsed splenocytes, while Ad-FMP-injected mice revealed the slightly lower killing activity (Figure 4a). Importantly, in agreement with the data of 51Cr-release assays (Figure 3), FMP-specific CTL activities were significantly observed in mice injected with lower doses (1.6 × 106, 1.6 × 105, and 1.6 × 104 TCID50) of Ad-SCT-H74L (Figure 4a,b). In contrast, any mice infected with the low doses of either Ad-SCT-HHD or Ad-FMP did not generate FMP-specific CTL responses (Figure 4a).
Figure 4.
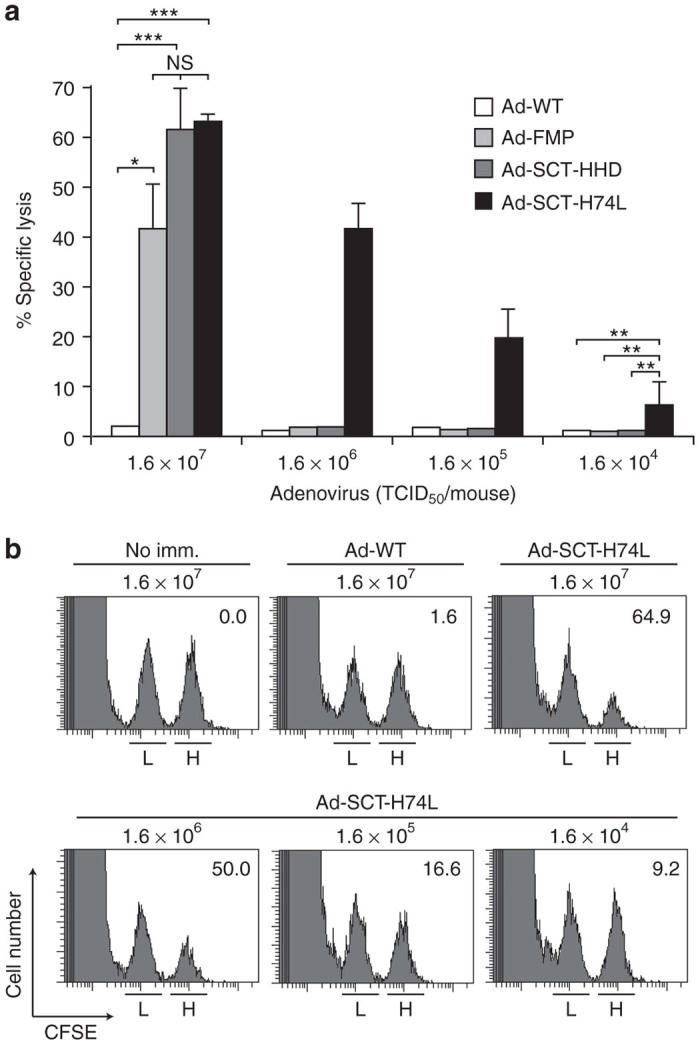
In vivo detection of enhanced FMP-specific cytotoxic T lymphocyte (CTL) killing activities in Ad-SCT-H74L-immunized mice. (a) HHD mice (five per group) were immunized with either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L at various doses. After 1 week following immunization, in vivo CTL assays were performed. The experiment was repeated twice with similar results. Data are shown as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant, One-way analysis of variance. (b) Representative data are shown. The numbers in the panels indicate the percentage of FMP-specific lysis. L: unpulsed spleen cells labeled with a low concentration of CFSE; H: peptide-pulsed spleen cells labeled with a high concentration of CFSE; no imm.: no immunization.
We also carried out the intracellular interferon (IFN)-γ staining of CD8+ T cells at the stimulation with FMP58-66. After 1 week following immunization with either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L at various doses, splenic lymphocytes of the immunized mice were prepared and cultured with FMP58–66 for 5 hours. Cells were then stained for their surface expression of CD8 and peptide-induced intracellular expression of IFN-γ. At an inoculation dose of 1.6 × 107 TCID50, all of the mice that had been infected with Ad-SCT-H74L generated substantial numbers of IFN-γ-secreting CD8+ T cells (Figure 5a,b). At the same dose, infection with either Ad-FMP or Ad-SCT-HHD induced high percentages of IFN-γ-producing CD8+ T cells in several mice tested as well. At a 10-fold lower dose (1.6 × 106 TCID50), however, any of the mice that had been injected with either Ad-FMP or Ad-SCT-HHD did not show significant percentages of IFN-γ-producing CD8+ T cells (Figure 5a). In contrast, Ad-SCT-H74L apparently induced peptide-driven, IFN-γ-producing CD8+ T cells in mice at 1.6 × 106 and 1.6 × 105 TCID50 (Figure 5a,b).
Figure 5.
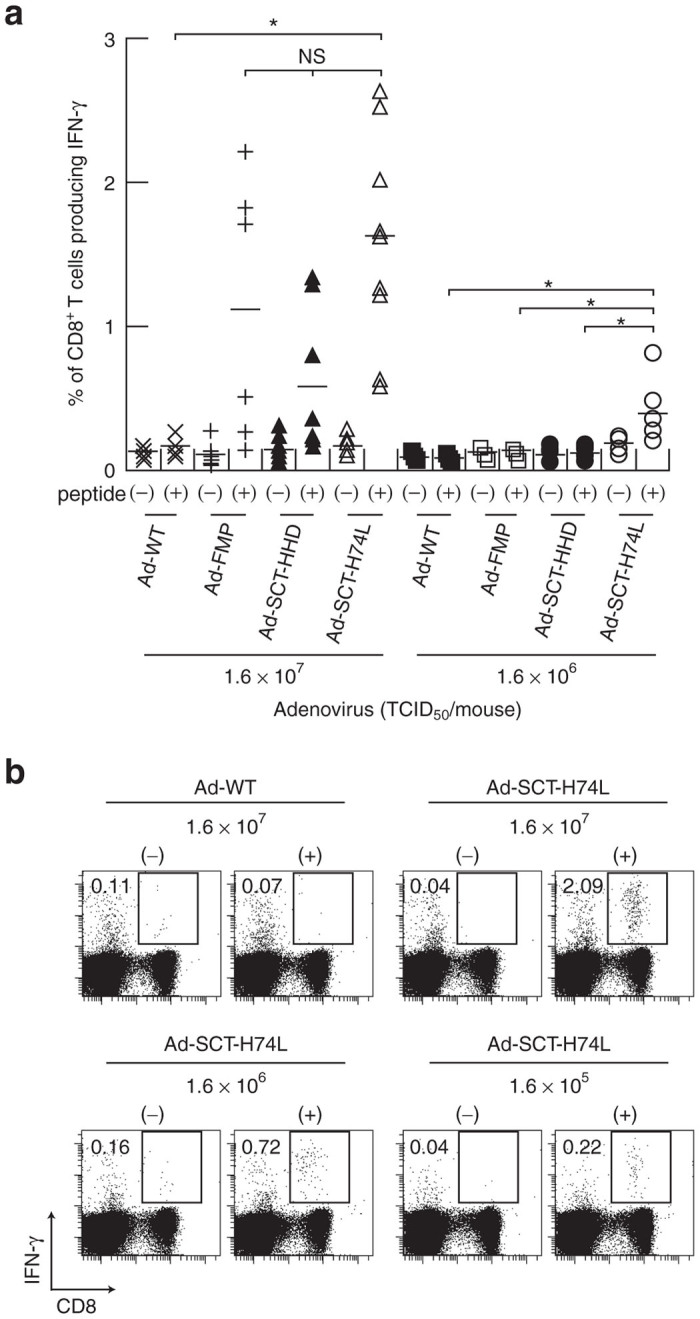
Intracellular IFN-γ staining of FMP-specific CD8+ T cells in mice immunized with Ad-SCT-H74L. HHD mice (five to nine per group) were immunized with Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L at various inoculation doses. After 1 week, splenic lymphocytes were prepared, and stimulated with (+) or without (−) FMP58–66 for 5 hours. Cells were then stained for their surface expression of CD8 and their intracellular expression of IFN-γ. The data indicate the percentages of INF-γ-producing cells within CD8+ T cells. (a) Each symbol represents an individual mouse. Horizontal bars represent the mean. *P < 0.05; NS, not significant, One-way analysis of variance. (b) Data are representative in mice immunized with Ad-WT or Ad-SCT-H74L. The numbers shown indicate the percentages of INF-γ-secreting cells within CD8+ T cells.
Taken all together, these data indicate that the H74L mutation in the HHD molecule significantly improves the efficacy of SCTs for priming peptide-specific CTLs in vivo.
Ad-SCT-H74L provides better protection from tumor challenge than Ad-SCT-HHD
We next performed tumor challenge experiments. After immunization with either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L, HHD mice were challenged by receiving the adoptive cell transfer of carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled RMA-HHD tumor cells expressing the full-length FMP (RMA-HHD-FMP) along with RMA-HHD cells as an internal control. Twelve hours later, % lysis specific for RMA-HHD-FMP cells in each spleen was measured by flow cytometry. At a virus inoculation dose of 1.6 × 107 TCID50, there was no statistical difference in % specific lysis between Ad-SCT-H74L-infected mice and either Ad-SCT-HHD- or Ad-FMP-injected mice (Figure 6a). However, a significant difference between them was observed at an inoculation dose of 1.6 × 106 TCID50. That is, vaccination with Ad-SCT-H74L gave significantly higher clearance of RMA-HHD-FMP tumor cells expressing endogenous FMP58-66 in mice than that with Ad-SCT-HHD or Ad-FMP (Figure 6a,b). We next determined whether Ad-SCT-H74L vaccination could mediate prophylactic protection against a lethal challenge of RMA-HHD-FMP tumor cells. After immunization with 1.6 × 106 TCID50 of either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L, mice were challenged intravenously (i.v.) with 5 × 106 cells/mouse of RMA-HHD-FMP cells. As shown in Figure 6c, all Ad-WT-infected mice died by day 29, whereas many of Ad-SCT-H74L-immunized mice (7 out of 10 mice) survived for over 60 days after tumor inoculation. On the other hand, vaccination with 1.6 × 106 TCID50 of either Ad-FMP or Ad-SCT-HHD did not induce sufficient protection against i.v. challenge with a high dose of tumor cells (Figure 6c). We also tested whether vaccination with Ad-SCT-H74L could inhibit the growth of tumor at the skin. To this end, HHD mice were immunized with various recombinant adenoviruses at a dose of 1.6 × 106 TCID50, and then inoculated subcutaneously with a low number (5 × 104/mouse) of RMA-HHD-FMP tumor cells. Vaccination with Ad-SCT-H74L inhibited tumor growth more efficiently than that with Ad-FMP or Ad-SCT-HHD, while immunization with Ad-WT had no effect on the reduction of tumor growth (Figure 6d). Overall, these data demonstrate that vaccination with Ad-SCT-H74L provides better protection from tumor challenge than that with Ad-FMP or Ad-SCT-HHD.
Figure 6.

Immunization with Ad-SCT-H74L induces strong antitumor protection in HHD mice. (a,b) In vivo cytotoxic T lymphocyte (CTL) assay with tumor cells. Mice (five to eight per group) were vaccinated with Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L at various doses. One week later, RMA-HHD and RMA-HHD-FMP cells were labeled with 0.25 and 2.5 μmol/l of CFSE, respectively, mixed at 1:1, and injected i.v. into each mouse (1 × 107 cells per mouse). After 12–14 hours, spleen cells were prepared and stained with PE-conjugated anti-H-2Db mAb. After washing, H-2Db+ cells were analyzed for their expression of CFSE by flow cytometry. (a) Each symbol represents an individual mouse. Horizontal bars represent the mean. ***P < 0.001; NS, not significant, One-way analysis of variance. (b) Representative data of the in vivo CTL assay are shown. The numbers in the panels indicate the percentage of FMP-specific lysis. L: RMA-HHD labeled with 0.25 μmol/l CFSE; H: RMA-HHD-FMP labeled with 2.5 μmol/l CFSE. (c) Survival of mice. After 1 week following immunization with 1.6 × 106 TCID50 of either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L, mice (ten per group) were challenged i.v. with 5 × 106 RMA-HHD-FMP cells. The experiment was repeated twice with similar results. (d) Inhibition of tumor growth. Mice (five per group) were immunized with 1.6 × 106 TCID50 of Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L. After 1 week, mice were challenged subcutaneously with 5 × 104 RMA-HHD-FMP cells/mouse and monitored for the tumor growth. Curves represent individual mice. The experiment was repeated twice with similar results.
Induction of FMP58–66-specific long-lasting memory CTLs in mice immunized with a low dose of Ad-SCT-H74L
It was also examined whether long-lasting FMP-specific CTLs could be elicited in HHD mice immunized with Ad-SCT-H74L at a low dose of 1.6 × 105 TCID50. At this inoculation dose, neither Ad-SCT-HHD nor Ad-FMP induced detectable FMP-specific CTL responses in mice (Figures 3 and 4). After 60 days following immunization, 51Cr-release assays were then performed at various effector to target cell (E:T) ratios. As shown in Figure 7, FMP58–66-specific CTLs were detected in mice even on day 60 after immunization with Ad-SCT-H74L, but not in mice injected with Ad-WT. These data indicate that immunization with Ad-SCT-H74L at a low dose can effectively generate long-lasting memory CTLs in mice.
Figure 7.
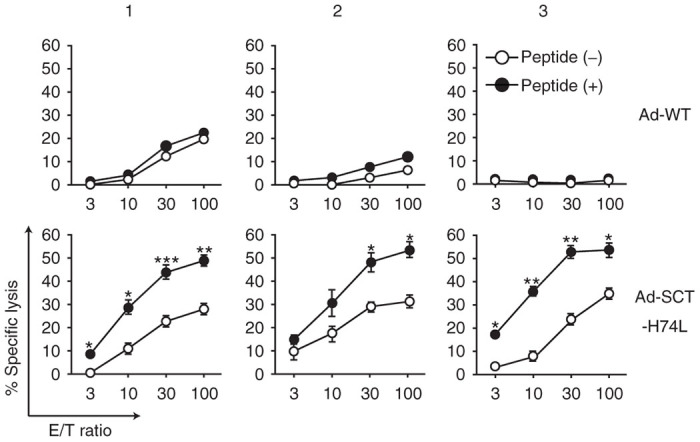
Induction of long-lasting memory CTLs. HHD mice (six per group) were immunized once with 1.6 × 105 TCID50 of Ad-WT or Ad-SCT-H74L. At day 60 after the immunization, spleen cells were prepared, and stimulated in vitro twice with peptide-pulsed syngeneic spleen cells. 51Cr-release assays were then carried out at various E:T ratios, using RMA-HHD cells pulsed with (+) or without (−) FMP58-66 as targets. The labels 1, 2, 3 indicate data from three different representative mice. Representative data are shown as the mean ± SD of triplicate wells. *P < 0.05; **P < 0.01; ***P < 0.001 compared to RMA-HHD pulsed without a peptide, Student’s t-test.
Discussion
The H74L mutation provided the HLA-A*02:01 molecule and its relevant HHD molecule with the unique ability to improve exogenous peptide presentation, leading to enhanced recognition by CTLs without loss of specificity (Figure 1).5,18 This phenomenon was not peptide-specific and came from the enhanced affinity of class I/peptide binding (Figure 1c,d).5,18 In this study, we have clearly shown that this outstanding property was advantageous for the in vivo priming of peptide-specific CTLs and the induction of antitumor immunity, using the SCT technology and recombinant adenovirus in the HHD system. In short, we demonstrated that Ad-SCT-H74L was more effective at priming FMP-specific CTL response than Ad-SCT-HHD or Ad-FMP in HHD mice (Figures 3–5). Furthermore, vaccination with Ad-SCT-H74L induced antitumor immunity better than that with Ad-SCT-HHD or Ad-FMP (Figure 6). Overall, these data strongly suggest that it may be possible to develop an efficient vaccine to enhance protective immunity against tumor and/or virus simply by introducing a point mutation into the MHC class I heavy chain of the SCT.
During the last two decades, a number of mutational analyses for MHC class I molecules have been performed by multiple groups, and several amino acid positions have been defined to be critical for antigen presentation and CTL recognition.3,5,21–23 These findings resulted from the data of CTL responses abolished or impaired by the introduction of a single amino acid substitution at a particular position into the MHC class I heavy chain. At the same time, there were a number of class I mutants that had no practical impact on peptide-specific CTL responses. On the other hand, a few mutants involving H74L have been reported to obviously enhance CTL recognition. Another example is the mutant HLA-A*0201 containing a mutation from glutamine to glutamic acid at position 115 (Q115E).24,25 The Q115E mutation resulted in the enhancement of CD8 binding that improved antigen recognition at the cell surface and CTL priming in vitro.24
The histidine at position 74 is located in the α1 helix and forms a portion of the wall of the C pocket that is one of the six binding pockets within the peptide-binding groove.2 According to the crystallographic analysis, however, this residue does not appear to engage in the direct interaction with peptides.26 Since position 74 is a histidine or an aspartic acid (D) in all HLA-A molecules except HLA-A*80:01 whose position 74 is an asparagine, this invariant residue may play an important role in the conformational stability common to the HLA-A. All HLA-A2 subtypes encode a histidine except HLA-A*02:11 which uses an aspartic acid. In addition, for all HLA-B molecules which have been sequenced, position 74 is an aspartic acid or a tyrosine (Y). Two different groups found that CTL recognition presented by H74D was slightly impaired.21,23 The same group showed that H74Y did not display an increase in CTL recognition.23 Interestingly, the H74D mutation had little effect on the CTL response but the H74Y mutation impaired CTL recognition in this study (Figure 1e). In addition to H74L, three more mutants, H74A, H74F and H74T enhanced FMP-specific CTL responses in the presence of an exogenous FMP58–66 peptide (Figure 1e). It is unclear at present why these four particular amino acid substitutions caused the enhancement of CTL recognition, but we could speculate that these mutations may provide more depth in the C pocket which is usually shallow in comparison with other pockets, leading to the increased affinity of peptide binding (Figure 1d). In contrast to the exogenous peptide, we previously showed that the endogenously processed peptide was not presented to CTL by HLA-A*02:01 with the H74L mutation.5,18 In accordance with these data, FMP-specific CTLs could not recognize either RMA-H74L, -H74A, -H74F or -H74T cells infected with influenza A virus (unpublished observations). These data suggest that the histidine at position 74 might plays a critical role related to the formation of the ER peptide-loading complex in the antigen processing. This idea might be able to explain why the human immune system has not evolved to incorporate these mutations at position 74.
To increase the efficiency of antigen presentation by MHC class I molecules, the SCTs have been developed by Hansen et al.6–8 Because the SCTs are immunogenic to stimulate peptide-specific CTLs in vivo, this technology may have applications as DNA vaccines against virus infection or tumors. Taking advantage of this attribute, we examined whether the H74L mutant had the potential as a vaccine platform for the immunotherapy. As shown in Figures 3–5, Ad-SCT-H74L stimulated FMP-specific CTLs in HHD mice at lower viral doses than Ad-SCT-HHD or Ad-FMP. These data strongly suggest that SCT-H74L molecules are more stable at the cell surface than SCT-HHD or native HHD molecules, and thereby, FMP-specific CTLs can be generated even by fewer MHC class I/peptide complexes per cell on the cell surface in mice vaccinated with Ad-SCT-H74L. As an SCT molecule retains a covalently attached peptide, the original SCTs are supposed to be steadier on the cell surface than are the equivalent native class I molecules, making them potentially more immunogenic.6 In the original SCTs, however, the linker extending from the C-terminus of the peptide would disrupt the F pocket anchoring of the peptide in the peptide-binding groove.27,28 A peptide with the high binding affinity may be little affected by this disruption. However, when a peptide with the low or medium binding affinity is incorporated in the SCTs, their assembly may be much less efficient. This might explain our current data showing that Ad-SCT-HHD was not drastically superior to Ad-FMP at the in vivo CTL priming and the antitumor protection (Figures 3–6). Since the H74L substitution enhances the peptide binding to HLA-A*02:01 (Figure 1d), this mutation would diminish the negative influence of the linker on the peptide anchoring.
To eliminate the detrimental effect of the peptide linker, Hansen et al. attempted to improve the design of the SCTs by engineering the MHC class I heavy chain. They focused on the tyrosine at position 84 (Y84) of the heavy chain because the conserved residue, Y84 creates a part of F pocket and plays a major role to accommodate the C-terminus of the peptide in the peptide-binding groove.27 Firstly, a mutation from tyrosine to alanine at position 84 of a heavy chain (Y84A) was introduced into the SCT comprises H-2Kb heavy chain, β2m, and an ovalbumin peptide (SCT-Y84A).27 It was speculated that Y84A would partially open the binding groove to allow a better fit of the C-terminal linker-attached peptide. Indeed, ovalbumin-specific CTLs killed target cells expressing SCT-Y84A somewhat better than those expressing the original SCT or native H-2Kb without the Y84A mutation.27 Unlike H74L (Figure 1d),5,18 however, noncovalently attached native H-2Kb mutant with the Y84A mutation exhibited very poor peptide binding in comparison with WT native H-2Kb,27 indicating the enhancement mechanism of SCT-Y84L in the CTL recognition is different from that of SCT-H74L. Notably, it is still unclear whether SCT-Y84A can efficiently elicit ovalbumin-specific CTLs in vivo. Secondary, the tyrosine at position 84 of the MHC class I heavy chain and the second residue of the first linker were changed to cysteine to form a new disulfide bond between the two cysteine residues, covalently trapping the peptide in the binding groove.28,29 The SCTs with the disulfide traps were particularly useful for relatively weak binding peptides. It was shown that DNA vaccines with disulfide traps enhanced the immune response to a low affinity peptide in vivo.30 However, there is no published information concerning the functional comparison between the SCT-Y84A and disulfide trap. In addition, the Q115E mutation described above24,25 was also introduced into SCT of H-2Kb (SCT-Q115E).30 However, they presented only a data of ELISPOT analysis to show the predominance of SCT-Q115E over its WT counterpart at in vivo priming peptide-specific CTLs in mice.30 Anyhow, the introduction of the Y84A mutation, the disulfide trap and/or the Q115E mutation into SCT-H74L might generate a synergistic effect on the in vivo priming of peptide-specific CTLs. This approach should also be beneficial for improving the instability of MHC class I multimers composed of lower affinity MHC/peptide complexes and enable them to make more comprehensive analyses of CTL responses to pathogens and tumors.
The most striking results in these experiments are that mice vaccinated with Ad-SCT-H74L even at a low dose of 1.6 × 106 TCID50 were protected against both subcutaneously and i.v. challenges with RMA-HHD-FMP tumor cells (Figure 6c,d). These data reveal the high efficiency of Ad-SCT-H74L for the induction of protective antitumor immunity, suggesting that SCT-H74L has the great potential to improve the current tumor vaccine strategy. In addition, the SCTs have a valuable advantage to bypass antigen processing,7,8 and therefore, the SCT-H74L may overcome several immune evasion strategies of tumors that block the antigen processing pathway. It was shown that the enhanced peptide loading by the H74L molecules was not peptide-specific (Figure 1c,d).18 However, it is necessary to confirm that other HLA-A*02:01-restricted peptides in the SCT-H74L format would behave in the same manner as FMP58–66 does. This is of particular importance in designing SCT-H74L as tumor vaccines because many tumor-derived peptides usually have low binding affinity to MHC class I molecules. The capacity of SCT-H74L to bind and present low affinity epitopes remains to be investigated. However, we have preliminary data showing that the H74L mutation enhanced binding and CTL recognition of tumor peptides including WT1, Her2/neu and MAGE-3 in comparison to the WT counterpart, using RMA-S-H74L and RMA-H74L. Hence, it is possible to assume that the SCT-H74L may improve to bind and present low affinity epitopes including tumor epitopes and self peptides. Another major concern is whether any H74L-like mutations can be found in other HLA class I alleles. On the bais of this data, the conformational change of the shallow C pocket might be of key importance.
In conclusion, we have clearly and extensively demonstrated for the first time that the introduction of a single mutation into the HLA-A*02:01 of the SCT induce enhancement of both in vivo priming of peptide-specific CTLs and protective antitumor immunity. This novel approach might serve as a driving force for the development of effective immunotherapy against tumors and pathogens.
Materials and Methods
Mice
We used the HLA-A*02:01-transgenic, and H-2Db−/− β2m−/− double knockout mice that express transgenic HLA-A*02:01 monochains, designated as HHD, in which human β2m is covalently linked to a chimeric heavy chain composed of HLA-A*02:01 (α1 and α2 domains) and H-2Db (α3, transmembrane and cytoplasmic domains) (Figure 1a,b).15 Seven- to ten-week-old mice were used for all experiments. Mice were housed in appropriate animal care facilities at Saitama Medical University, and were handled according to the international guideline for experiments with animals. This study was approved by the Animal Research Committee of Saitama Medical University.
Synthetic peptides
HLA-A*02:01-restricted epitopes used were as follows: FMP58–66 (sequence: GILGFVFTL),31 HPV-E629–38 (sequence: TIHDIILECV),32 HTLV-tax11–19 (sequence: LLFGYPVYV),33 and HIV-pol476–484 (sequence: ILKEPVHGV).34 Synthetic peptides corresponding to the above epitopes were synthesized by Operon Biotechnologies (Tokyo, Japan).
Cell lines
Mouse lymphoma cell lines, RMA (H-2b) and TAP-2-deficient RMA-S (H-2b),19 were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO) with 10% FCS (Sigma-Aldrich) (R-10). The HHD gene-transfected cell lines, RMA-HHD and RMA-S-HHD, were previously described,15 and maintained in R-10 containing 500 μg/ml G418 (Nacalai Tesque, Kyoto, Japan). A human kidney cell line, 293 cell lines were obtained from the American Type Culture Collection (ATCC) (Rockville, MD). The Ad-293 cell line was provided with the AdEasy adenoviral vector system (Agilent Technologies, La Jolla, CA). 293 and Ad-293 cells were maintained in DMEM (Sigma-Aldrich) supplemented with 10% FCS (Sigma-Aldrich).
Generation of peptide-specific CTL lines
Spleen cells of naive HHD mice were prepared, pulsed with 10 μmol/l of an appropriate peptide for 1 hour at 37 °C, and irradiated at 20 grays. The peptide-pulsed cells were then adoptively transferred i.v. into mice (2 × 107 cells/mouse). After 2 weeks, spleen cells of immunized mice were prepared, and stimulated twice or three times with peptide-pulsed, irradiated (20 grays) syngeneic spleen cells. After the second stimulation, human recombinant IL-2 (Cetus, Barkley, CA) was added to the culture media at a final concentration of 100 U/ml. The peptide-specific killing activities of CTL lines were checked by 51Cr-release assays prior to experiments, using RMA-HHD cells pulsed with or without an appropriate peptide as targets. Peptide-specific CTL lines were then used to investigate for their killing activities against RMA, RMA-HHD and RMA-HHD mutants in the presence or absence of an appropriate peptide at various concentrations at an E:T ratio of 1 in standard 51Cr-release assays.
Plasmid constructs
The cDNA encoding HHD gene was generated by reverse transcriptase PCR from total RNA of RMA-HHD cells. After addition of restriction site sequences at the 5′ and 3′ ends, HHD cDNA was subcloned into the NheI-HindIII sites of the pcDNA3.1 (+) (Invitrogen, Carlsbad, CA) expression vector (pcDNA3.1-HHD). pcDNA3.1-HHD was then used as a template to construct a library of mutant HHD genes ligated with pcDNA3.1. The inverse PCR-based site-directed mutagenesis35 was performed using KOD-Plus mutagenesis kit (Toyobo, Osaka, Japan) according to the manufacture’s instructions. In brief, this method requires two back-to-back primers oriented in the reverse direction to amplify the entire plasmid. One of the primers contained a nucleotide change that caused a single amino acid substitution at position 74 in the α1 domain of HHD. After the reaction, the template plasmid DNA was digested by DpnI that recognizes the Gm6ATC (m6-methylated) sites of plasmid DNA purified from ordinary host E. coli. The indigested PCR product containing a nucleotide substitution was then recircularized by T4 polynucleotide kinase and ligase, and the resulting plasmid was transformed. We generated 15 different kinds of pcDNA3.1-mutant HHD plasmids with point mutations at position 74 from histidine to alanine (H74A), cysteine (H74C), aspartic acid (H74D), glutamic acid (H74E), phenylalanine (H74F), glycine (H74G), isoleucine (H74I), lysine (H74K), leucine (H74L), asparagine (H74N), glutamine (H74Q), serine (H74S), threonine (H74T), valine (H74V), and tyrosine (H74Y). The whole nucleotide sequence of each mutant HHD gene was confirmed by the DNA sequencing services (Operon Biotechnologies).
An artificial gene encoding SCT of HHD linked to FMP58–66 peptide (SCT-HHD) (Figure 2a,b) was synthesized by Operon Biotechnologies. This gene consists of the leader sequence of human β2m followed immediately by the FMP58–66 sequence and then a linker of 15 residues ((G4S)3). This first linker is followed by the human mature β2m sequence, the second linker of 15 residues ((G4S)3), and then the HHD sequence. In the process of the SCT gene synthesis, the XhoI and HindIII site sequences were added at the 5′ and 3′ ends, respectively. The SCT gene was subcloned into the XhoI and HindIII sites of the expression vector, pcDNA3.1 (−) (Invitrogen) (pcDNA3.1-SCT-HHD). A nucleotide substitution that causes the H74L mutation in the α1 domain of HHD was then created in pcDNA3.1-SCT-HHD (pcDNA3.1-SCT-H74L) using the KOD-Plus mutagenesis kit (Toyobo) as described above. The construct was confirmed by DNA sequencing (Operon Biotechnologies).
The gene encoding the whole FMP of influenza A virus (A/PR8/34) was synthesized by Operon Biotechnologies, and was inserted into the BglII-XhoI sites of the pAcGFP1-Hyg-N1 expression vector (Clontech Laboratories, Mountain View, CA) (pAcGFP1-FMP). This vector allows FMP to be fused to GFP protein at C-terminus.
Transfectants
RMA cells were transfected with each of pcDNA3.1-mutant HHD plasmids by electroporation (Gene Pulser, Bio-Rad Laboratories, Hercules, CA) as described before.36 RMA-S cells were also electroporated with pcDNA3.1-H74L, pcDNA3.1-SCT-HHD, or pcDNA3.1-SCT-H74L. After selection with G418 (Nakarai Tesque) at a final concentration of 1.2 mg/ml, transfectants were analyzed for their surface HHD expression by flow cytometry (FACSCanto II, BD Biosciences, Franklin Lakes, NJ) using indirect immunofluorescence with anti-HLA-A2 monoclonal antibody, BB7.2.37 Transfectants expressing multiple positive peaks were cloned by limiting dilution. The expression of mutant HHD molecules on each transfectant was almost equivalent to that of RMA-HHD or RMA-S-HHD cells.
RMA-HHD cells were transfected with pAcGFP1-FMP by electroporation (RMA-HHD-FMP). After selection with Hygromycin B (Nakarai Tesque) at a final concentration of 1.2 mg/ml, cells were cloned and confirmed for their expression of GFP-fused FMP by flow cytometry (FACSCantoTM II, BD Biosciences) (data not shown).
Construction of recombinant adenoviruses
Recombinant adenovirus expressing SCT molecules of FMP58–66 peptide-linked HHD (Ad-SCT-HHD) or -H74L (Ad-SCT-H74L) was generated using the AdEasy Adenoviral Vector System (Agilent Technologies) as described before.38 Briefly, the synthesized SCT gene encoding SCT of HHD linked to FMP58–66 peptide (Operon Biotechnologies) described above was inserted into the XhoI-HindIII sites of pShuttle-CMV vector (pShuttle-CMV-SCT-HHD). The H74L point mutation in this construct (pShutle-CMV-SCT-H74L) was created using the KOD-Plus mutagenesis kit (Toyobo) as described above. The construct was confirmed by DNA sequencing (Operon Biotechnologies). The shuttle vector, pShuttle-CMV-SCT-HHD or pShuttle-CMV-SCT-H74L, was linearized with PmeI (New England Biolabs, Herts, UK) and transformed into the BJ5183-Ad-1 competent cells containing the adenoviral backbone plasmid vector, pAdEasy-1. Recombinant adenovirus plasmids were digested with PacI (New England Biolabs), and transfected into Ad-293 cells by Lipofectamine 2000 (Invitrogen). After 10–14 days following transfection, adenoviruses were released from the cells by three cycles of rapid freezing and thawing. Virus was amplified in 293 cells, and viral titers were determined by calculating TCID50 on 293 cells.
The synthesized FMP gene used for the construction of pAcGFP1-FMP was inserted into the he BglII-XhoI sites of pShuttle-IRES-hrGFP-1 vector (Agilent Technologies). Recombinant adenovirus expressing FMP (Ad-FMP) was then generated using the AdEasy system as described above. The pShuttle-IRES-hrGFP-1 vector allows the FMP protein to be fused to the 3×FLAG tag sequence at C-terminus, and therefore, Western blotting was performed as described previously38 to detect expression of the FMP protein in cells infected with Ad-FMP using anti-FLAG M2 mAb (Sigma-Aldrich) (data not shown).
For the immunization, mice were injected intraperitoneally (i.p.) once with either 1.6 × 104, 1.6 × 105, 1.6 × 106, 1.6 × 107 or 1.6 × 108 TCID50 of adenovirus in 0.5 ml of phosphate-buffered saline.
51Cr-release assay
51Cr-release assays were carried out as described before.39 Briefly, after 2 weeks following immunization, spleen cells of immunized mice were stimulated once with gamma-irradiated, syngeneic spleen cells pulsed with 10 μmol/l of an appropriate peptide for 1 week, and used as effector cells (Figure 3). For the detection of memory CTLs (Figure 7), spleen cells were prepared on day 60 after the immunization, and stimulated twice with gamma-irradiated, peptide-pulsed syngeneic spleen cells. At the second stimulation, human recombinant IL-2 (Cetus) was added to the culture media at a final concentration of 100 U/ml. To examine various HHD mutants, peptide-specific CTLs were employed (Figure 1). For targets, RMA, RMA-HHD and RMA-HHD mutant cells were labeled with 100 μCi of Na251CrO4. After a 4-hour incubation, the radioactivity in each supernatant was counted. Results were calculated as the mean of a triplicate assay. Percent-specific lysis was calculated according to the formula: % specific lysis = [(cpmsample − cpmspontaneous)/(cpmmaximum − cpmspontaneous)] × 100. Spontaneous release represents the radioactivity released by target cells in the absence of effectors, and maximum release represents the radioactivity released by target cells lysed with 5% Triton X-100 (Sigma-Aldrich). FMP-specific CTL responses against various RMA-HHD mutant cells were standardized as the percent relative lysis by the following formula: % relative lysis = [(% specific lysis against RMA-HHD mutants − % specific lysis against RMA)/(% specific lysis against RMA-HHD − % specific lysis against RMA)] × 100.
Peptide-binding assay
Binding of peptides to the HHD and H74L molecules was investigated by the cell surface stabilization assay as described,36 using RMA-S-HHD and RMA-S-H74L. In brief, RMA-S transfectants were cultured for 16 hours at 26 °C in a CO2 incubator, and were then pulsed with peptides at various concentrations for 1 hour at 26 °C. After incubation for 3 hours at 37 °C, peptide-pulsed cells were stained with BB7.2, followed by FITC-labeled goat antimouse IgG antibody (Sigma-Aldrich). The mean fluorescence intensity (MFI) of each cell line was measured by flow cytometry (FACSCantoTM II, BD Biosciences), and standardized as the percent cell surface expression relative to RMA-S transfectants incubated at 26 °C overnight by the following formula: % relative binding = [{(MFI of cells pulsed with a peptide) – (MFI of cells incubated at 37 °C without a peptide)}/{(MFI of cells incubated at 26 °C without a peptide) – (MFI of cells incubated at 37 °C without a peptide)}] × 100.
Intracellular cytokine staining
Intracellular cytokine staining was performed as described.40 In brief, after 1 week following immunization, spleen cells were incubated with 10 μmol/l of FMP58–66 for 5 hours at 37 °C in the presence of brefeldin A (GolgiPlugTM, BD Biosciences). After blocking Fc receptors, cells were stained with FITC-conjugated antimouse CD8 smonoclonal antibody (mAb) (BioLegend, San Diego, CA), followed by the staining of intracellular IFN-γ with PE-conjugated anti-IFN-γ mAb (BioLegend). After washing the cells, flow cytometric analyses were performed.
In vivo CTL assay
In vivo CTL assays were carried out as described before.39 In brief, naive spleen cells were prepared and split into two populations. One population was pulsed with 10 μmol/l of FMP58–66 and labeled with 2.5 μmol/l of CFSE (Molecular Probes, Eugene, OR), whereas the other population was just labeled with 0.25 μmol/l of CFSE. An equal number of cells from each population was mixed together, and adoptively transferred i.v. into mice (1 × 107/mouse) that had been immunized with adenovirus 1 week earlier. After 12–14 hours, spleen cells were prepared and analyzed by flow cytometry (FACSCantoTM II, BD Biosciences). To calculate specific lysis, the following formula was used: % specific lysis = [1 − {(number of CFSElow cells in normal mice)/(number of CFSEhigh cells in normal mice)}/{(number of CFSElow cells in immunized mice)/(number of CFSEhigh cells in immunized mice)}] × 100.
Tumor challenge experiments
(i) In vivo CTL assay with tumor cells: RMA-HHD and RMA-HHD-FMP cells were labeled with 0.25 and 2.5 μmol/l of CFSE, respectively, and an equal number of cells from each cell line was mixed together. A total of 1 × 107 cells was injected i.v. into each mouse that had been immunized with adenovirus 1 week previously. After 12–14 hours, spleen cells were prepared, treated with anti-Fc receptor mAb (BioLegend), and stained with PE-conjugated anti-H-2Db mAb (BioLegend). After washing, H-2Db+ cells were analyzed for their expression of CFSE by flow cytometry (FACSCantoTM II, BD Biosciences). Percent-specific lysis was calculated according to the formula of standard in vivo CTL assay described above. (ii) Survival of mice: Ten HHD mice per group were immunized with 1.6 × 106 TCID50 of either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L. After 1 week following immunization, mice were challenged i.v. with 5 × 106 RMA-HHD-FMP cells. Mice were observed for survival for more than 60 days. (iii) Tumor growth: Mice (five per group) were immunized with 1.6 × 106 TCID50 of either Ad-WT, Ad-FMP, Ad-SCT-HHD or Ad-SCT-H74L. One week later, mice were inoculated subcutaneously with 5 × 104 RMA-HHD-FMP cells on the right abdominal flank. Tumor size was measured every 2–3 days using a caliper in the two perpendicular diameters. The tumor volume (mm3) was calculated using the following formula: tumor volume = (D × d2)/2, where d is the smaller of the two diameters. Mice were killed when the tumor volume grew up to 800 mm3.
Statistical analyses
Statistical analyses between two groups were performed with Student’s t-test. One-way analysis of variance followed by post hoc tests was carried out for statistical analyses between multiple groups using Graph Pad Prism 5 software (GraphPad software, San Diego, CA) . A value of P < 0.05 was considered statistically significant.
Acknowledgments
The authors are grateful to Professor François A. Lemonnier (Pasteur Institute, Paris, France) for providing HHD mice, RMA, RMA-S, RMA-HHD and RMA-S-HHD cells. We also thank Professor Jeffrey. A. Frelinger (The University of Arizona, Tucson, AZ) for helpful comments. The initial idea and observation in this work were made in Frelinger’s lab at UNC-Chapel Hill. This work was supported by a Grant-in-Aid for Scientific Research (C) (No. 23590550) to M. M. from Japan Society for the Promotion of Science. M.M. conceived and designed the experiments. M.M. and M.K. performed the experiments and analyzed the data. S.M. and T.A. contributed reagents/materials/analysis tools. M.M. wrote the article.
The authors declare no conflict of interest.
References
- Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- Saper MA, Bjorkman PJ, Wiley DC. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J Mol Biol. 1991;219:277–319. doi: 10.1016/0022-2836(91)90567-p. [DOI] [PubMed] [Google Scholar]
- Matsui M, Hioe CE, Frelinger JA. Roles of the six peptide-binding pockets of the HLA-A2 molecule in allorecognition by human cytotoxic T-cell clones. Proc Natl Acad Sci USA. 1993;90:674–678. doi: 10.1073/pnas.90.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Moots RJ, Warburton RJ, Peace-Brewer AL, Tussey LG, Quinn DG. Genetic evidence for difference between intracellular and extracellular peptides in influenza A matrix peptide-specific CTL recognition. J Immunol. 1995;154:1088–1096. [PubMed] [Google Scholar]
- Tussey LG, Matsui M, Rowland-Jones S, Warburton R, Frelinger JA, McMichael A. Analysis of mutant HLA-A2 molecules. Differential effects on peptide binding and CTL recognition. J Immunol. 1994;152:1213–1221. [PubMed] [Google Scholar]
- Yu YY, Netuschil N, Lybarger L, Connolly JM, Hansen TH. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol. 2002;168:3145–3149. doi: 10.4049/jimmunol.168.7.3145. [DOI] [PubMed] [Google Scholar]
- Hansen TH, Connolly JM, Gould KG, Fremont DH. Basic and translational applications of engineered MHC class I proteins. Trends Immunol. 2010;31:363–369. doi: 10.1016/j.it.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsiou E, Brzostek J, Gould KG. Properties and applications of single-chain major histocompatibility complex class I molecules. Antioxid Redox Signal. 2011;15:645–655. doi: 10.1089/ars.2010.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Peng S, He L, Tsai YC, Boyd DA, Hansen TH. Cancer immunotherapy using a DNA vaccine encoding a single-chain trimer of MHC class I linked to an HPV-16 E6 immunodominant CTL epitope. Gene Ther. 2005;12:1180–1186. doi: 10.1038/sj.gt.3302519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CF, Calizo R, Tsai YC, He L, Wu TC. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine. 2007;25:127–135. doi: 10.1016/j.vaccine.2006.06.087. [DOI] [PubMed] [Google Scholar]
- Kim S, Li L, McMurtrey CP, Hildebrand WH, Weidanz JA, Gillanders WE. Single-chain HLA-A2 MHC trimers that incorporate an immundominant peptide elicit protective T cell immunity against lethal West Nile virus infection. J Immunol. 2010;184:4423–4430. doi: 10.4049/jimmunol.0903955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zuiani A, Carrero JA, Hansen TH. Single chain MHC I trimer-based DNA vaccines for protection against Listeria monocytogenes infection. Vaccine. 2012;30:2178–2186. doi: 10.1016/j.vaccine.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JL, Morris CR, Solheim JC. Virus evasion of MHC class I molecule presentation. J Immunol. 2003;171:4473–4478. doi: 10.4049/jimmunol.171.9.4473. [DOI] [PubMed] [Google Scholar]
- Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172–1187. doi: 10.1093/jnci/djt184. [DOI] [PubMed] [Google Scholar]
- Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Pérarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J Exp Med. 1997;185:2043–2051. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucherma R, Kridane-Miledi H, Bouziat R, Rasmussen M, Gatard T, Langa-Vives F. HLA-A*01:03, HLA-A*24:02, HLA-B*08:01, HLA-B*27:05, HLA-B*35:01, HLA-B*44:02, and HLA-C*07:01 monochain transgenic/H-2 class I null mice: novel versatile preclinical models of human T cell responses. J Immunol. 2013;191:583–593. doi: 10.4049/jimmunol.1300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolo S. HLA class I transgenic mice: development, utilisation and improvement. Expert Opin Biol Ther. 2005;5:919–938. doi: 10.1517/14712598.5.7.919. [DOI] [PubMed] [Google Scholar]
- Caley RR, Peace-Brewer AL, Matsui M, Frelinger JA. Analysis of the mutant HLA-A*0201 heavy chain H74L: impaired TAP-dependent peptide loading. Hum Immunol. 1999;60:743–754. doi: 10.1016/s0198-8859(99)00022-1. [DOI] [PubMed] [Google Scholar]
- Powis SJ, Townsend AR, Deverson EV, Bastin J, Butcher GW, Howard JC. Restoration of antigen presentation to the mutant cell line RMA-S by an MHC-linked transporter. Nature. 1991;354:528–531. doi: 10.1038/354528a0. [DOI] [PubMed] [Google Scholar]
- Palmowski MJ, Parker M, Choudhuri K, Chiu C, Callan MF, van der Merwe PA. A single-chain H-2Db molecule presenting an influenza virus nucleoprotein epitope shows enhanced ability at stimulating CD8+ T cell responses in vivo. J Immunol. 2009;182:4565–4571. doi: 10.4049/jimmunol.0803893. [DOI] [PubMed] [Google Scholar]
- McMichael AJ, Gotch FM, Santos-Aguado J, Strominger JL. Effect of mutations and variations of HLA-A2 on recognition of a virus peptide epitope by cytotoxic T lymphocytes. Proc Natl Acad Sci USA. 1988;85:9194–9198. doi: 10.1073/pnas.85.23.9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CC, Carreno BM, Turner RV, Koenig S, Biddison WE. The 45 pocket of HLA-A2.1 plays a role in presentation of influenza virus matrix peptide and alloantigens. J Immunol. 1991;146:3508–3512. [PubMed] [Google Scholar]
- Teng JM, Hogan KT. Both major and minor peptide-binding pockets in HLA-A2 influence the presentation of influenza virus matrix peptide to cytotoxic T lymphocytes. Mol Immunol. 1994;31:459–470. doi: 10.1016/0161-5890(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Wooldridge L, Lissina A, Vernazza J, Gostick E, Laugel B, Hutchinson SL. Enhanced immunogenicity of CTL antigens through mutation of the CD8 binding MHC class I invariant region. Eur J Immunol. 2007;37:1323–1333. doi: 10.1002/eji.200636765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge L, van den Berg HA, Glick M, Gostick E, Laugel B, Hutchinson SL. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J Biol Chem. 2005;280:27491–27501. doi: 10.1074/jbc.M500555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden DR, Garboczi DN, Wiley DC. The antigenic identity of peptide-MHC complexes: a comparison of the conformations of five viral peptides presented by HLA-A2. Cell. 1993;75:693–708. doi: 10.1016/0092-8674(93)90490-h. [DOI] [PubMed] [Google Scholar]
- Lybarger L, Yu YY, Miley MJ, Fremont DH, Myers N, Primeau T. Enhanced immune presentation of a single-chain major histocompatibility complex class I molecule engineered to optimize linkage of a C-terminally extended peptide. J Biol Chem. 2003;278:27105–27111. doi: 10.1074/jbc.M303716200. [DOI] [PubMed] [Google Scholar]
- Truscott SM, Lybarger L, Martinko JM, Mitaksov VE, Kranz DM, Connolly JM. Disulfide bond engineering to trap peptides in the MHC class I binding groove. J Immunol. 2007;178:6280–6289. doi: 10.4049/jimmunol.178.10.6280. [DOI] [PubMed] [Google Scholar]
- Truscott SM, Wang X, Lybarger L, Biddison WE, McBerry C, Martinko JM. Human major histocompatibility complex (MHC) class I molecules with disulfide traps secure disease-related antigenic peptides and exclude competitor peptides. J Biol Chem. 2008;283:7480–7490. doi: 10.1074/jbc.M709935200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Herndon JM, Truscott SM, Hansen TH, Fleming TP, Goedegebuure P. Engineering superior DNA vaccines: MHC class I single chain trimers bypass antigen processing and enhance the immune response to low affinity antigens. Vaccine. 2010;28:1911–1918. doi: 10.1016/j.vaccine.2009.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotch F, Rothbard J, Howland K, Townsend A, McMichael A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature. 1987;326:881–882. doi: 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- Ressing ME, Sette A, Brandt RM, Ruppert J, Wentworth PA, Hartman M. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154:5934–5943. [PubMed] [Google Scholar]
- Kannagi M, Shida H, Igarashi H, Kuruma K, Murai H, Aono Y. Target epitope in the Tax protein of human T-cell leukemia virus type I recognized by class I major histocompatibility complex-restricted cytotoxic T cells. J Virol. 1992;66:2928–2933. doi: 10.1128/jvi.66.5.2928-2933.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsomides TJ, Walker BD, Eisen HN. An optimal viral peptide recognized by CD8+ T cells binds very tightly to the restricting class I major histocompatibility complex protein on intact cells but not to the purified class I protein. Proc Natl Acad Sci USA. 1991;88:11276–11280. doi: 10.1073/pnas.88.24.11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer WP, Morris SK. Enzymatic inverse PCR: a restriction site independent, single-fragment method for high-efficiency, site-directed mutagenesis. Biotechniques. 1992;13:214–220. [PubMed] [Google Scholar]
- Kohyama S, Ohno S, Suda T, Taneichi M, Yokoyama S, Mori M. Efficient induction of cytotoxic T lymphocytes specific for severe acute respiratory syndrome (SARS)-associated coronavirus by immunization with surface-linked liposomal peptides derived from a non-structural polyprotein 1a. Antiviral Res. 2009;84:168–177. doi: 10.1016/j.antiviral.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham P, Brodsky FM. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum Immunol. 1981;3:277–299. doi: 10.1016/0198-8859(81)90065-3. [DOI] [PubMed] [Google Scholar]
- Suda T, Kawano M, Nogi Y, Ohno N, Akatsuka T, Matsui M. The route of immunization with adenoviral vaccine influences the recruitment of cytotoxic T lymphocytes in the lung that provide potent protection from influenza A virus. Antiviral Res. 2011;91:252–258. doi: 10.1016/j.antiviral.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Kawano M, Morikawa K, Suda T, Ohno N, Matsushita S, Akatsuka T. Chimeric SV40 virus-like particles induce specific cytotoxicity and protective immunity against influenza A virus without the need of adjuvants. Virology. 2014;448:159–167. doi: 10.1016/j.virol.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Matsui M, Moriya O, Yoshimoto T, Akatsuka T. T-bet is required for protection against vaccinia virus infection. J Virol. 2005;79:12798–12806. doi: 10.1128/JVI.79.20.12798-12806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]