

Abstract
A preclinical safety study was conducted to evaluate the short- and long-term toxicity of a recombinant adeno-associated virus serotype 8 (AAV2/8) vector that has been developed as an immune-modulatory adjunctive therapy to recombinant human acid α-glucosidase (rhGAA, Myozyme) enzyme replacement treatment (ERT) for patients with Pompe disease (AAV2/8-LSPhGAApA). The AAV2/8-LSPhGAApA vector at 1.6 × 1013 vector particles/kg, after intravenous injection, did not cause significant short- or long-term toxicity. Recruitment of CD4+ (but not CD8+) lymphocytes to the liver was elevated in the vector-dosed male animals at study day (SD) 15, and in group 8 animals at SD 113, in comparison to their respective control animals. Administration of the vector, either prior to or after the one ERT injection, uniformly prevented the hypersensitivity induced by subsequent ERT in males, but not always in female animals. The vector genome was sustained in all tissues through 16-week postdosing, except for in blood with a similar tissue tropism between males and females. Administration of the vector alone, or combined with the ERT, was effective in producing significantly increased GAA activity and consequently decreased glycogen accumulation in multiple tissues, and the urine biomarker, Glc4, was significantly reduced. The efficacy of the vector (or with ERT) was better in males than in females, as demonstrated both by the number of tissues showing significantly effective responses and the extent of response in a given tissue. Given the lack of toxicity for AAV2/8LSPhGAApA, further consideration of clinical translation is warranted in Pompe disease.
Introduction
Infantile-onset GSD-II causes death early in childhood from cardiorespiratory failure related to an underlying hypertrophic cardiomyopathy. Pilot studies of enzyme replacement treatment (ERT) with recombinant human GAA (rhGAA, Myozyme) improved cardiomyopathy and prolonged survival in all subjects beyond 1 year. Pompe disease patients who lack any residual GAA protein are deemed crossreactive immunological material (CRIM)-negative. CRIM-negative Pompe disease subjects in these clinical trials formed high, sustained anti-hGAA antibodies and demonstrated markedly reduced efficacy from ERT. Taken together, these data suggest that immune tolerance to ERT is absent in CRIM-negative patients, and high titer antibody formation reduces any clinical benefit from ERT. At present, there is no successful immune modulation or tolerization protocol for patients that maintain the efficacy of ERT following the formation of anti-GAA antibodies.
Like CRIM-negative patients with Pompe disease, GAA-knockout (KO) mice lack immune tolerance to hGAA. In these GAA-KO mice, ERT had no efficacy and provoked fatal anaphylaxis. A strategy for inducing immune tolerance was developed in GAA-KO mice by administering a low copy number of the candidate vector (AAV2/8-LSPhGAApA; 2 × 1010 vector particles (vp)) prior to the initiation of ERT.1 The mechanism for inducing immune tolerance required liver-specific hGAA expression and was thought to involve T-regulatory cell activation. An immunomodulatory gene therapy strategy could be an important adjunct to ERT in CRIM-negative Pompe disease patients. The efficacy of ERT would be enhanced by preventing or suppressing antibody responses, and safety would be enhanced by the low number of vector particles needed to induce immune tolerance. The current study is designed to evaluate the toxicity of AAV2/8-LSPhGAApA vector in GAA-KO mice and to support the potential clinic use of the immunomodulatory gene therapy as an adjunct therapy to rhGAA ERT in Pompe disease, which could ultimately lead to curative therapy for the Pompe disease.
The potential toxicity, biodistribution, and efficacy of AAV2/8-LSPhGAApA were assessed at a dose of 1.6 × 1013 vp/kg. This dose is 40 times greater than the intended clinical dose in humans.
Results
The group designation and the dosing schedule for each group are summarized in Table 1. The initial dosing day was defined as study day (SD) 1.
Table 1. Study group designation.
| Dose group | Number animals (M/F) | Study day 1 |
Additional dosing days |
Study day 15 | 8-week study day 57 | 16-week study day 113 | Experimental goal | |
|---|---|---|---|---|---|---|---|---|
| Vectorb | rhGAA | |||||||
| 1 | 10/10a | Vehicle | x | Assessment of potential toxicity at an early time point | ||||
| 2 | 10/10a | Vectorb | x | |||||
| 3 | 10/10a | Vehicle | x | Assessment of potential toxicity at a late time point | ||||
| 4 | 10/10a | Vectorb | x | |||||
| 5 | 10/10a | Vectorb | Day 4, 18c | x | ||||
| 6 | 10/10a | Day 4, 18c | x | |||||
| 7 | 10/10a | Vectorb | Day 4, 18c, 32, 46, 60, 74, 88, and 102 | x | Efficacy of therapy to reduce immune response to rhGAA; toxicity evaluation of proposed therapeutic strategy. | |||
| 8d | 10/10a | rhGAA | Day 22 | Day 46e, 60, 74, 88, and 102 | x | Assessment of prevention of hypersensitivity by immunomodulatory gene therapy | ||
| 9 | 5/5 | Vehicle | x | Biodistribution | ||||
| 10 | 5/5 | Vectorb | x | |||||
| 11 | 5/5 | Vehicle | x | |||||
| 12 | 5/5 | Vectorb | x | |||||
| 13 | 5/5 | Vehicle | x | |||||
| 14 | 5/5 | Vectorb | x | |||||
Five male/five female animals will be “hematology mice”, the other five male/five female were “chemistry mice”.
AAV2/8 LSPhGAApA vector were dosed via tail vein injection.
On study day 18, prior to rhGAA administration by ~10 minutes, animals in groups 3 and 5–8 were intraperitoneally pretreated with diphenhydramine (20 mg/kg), an antihistamine, to prevent animal from anaphylaxis.
Blood was collected by submandibular puncture from this group of animals on study day 21 and study day 43 postinitial dosing for anti-GAA antibody analysis.
Assessment of hypersensitivity within 30 minutes post-rhGAA injection.
AAV, adeno-associated virus; rhGAA, recombinant human acid α-glucosidase; x, time when euthanized.
Assessment of toxicity
The AAV2/8-LSPhGAApA vector (1.6 × 1013 vp/kg), either alone or in combination with ERT, did not affect the body weights of mice as compared to their respective control group at the same SD time point (Figure 1a). The weekly measurement of food consumption did not reveal any significant reduction of appetite in animals administered vector, ERT, or vector plus ERT, compared with control animals (Figure 1b). No gross pathological lesions were attributable to vector administration, and only minor sporadic anomalies were identified upon postmortem examination.
Figure 1.

Growth and feeding. (a) Body weights for each experimental group by sex. Each bar represents the mean value of group body weight (n = 10 except that n = 9 for group 8 males and n = 7 for group 8 females). (b) Weekly food consumption by sex for mice euthanized at study day (SD) 15; b, groups 3–8 animals were euthanized at SD 113, and showed no difference for food consumption between groups (data not shown). (c) Blood chemistry testing. Units are as follows, for sodium (mmol/l), alkaline phosphatase (U/l), and cholesterol (mg/dl). Mean ± SD is shown; *P < 0.05. Group number indicated on the horizontal axes. Group: 1, vehicle; 2, vector; 3, vehicle; 4, vector; 5, vector, ERT ×2; 6, ERT ×2; 7, vector, ERT ×8; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
Vector toxicity was assessed at the early time point of SD 15 by standard blood chemistry and hematology testing. Only alkaline phosphatase in males, and cholesterol and sodium in females were significantly different between the vector-treated (group 1) animals and the vehicle-treated (group 2) animals (Figure 1c). Since the differences only existed in one gender, and no lesion was identified in the same group by microscopic examination (data not shown), the biological significance was deemed to be minimal.
Hypersensitivity in response to rhGAA administration has been reported following anti-GAA IgG formation,2,3 although hypersensitivity is rare among patients treated with rhGAA ERT.4,5 AAV vector administration has prevented hypersensitivity reactions, when liver-specific GAA expression prevented anti-GAA IgG formation.1,3 The vector, either dosed prior to ERT (group 7) or after a single rhGAA injection and followed by ERT (group 8), was uniformly effective in prevention of anaphylactic reaction occurrence in male animals, but was not always effective in females. In group 8, three out of 10 females showed hypersensitivity as evidenced by lower body temperature and signs of labored breathing and inactivity (allergy score = 3 or 4). Moreover, at SD 60, two female mice died within 30 minutes after rhGAA administration. In addition, signs of hypersensitivity were observed in several females from group 7. Subsequently, all the vector-treated female mice in groups 7 and 8 were given diphenhydramine prior to rhGAA administration on SD 74, 88, and 108. No hypersensitivity was observed on those days when diphenhydramine was administered.
Biochemical correction
GAA activity was elevated in the serum of GAA-KO mice following vector administration (Figure 2a; groups 4, 5 and 8), in comparison with the administration of vehicle (group 3). GAA activity was also detected in vehicle-treated serum, consistent with a prior report.6 A sex-determined response was detected, when serum GAA activity was significantly elevated in male, vector-treated mice in comparison with female mice (Figure 2b; groups 2, 4, 5, 7 and 8). The serum GAA was significantly elevated in male mice from vector-treated groups, in comparison with female mice (Figure 2b; groups 4, 5, 7, and 8). GAA activity was slightly, but significantly elevated in male vehicle-treated mice, in comparison to female mice without explanation (Figure 2b; group 3). However, the much higher elevations of GAA activity in vector-treated groups of male mice were attributable to GAA expression from the vector.
Figure 2.

GAA activity. (a) Serum. (b) Serum by sex. Differences indicated between male and female mice indicated (two-tailed t-test). GAA activity on study day (SD) 15 for (c)male and (d) female mice in each group. GAA on SD 113 for (e) male and (f) female in each group. Mean ± SD is shown. #P = 0.05; *P < 0.05; **P < 0.01; and ***P < 0.001. Group number indicated on the horizontal axes. Group: 1, vehicle; 2, vector; 3, vehicle; 4, vector; 5, vector, ERT ×2; 6, ERT ×2; 7, vector, ERT ×8; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
The GAA activity of liver was elevated for vector-treated male mice at SD 15 in group 2, in comparison with vehicle-treated mice in group 1 (Figure 2c). The GAA activities of liver and heart were elevated for vector-treated female mice in group 2 at SD 15, in comparison with vehicle-treated mice in group 1 (Figure 2d). The GAA activities of all tissues examined, except for in the brain, were elevated for all groups of vector-treated male mice at SD 113 (Figure 2e; groups 4, 5, 7, and 8) in comparison with vehicle-treated mice in group 3. The GAA activities of liver and diaphragm were elevated for all groups of vector-treated female mice at SD 113 (Figure 2f; groups 4, 5, 7, and 8), in comparison with vehicle-treated mice in group 3. Quadriceps featured elevated GAA only in vector-treated female mice from group 8 at SD 113, in comparison with vehicle-treated females in group 3 (Figure 2f).
The glycogen content of all tissues examined remained elevated in vector-treated mice of both sexes at SD 15 (Figure 3a,b). Glycogen content was decreased following vector administration by SD 113 in the heart, diaphragm, and quadriceps (Figure 3c; groups 4, 5, 7, and 8), in comparison with vehicle-treated male mice in group 3. Glycogen content was reduced in liver for vector-treated male mice in group 8, in comparison with vehicle-treated mice in group 3 (Figure 3c). Vector administration reduced the glycogen content in heart for three of four groups of female mice at SD 113 (Figure 3d, groups 4, 7, and 8 versus group 3). Female mice had consistently lower glycogen content in the diaphragm and quadriceps following vector administration at SD 113 (Figure 3d; groups 4, 5, 7, and 8 versus group 3). Administration of rhGAA and vector reduced the glycogen content of liver in female mice at SD 113, in comparison with vehicle alone (Figure 3d; groups 7 and 8 versus group 3). Finally, brain glycogen content was slightly reduced among female mice in group 8 (Figure 3d).
Figure 3.

Glycogen content and urinary biomarker. Glycogen content on study day (SD) 15 for (a) male and (b) female mice in each group. Glycogen content on SD 113 for (c) male and (d) female in each group. (e) Urinary biomarker, Glc4, on SD 15 for male and female mice in each group. (f) Glc4 for on SD 113 for male and female mice in each group. Mean ± SD is shown. *P < 0.05; **P < 0.01; and ***P < 0.001. Group number indicated on the horizontal axes. Group: 1, vehicle; 2, vector; 3, vehicle; 4, vector; 5, vector, ERT ×2; 6, ERT ×2; 7, vector, ERT ×8; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
The urinary biomarker, Glc4, was reduced following vector administration by SD 15 in male mice, but not in female mice (Figure 3e). Vector administration consistently reduced urinary Glc4, in comparison with vehicle at SD 113 in both sexes (Figure 3f; groups 4, 5, 7, and 8 versus group 3).
T cell immune responses in liver
There were significantly greater numbers of CD4+ and CD8+ lymphocytes in the hepatic sinusoids/space of Disse (SOD) in the vehicle-treated control females, in comparison with the control males. This phenomenon was consistently observed both in group 1 (SD 15) and group 3 (SD 113) vehicle-treated animals (Figure 4a,b). Vector administration was associated with increased CD4+ lymphocyte at early and late time points. On SD 15, the vector-treated male animals in group 2 had significantly increased recruitment of CD4+ lymphocytes to hepatic sinusoids/SOD and portal triads in comparison with the vehicle-treated control males group 1 (data not shown), which was also seen for group 8 males on SD 113 (Figure 4c–h). A significantly increased recruitment of CD4+ lymphocytes to hepatic triads was detected in the vector-dosed male animals in comparison with the vehicle control male animals (Figure 4c). However, the number of CD4+ lymphocytes in the foci from vector and vehicle-treated mice were not significantly different (Figure 4d). Vector-treated group 2 males had rare CD4+ lymphocytes on SD 15 (Figure 4e,f), whereas vector-treated group 8 animals had multiple CD4+ lymphocytes detected on SD 113 (Figure 4g,h). The elevation of CD4+ lymphocytes has been previously reported to be associated with induction of immune tolerance to GAA.7 No significant increases in CD4+ lymphocytes were observed in other groups of vector mice, in comparison with control mice. In general, the vector (alone or with ERT) treatment had no significant effect on CD8+ lymphocyte recruitment to the liver, indicating a lack of cytotoxic T cell responses (data not shown).
Figure 4.
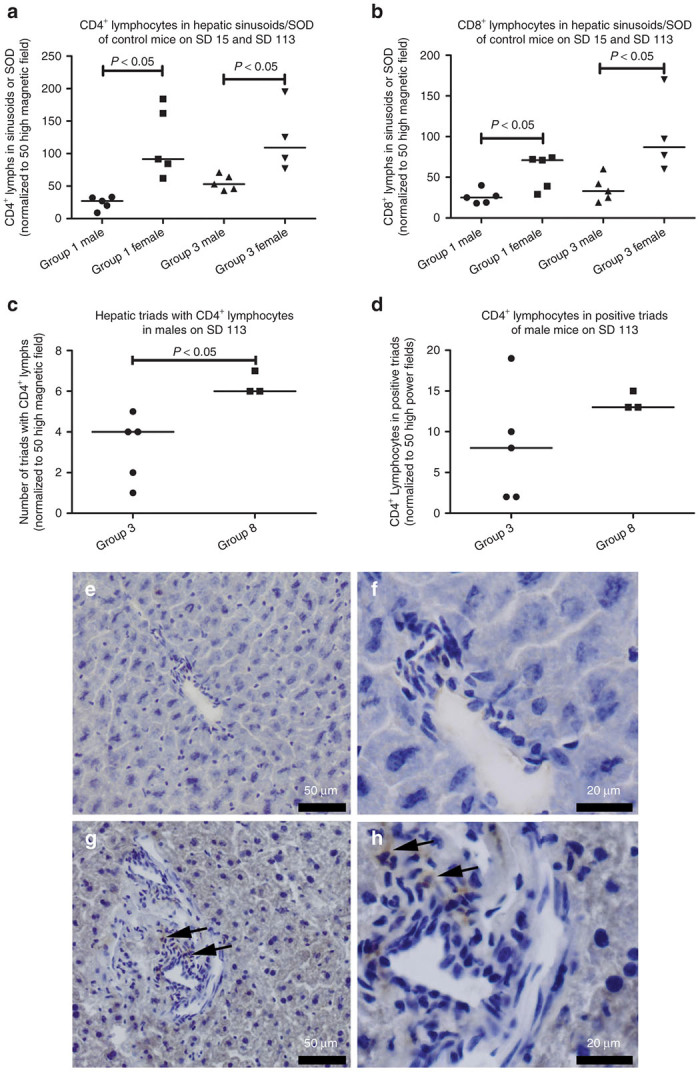
Immunodetection of CD4+ lymphocytes in liver. (a) CD4+ lymphocytes quantified from hepatic sinusoids/space of Disse (SOD) for vehicle-treated mice on day 15 and day 113 (groups 1 and 3). (b) CD8+ lymphocytes in hepatic sinusoids/SOD for control mice. (c) Hepatic triads containing CD4+ lymphocytes quantified from vehicle-treated (group 3) and vector-treated (group 8) mice, (d) CD4+ lymphocytes quantified within positive triads. P values calculated with two-tailed t-test. (e–h) Photomics of CD4-stained sections from male mice, focused on the triad regions. On study day (SD) 15, (e) and (f) are from group 2. On SD 113, (g) and (h) are from group 8. (e) and (g) are ×200, and (f) and (h) are ×600 magnification. Arrows indicate CD4+ lymphocytes. Group number indicated on the horizontal axes. Group: 1, vehicle; 2, vector; 3, vehicle; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
Vector administration was associated with lower serum creatine kinase (Figure 5a), and aspartate aminotransferase (Figure 5b), consistent with effective correction of Pompe disease. At SD 113, the serum aspartate aminotransferase level was found significantly lower in vector-treated males from groups 4, 5, 7, and 8, and also in group 7 and 8 females as compared to the vehicle-treated group 3 control females. Serum creatine kinase was significantly reduced in the vector-treated females in group 7 and males in group 8. Since the elevation of creatine kinase and aspartate aminotransferase is listed as clinical signs in infants with Pompe disease,8 the reductions in creatine kinase and aspartate aminotransferase were deemed the result of the therapeutic effect of the vector administration ± ERT in those groups. In addition, the glucose level was significantly lower in vector-treated female mice from groups 5, 6, 7, and 8, and also ERT-treated male mice from group 6, in comparison to vehicle-treated mice in group 3 (Figure 5c). The glucose level change is speculated to be associated with response to the treatment of a GAA supplement, although the glucose elevation was not seen in all male mice following ERT. Finally, triglycerides were significantly lower in vehicle-treated mice of both sexes from group 8 (Figure 5d), suggesting a treatment effect.
Figure 5.
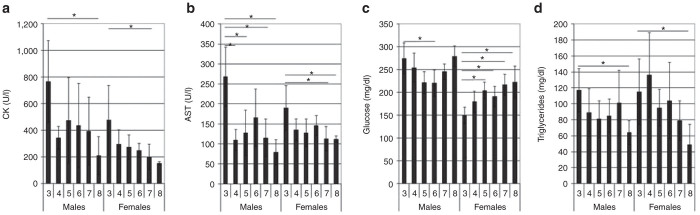
Serum biomarkers. (a) Creatine kinase. (b) Aspartate aminotransferase. (c) Glucose. (d) Triglycerides. Mean ± SD is shown. *P < 0.05. Group: 3, vehicle; 4, vector; 5, vector, ERT ×2; 6, ERT ×2; 7, vector, ERT ×8; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
Antibody responses
Neutralizing anti-AAV8 antibodies developed in all vector-dosed animals with a titer range of 40–640 at 2 weeks and 320–1,280 at 16 weeks. In group 7 (vector plus 8× ERT), the anti-AAV8 antibody titer was statistically significantly higher in the female mice than in male mice (Figure 6a).
Figure 6.
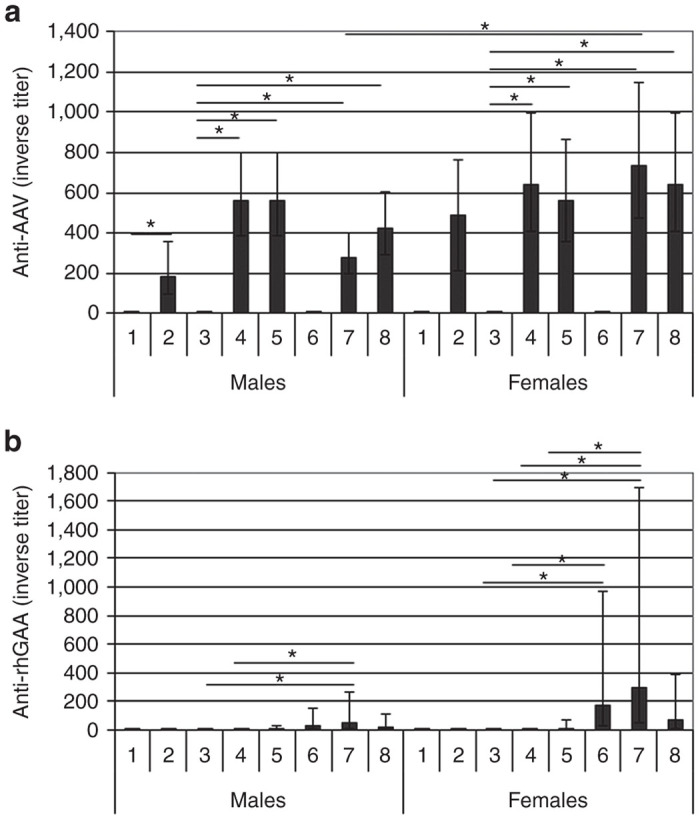
Antibody responses. Immunoglobulin G (IgG) against (a) AAV. (b) GAA. The data were presented as geometric mean (n = 5). The positive error bar represents the upper limit of 95% confidence interval, and the negative error bar the lower limit of 95% confidence interval. Group number indicated on the horizontal axes. Group: 1, vehicle; 2, vector; 3, vehicle; 4, vector; 5, vector, ERT ×2; 6, ERT ×2; 7, vector, ERT ×8; 8, ERT, vector, ERT ×5. ERT, enzyme replacement treatment.
Low titers of anti-hGAA antibodies (group geomean from 1:20 to 1:290) developed in some animals in the groups given ERT (Figure 6b). The antibody titer was variable among individual animals within the same group. There was a consistent trend that female animals had slightly higher titers than males.
Biodistribution
The AAV2/8-LSPhGAApA vector, after tail vein injection, was detected at 2 weeks in all the main tissues and blood examined (Figure 7) with a highest level in the liver (~107 vg/μg DNA) and lowest in the brain (~102 vg/μg DNA). The vector persisted in all the examined tissues up to 16 weeks without significant reduction except that in blood the vector level was decreased to around unquantifiable level (the low limit of quantification for the assay is 38 vg/μg DNA). The vector distribution pattern was similar between male and female animals (data not shown).
Figure 7.

Biodistribution of AAV2/8-LSPhGAApA vector in mouse tissues. Tissues were collected from mice administered AAV2/8-LSPhGAApA vector at 1.6 × 1013 vp/kg via tail vein injection at 2, 8, and 16 weeks postdosing. The vector genome copies were analyzed by quantitative polymerase chain reaction (qPCR) as copies/µg genomic DNA. The lowest limit of quantitation of the qPCR assay was 38 copies/µg genomic DNA. The data were presented as geometric mean (n = 5). The positive error bar represents the upper limit of 95% confidence interval, and the negative error bar the lower limit of 95% confidence interval. M tissue was from male mice; F tissue was from female mice. Note that only data from vector-dosed groups (group 10 for 2 weeks, group 12 for 8 weeks, and group 14 for 16 weeks) were presented, and vector genome copy numbers in the vehicle control animal tissues from group 9, 11, and 13 were all below the lower limit of quantification of the qPCR assay (data not shown).
Discussion
The AAV2/8-LSPhGAApA vector at 1.6 × 1013 vp/kg, after intravenous injection, did not cause significant short- or long-term toxicity. Administration of the vector, either prior to or after the one ERT injection, uniformly prevented the hypersensitivity induced by subsequent ERT in males, but not always in female animals, suggesting that antibody formation and anaphylaxis should still be monitored when the vector is used in conjunction with Myozyme. The vector genome was sustained in all tissues through 16-week postdosing, except for in blood with a similar tissue tropism between males and females. The vector genome were highest in the liver (~107 vg/μg DNA) and lowest in the brain (~102 vg/μg DNA). Administration of the vector alone, or combined with the ERT, was effective in producing significantly increased GAA activity and consequently decreased glycogen accumulation in multiple tissues, and the urinary Glc4 was reduced in association with the correction of glycogen accumulations.
At 2 weeks, the vector alone treatment had already lead to significant increases in GAA activity in the liver and diaphragm (males) or heart (females), as well as a significant reduction in urine Glc4 marker in males. However, correction of glycogen accumulation was not observed in any tissues at this early time point. By 16 weeks, following vector administration either with or without ERT, GAA activity was increased and glycogen accumulation was reduced in multiple tissues, and the urine biomarker, Glc4, was significantly reduced. The efficacy of the vector (or with ERT) was better in males than in females, as demonstrated both by the number of tissues showing significantly effective responses and the extent of response in a given tissue.
The immunohistochemical staining also demonstrated that there were greater numbers of CD4+ and CD8+ lymphocytes in the hepatic sinusoids/SOD of female control mice than male control mice. The higher level of baseline immune response in the liver in female animals could contribute to the sex-determinant, male-advantage effect of AAV vector transduction efficacy in the liver in the current and previous studies.9–11 In addition, on SD 15 and SD 113, some groups of vector-treated male animals (groups 2 and 8) had significantly increased recruitment of CD4+ lymphocytes to hepatic sinusoids/SOD and portal triads in comparison with the vehicle-treated control animals. Since no reduced efficacy or explicit adverse effect in the liver was identified in the group 2 or 8 males, the biological function of CD4+ lymphocyte recruitment is unclear. However, recent published data indicated that CD4+ lymphocytes were increased following induction of immune tolerance to hGAA.7
The presence of CD4+ lymphocytes has been associated with immune tolerance to the transgene product following AAV vector administration. Immune tolerance to hGAA following AAV vector or ERT administration has been demonstrated by the absence of anti-GAA IgG.12 Recently, increased CD4+ lymphocytes in the liver were associated with immune tolerance defined as lack of anti-GAA antibody formation following AAV vector administration and a subsequent immune challenge with rhGAA plus adjuvant; moreover, adoptive transfer of CD4+ lymphocytes conferred immune tolerance in wild-type mice.7 Further evidence for CD4+ cell involvement in mediating immune tolerance was demonstrated by the increased number of CD4+CD25+FoxP3+ splenocytes following AAV2/8-LSPhGAApA administration.13 Similarly, the depletion of CD25+ lymphocytes broke immune tolerance and precipitated anti-GAA antibody formation following AAV2/8-LSPhGAApA administration in GAA-KO mice.1 Adoptive transfer of CD4+CD25+ lymphocytes also conferred immune tolerance in mice with hemophilia B14 or Fabry disease.15 Taken together, the above-mentioned results support a tolerogenic effect of CD4+ lymphocytes in the liver or spleen, in absence of accompanying increased CD8+ lymphocytes that reflected a cytotoxic response to transgene expression.16 Therefore, the demonstration of increased CD4+ T cells in the liver correlated with immune tolerance and transgene-mediated efficacy in the current study was anticipated by existing gene therapy literature. Furthermore, vector-mediated immune tolerance can be induced following an initial exposure to ERT (group 8), which has been achieved previously in preclinical1 and clinical17 studies.
The data from this pharmacology–toxicology support the safety of the AAV2/8-LSPhGAApA vector for liver-targeted gene therapy in Pompe disease. Additionally, biochemical data demonstrated efficacy either from the vector alone or combination with ERT. Therefore, further consideration of clinical translation is warranted for AAV2/8-LSPhGAApA in Pompe disease.
Materials and Methods
Study design
Groups of mice and the dosing schedule for each group are summarized in Table 1. The potential toxicity of the AAV2/8-LSPhGAApA vector at an early time point was evaluated in group 2, in which 10 male/10 female mice were dosed the vector and euthanized at 2 weeks postdosing. Group 1 served as the control group for the early time point toxicity assessment. Mice were treated at 9–29 weeks of age. The study was approved by institutional animal care and use committees at Lovelace Respiratory Research Institute and Duke University.
For groups 1–6 mice, evaluated endpoints included clinical signs of toxicity and body weight, survival, hematology, serum chemistry, T-cell–medicated immune response in liver, organ weights at necropsy, and histopathology. Efficacy associated parameters, including urine Glucose Tetrasaccharide marker (Glc4), acid α-glucosidase (GAA) activity and glycogen content in several tissues, and serum anti-GAA and anti-AVV antibodies, were evaluated.
The safety of the intended AAV2/8-LSPhGAApA treatment strategy in humans was assessed in group 7. Group 7 was administered vector and then rhGAA ERT (20 mg/kg) beginning 3 days after vector treatment. The ERT was performed every other week for a total of eight injections. Group 7 mice were euthanized at week 16. Endpoints were the same as described for groups 1–6. Results were directly compared to in-life and postmortem data obtained from groups 3 to 6.
Group 8 was designed to assess prevention of hypersensitivity by AAV2/8-LSPhGAApA treatment. The group of mice was injected with rhGAA(20 mg/kg) at SD 1, and then AAV2/8-LSPhGAApA vector (1.6 × 1013 vp/kg) at 3 weeks postinitial rhGAA, and followed by rhGAA ERT (20 mg/kg) starting at SD 46 biweekly for five injections. Blood was collected by submandibular puncture at SD 21 and SD 43 for serum anti-GAA analysis. Hypersensitivity was assessed within 30 minutes after rhGAA injection at SD 46. Animals in group 8 were euthanized at week 16, and evaluated endpoints were the same as groups 1–7.
Vector production
AAV2/8-LSPhGAApA has been described.1 The LSP regulatory cassette in AAV2/8-LSPhGAApA (subcloned from pAV-LSP-cFIX, courtesy of Dr. Inder Verma, Salk Institute, La Jolla, CA; sequence available upon request) contains a thyroid hormone–binding globulin promoter sequence downstream from two copies of a α1-microglobulin/bikunin enhancer sequence,18 and previously achieved long-term efficacy in hemophilia B mice within an AAV vector–encoding coagulation factor IX.19 Production and characterization of the AAV2/8-LSPhGAApA vector was performed by the Gene Therapy Resources Program, Clinical Grade AAV Vector Core at Children’s Hospital of Philadelphia. The test articles used for the current toxicity study were manufactured in the same way as the intended clinical product.
GAA activity
GAA activity was measured on frozen tissues following homogenization and sonication of tissue samples in distilled water. Depending upon the tissue size, 10–50 mg tissue was weighed and homogenized, the homogenates were sonicated at 4 °C three times for 15 seconds, then centrifuged for 3 minutes at 15,000 RPM. The reaction was set up with 10 µl of supernatant and 20 µl of substrate—4MUα-D-glucoside, in a 96-well plate (VWR62402-970). The reaction mixture was incubated at 37 °C for 1 hour, and was stopped by adding 130 µl of sodium carbonate buffer of pH 10.5. A standard curve (0–1,000 pmol/µl of 4MU) was used to measure released fluorescent 4MU from individual reaction mixture, using TECAN GENios microplate reader at 465 nm (Emission) and 360 nm (excitation). The protein concentrations of the clarified supernatants were quantified via the Bradford assay (Bio-Rad Laboratories, Hercules, CA; Cat No. 500-0006). GAA activity was measured in the tissue homogenates by conversion of the artificial substrate 4-methylumbelliferyl (4-MU) α-D-glucoside to the fluorescent product umbelliferone at acidic pH 4.3 as described.20 To calculate the GAA activity, released 4MU concentration was divided by the sample protein concentration and activity was reported as nmol/hour/mg protein. Quality assurance and quality control samples were run on the same plate for experimental assay controls.
Glycogen content
Glycogen content of tissues was measured indirectly as the glucose released after total digestion by amyloglucosidase of the tissue homogenates described above using the Aspergillus niger assay system and the glucose reagent (Infinity Glucose; cat. no. TR15421, Thermo Scientific, Waltham, MA) in a standardized reaction using the Aspergillus niger assay system, as described.21 The same tissue homogenates used above (see GAA activity section ) were used to measure total glycogen content in each tissue. The reaction was set up with 20 µl of supernatant and 55 µl distilled water. Samples were boiled for 3 minutes and immediately cooled on ice for 10 minutes. Twenty-five microliters of amyloglucosidase (1:50 in 0.1 mol/l potassium acetate pH 5.5) was added to each reaction tube. A reaction control tube without any amyloglucosidase (homogenate + water) was also set up for each reaction tube. Both sample tube and control sample reaction tubes were incubated at 37 °C for 90 minutes and reaction, was stopped by boiling the tubes for 3 minutes followed by centrifugation for 3 minutes at 15,000 RPM. The glucose released from each reaction tube was determined by adding 30 µl of supernatant into 1 ml of Infinity glucose reagent (TR15421), and measuring on spectrophotometer at 340 nm (Shimadzu UV-1700).
Glc4 urine marker analysis
Glcα1-6Glcα1-4Glcα1-4Glc (Glc4), previously termed Hex4 when detected by the method used previously, was quantified as follows. Fifty microliters of urine diluted with water based on creatinine concentration was mixed with a fixed amount internal standard (13C6-Glc4 and 13C6-IMIM) was derivatized with butyl-4-aminobenzoate and cleaned by SPE using a previously published method.22 The samples were reconstituted in acetonitrile:H2O (80:20, v:v) with 10 mmol/l amino acetate, then analyzed by UPLC-MS/MS on an Acquity UPLC system equipped with Xevo TQ mass spectrometer (Waters Corporation, Milford, MA) with electrospray ionization in positive ion mode. Five microliters of sample were injected to mass spectrometer through an ACQUITY UPLC BEH amide 2.1 × 50 mm, 1.7 µm column for separation with gradient elution. Selected reaction monitoring mode was used to monitor transitions: m/z 844>358 for Glc4 and isomers derivatives and m/z 850>364 for 13C6-Glc4 and 13C6-IMIM derivatives. Calibrators with 2.5, 5, 20, 40, and 70 µmol/l of Glc4 standard spiked in control urine were prepared and analyzed in the same method as urine samples. After analysis by UPLC-MS/MS, the ratio of signals corresponding to Glc4 to the signals of 13C6-Glc4 were plotted against the added concentration of the standards. The slope of the curve, determined by linear regression, was used to quantify Glc4 in unknown urine samples.
Antibody detection
The neutralizing anti-AAV8 antibody was analyzed using a transduction inhibition-based neutralizing antibody (NAb) assay, which analyzes and quantifies the presence of antibodies in test serum capable of blocking AAV8 vector, carrying a reporter LacZ transgene, transduction of a human hepatocellular carcinoma cell line (Huh7).
A validation of the neutralizing antibody assay was performed before any study sample serum was analyzed. The objectives of the AAV NAb assay validation included:
Estimate the intra- and interoperator reproducibility of transduction controls (mouse serum) and cells only (negative control).
Estimate the intra- and interoperator reproducibility of the AAV NAb titer obtained in six human test samples.
Estimate the intra- and interoperator reproducibility of the AAV NAb titer obtained in a positive control sample from a rabbit immunized with the AAV serotype used in the NAb testing (rabbit antiserum control).
Anti-rhGAA was detected by a standard enzyme-linked immunosorbent assay. Briefly, the study serum samples were serially diluted starting at 1:20, and then added to a 96-well plate precoated with rhGAA and incubated for 1–3 hours at room temperature. After being washed, the plate was added and incubated with the alkaline phosphatase–conjugated goat anti-mouse IgG second antibody (1:2,500, Cat# 115-055-205; Jackson ImmunoResearch, West Grove, PA) for 1–3 hours at room temperature. The plate was then washed and incubated with the substrate, 4-nitrophenyl phosphate disodium salt hexahyate (Cat# N9389-100TAB; Sigma, St Louis, MO) for 20 ± 3 minutes for color development, and then read for optical density at a wavelength of 405 nm using a µQuant Plate Reader (BioTek, Winooski, VT). Each dilution of a study sample was run in triplicate. Blank, negative, and positive controls were included in each plate. The cutoff value for a positive titer was set as a net optical density reading of 0.1, after the blank value was subtracted out. The titer of a study sample is expressed as the reciprocal value of the highest dilution at which the average net optical density of the triplicates was ≥ 0.1. In the case where the highest dilution of a sample was still positive, the titer of the sample was expressed as greater than the highest dilution analyzed (e.g., > 6,400) and vice versa, if the lowest dilution of a sample was still negative, the titer of the sample was expressed as less than the lowest dilution analyzed (e.g., < 20).
Biodistribution
The biodistribition of the AAV2/8-LSPhGAApA vector genome DNA was analyzed in following tissues: blood, brain, liver, heart, spleen, lungs, kidneys, diaphragm, quadriceps, testes/ovaries, and tail (injection site). Tissues were collected from mice in biodistribution groups 9–14 at necropsy performed at weeks 2, 8, and 16. The tissue samples were aliquoted to small pieces/volume, snap frozen in liquid nitrogen, and stored at −80 ± 10 °C until the time of DNA extraction. A clean procedure was used and attention was paid to avoid crosscontamination during tissue collection and aliquoting.
DNA was extracted from the tissues and blood using a QIAGEN DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) per Lovelace Respiratory Research Institute SOPs. The extracted DNA was quantified using a spectrophotometer (BioTek, Broadview, IL).
Quantitation of the AAV2/8-LSPhGAApA vector genome in tissues was analyzed by a quantitative polymerase chain reaction (qPCR) assay using a Stratagene Model Mx3005P system. The qPCR assay was validated (VP11-069) prior to being used for study sample analyses. The lower limit of quantitation of the qPCR assay was determined to be 38 copies/µg DNA. Briefly, the qPCR employed a SYBR green-based real-time PCR reaction. Linearized plasmid pAAV-LSPhGAApa (provided by the investigator, Dr. Koeberl, at Duke University) was used to construct a seven-point standard curve to quantify the study samples analyzed in the same plate. Each study sample was analyzed in triplicate. Both negative and positive controls were included in each plate. The quantitation of the vector genome was normalized by the housekeeping gene, β-actin. The primers used for the vector genome analysis were forward-AGTGCCCACACAGTGCGACGT and reverse-CCTCGTAGCGCCTGTTAGCTG, and the primers for the β-actin analysis were forward-AGAGGGAAATCGTGCGTGAC and reverse-CAATAGTGATGACCTGGCCGT. The thermal cycling conditions used for the qPCR reaction were: 95 °C for 15 minutes, 40 cycles of 94 °C for 30 seconds, 60 °C for 30 seconds, 72 °C for 45 seconds, 72 °C for 10 minutes, and a melting curve analysis of 55–95 °C range. All specified tissues from 2-, 8-, and 16-week biodistribution animals were analyzed. The remaining biodistribution tissues have been stored at −80 ± 10 °C at Lovelace Respiratory Research Institute, and per the sponsor representative’s decision, they were shipped to National Gene Vector Biorepository (www.ngvb.org) located at the Indiana University School of Medicine for archive.
T cell immune responses in liver
Each liver section was frozen in dry ice in an optimal cutting temperature compound in a cryomold and then stored at −80 ± 10 °C. The frozen sections (4–7-μm thickness) were prepared on a microtome and subjected to immunohistochemical staining for CD4 and CD8. Briefly, the liver sections were thawed, then fixed in 10% neutral-buffered formalin, and then incubated with a series of blocking solutions. The sections were then incubated with the purified rat anti-mouse CD4 (Cat#550280, 1:10 dilution; BD Biosciences, San Jose, CA) or CD8 (Cat#550281, 1:10 dilution; BD Biosciences) primary antibody and followed by Rabbit Anti-Rat IgG biotinylated secondary antibody (Cat#BA-4001, 1:200 dilution; Vector Laboratories, Burlingame, CA). The immunostaining reaction was detected by incubation with Horseradish Peroxidase Avidin D solution (Cat#A-2704; Vector Laboratories) and then developed using 3,3-diaminobenzidine (Cat#K3468; Dako, Carpinteria, CA). The sections were counterstained with hematoxylin (Cat#H-3404; Vector Laboratories) and then subjected to dehydration and coverslipping. Two sets (liver and spleen) of positive and negative control slides were included in each batch of staining.
Slides were evaluated in a blinded manner, without any knowledge of the group designation for each mouse. Briefly, positively stained CD4 or CD8 cells were counted in three locations for each slide: (i) the number of CD4+ or CD8+ lymphocytes was counted in the hepatic sinusoids or SOD in ×50 (where available), ×600 magnification fields; (ii) the number of portal triads containing positively stained CD4 or CD8 lymphocytes and the number of positively stained lymphocytes in each positive triad were counted in ×50 (where available), ×600 magnification fields; and (iii) the number of cellular foci containing positively stained CD4, CD8, or polymorphonuclear/neutrophil cells were counted in the liver parenchyma throughout the entire specimen as three categories: the number of foci consisting of pure mononuclear (Mono) cells (monocytes/macrophages and lymphocytes), the number of foci consisting of mixed Mono and polymorphonuclear cells, and the number of foci consisting of pure polymorphonuclear cells. The number of positively stained cells in each foci was also counted. For the sections where less than 50 fields were available, the numbers were normalized to 50 fields when needed during the data analysis.
Statistical analysis
The efficacy parameters, GAA activity, glycogen content, and Glc4, were assessed. T-tests were used to assess parameter differences between groups 1 and 2 at the early time point assessment for males and females separately. In addition, one-way analysis of variance was used to assess the group effect of the parameters in groups 3–7 near the end of the 16-week study for males and females separately. When there was a significant group effect (P ≤ 0.05), Tukey’s multiple comparison test was used to assess differences between groups. A t-test was also used to assess differences between groups 3 and 8 for males and females separately.
In order to better comply with the normality and constant variance assumptions, logarithmic transformations were applied prior to analysis. Statistical calculations were performed using the SAS software system, versions 9.2 and 9.3 (SAS, Cary, NC). All reported P values are two sided, and statistical significance was assessed at P ≤ 0.05.
Acknowledgments
This study was funded by the Gene Therapy Resources Program at the National Heart, Lung, and Blood Institute. D.D.K. was supported by National Institutes of Health (NIH, Bethesda, MD) Grants R01HL081122 from the National Heart, Lung, and Blood Institute and R01HD054795 from the National Institute of Child Health and Human Development. GAA-KO mice were provided courtesy of Nina Raben at the NIH. We thank Andrew Bird and Jian Dai for technical support. Statistical analysis was provided by Yushi Liu. Vector production was supervised by J. Fraser Wright.
This study was funded by The National Institutes of Health.
References
- Sun B, Kulis MD, Young SP, Hobeika AC, Li S, Bird A. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther. 2010;18:353–360. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- Sun B, Zhang H, Benjamin DK, Jr, Brown T, Bird A, Young SP. Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid alpha-glucosidase in glycogen storage disease type II. Mol Ther. 2006;14:822–830. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Luo X, Bird A, Li S, Koeberl DD. Deficiency in MyD88 Signaling Results in Decreased Antibody Responses to an Adeno-Associated Virus Vector in Murine Pompe Disease. Biores Open Access. 2012;1:109–114. doi: 10.1089/biores.2012.0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff AM, Ng CY, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, Mizukami H, Ogura T, Kure S, Ichinohe A, Kojima K. Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 2004;11:1081–1086. doi: 10.1038/sj.gt.3302262. [DOI] [PubMed] [Google Scholar]
- Chulay JD, Ye GJ, Thomas DL, Knop DR, Benson JM, Hutt JA. Preclinical evaluation of a recombinant adeno-associated virus vector expressing human alpha-1 antitrypsin made using a recombinant herpes simplex virus production method. Hum Gene Ther. 2011;22:155–165. doi: 10.1089/hum.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl DD, Kishnani PS. Immunomodulatory gene therapy in lysosomal storage disorders. Curr Gene Ther. 2009;9:503–510. doi: 10.2174/156652309790031094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sun B, Osada T, Rodriguiz R, Yang XY, Luo X. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther. 2012;23:460–472. doi: 10.1089/hum.2011.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao O, Dobrzynski E, Wang L, Nayak S, Mingle B, Terhorst C. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler RJ, Cherry M, Barbon CM, Li C, Bercury SD, Armentano D. Correction of the biochemical and functional deficits in fabry mice following AAV8-mediated hepatic expression of alpha-galactosidase A. Mol Ther. 2007;15:492–500. doi: 10.1038/sj.mt.6300066. [DOI] [PubMed] [Google Scholar]
- Franco LM, Sun B, Yang X, Bird A, Zhang H, Schneider A. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ill CR, Yang CQ, Bidlingmaier SM, Gonzales JN, Burns DS, Bartholomew RM. Optimization of the human factor VIII complementary DNA expression plasmid for gene therapy of hemophilia A. Blood Coagul Fibrinolysis. 1997;8 2:S23–S30. [PubMed] [Google Scholar]
- Wang L, Takabe K, Bidlingmaier SM, Ill CR, Verma IM. Sustained correction of bleeding disorder in hemophilia B mice by gene therapy. Proc Natl Acad Sci USA. 1999;96:3906–3910. doi: 10.1073/pnas.96.7.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalfitano A, McVie-Wylie AJ, Hu H, Dawson TL, Raben N, Plotz P. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci USA. 1999;96:8861–8866. doi: 10.1073/pnas.96.16.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Yang HW, Pennybacker M, Ichihara N, Mizutani M, Van Hove JL. Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail. J Clin Invest. 1998;101:827–833. doi: 10.1172/JCI1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y, Young SP, Hillman SL, Van Hove JL, Chen YT, Millington DS. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem. 2000;287:136–143. doi: 10.1006/abio.2000.4838. [DOI] [PubMed] [Google Scholar]