

Abstract
Aim
TRPC3 is a non-selective cation channel, which forms a Ca2+ entry pathway involved in cardiac remodelling. Our aim was to analyse acute electrophysiological and contractile consequences of TRPC3 activation in the heart.
Methods and results
We used a murine model of cardiac TRPC3 overexpression and a novel TRPC3 agonist, GSK1702934A, to uncover (patho)physiological functions of TRPC3. GSK1702934A induced a transient, non-selective conductance and prolonged action potentials in TRPC3-overexpressing myocytes but lacked significant electrophysiological effects in wild-type myocytes. GSK1702934A transiently enhanced contractility and evoked arrhythmias in isolated Langendorff hearts from TRPC3-overexpressing but not wild-type mice. Interestingly, pro-arrhythmic effects outlasted TRPC3 current activation, were prevented by enhanced intracellular Ca2+ buffering, and suppressed by the NCX inhibitor 3′,4′-dichlorobenzamil hydrochloride. GSK1702934A substantially promoted NCX currents in TRPC3-overexpressing myocytes. The TRPC3-dependent electrophysiologic, pro-arrhythmic, and inotropic actions of GSK1702934A were mimicked by angiotensin II (AngII). Immunocytochemistry demonstrated colocalization of TRPC3 with NCX1 and disruption of local interaction upon channel activation by either GSK1702934A or AngII.
Conclusion
Cardiac TRPC3 mediates Ca2+ and Na+ entry in proximity of NCX1, thereby elevating cellular Ca2+ levels and contractility. Excessive activation of TRPC3 is associated with transient cellular Ca2+ overload, spatial uncoupling between TRPC3 and NCX1, and arrhythmogenesis. We propose TRPC3-NCX micro/nanodomain communication as determinant of cardiac contractility and susceptibility to arrhythmogenic stimuli.
Keywords: TRPC3, NCX1, Ca2+ handling, Cardiac contractility, Arrhythmias
1. Introduction
Hypertrophic remodelling of the myocardium is typically initiated by stimuli originating from either release of neuroendocrine factors such as angiotensin II (AngII),1 endothelin I,2 and noradrenalin3 or by exaggerated mechanical strain.4 These pathogenic stimuli trigger cellular Ca2+ signals that modify gene expression to restructure the myocardium. The pivotal signalling role of Ca2+ in cardiac remodelling is well established5,6 and involves spatial linkage between Ca2+ entry channels and Ca2+-dependent signalling molecules, such as calmodulin-dependent protein kinase II (CaMKII),7 PKC,8 and calcineurin (CaN).9,10 A critical step in the early stage of cardiac remodelling is up-regulation of the expression of certain Ca2+ channel proteins to modify cardiac Ca2+ homeostasis and Ca2+-transcription coupling. Some Ca2+ channels, such as voltage-gated l-type channels (CaV1.2), are capable of serving both normal excitation–contraction as well as excitation–transcription coupling. In contrast, voltage-insensitive channels are considered to control preferentially Ca2+ signals that activate gene transcription. Initiation of Ca2+-dependent processes of transcriptional regulation is likely to take place in specialized regulatory microdomains that are somehow segregated from the oscillatory rise and fall in cytoplasmic Ca2+, as associated with excitation–contraction coupling.1,5,11–14 TRPC channels were found up-regulated in response to mechanical stress-associated afterload2,15–17 or increased neurohumoural stimuli.1,3,15,16 TRPC3 has been demonstrated to supply Ca2+ in a highly efficient and selective manner for CaN regulation,4,12 which requires minute membrane currents and is not associated with global cytoplasmic Ca2+. Nonetheless, non-selective cation conductances, as generated by TRPC channels, are expected to impact on excitability and contractile function of cardiomyocytes. Nonetheless, the consequences of acute activation of cardiac TRPC channels, especially in conditions of enhanced expression such as cardiac hypertrophy or heart failure, are so far unclear, and the role of non-selective cation conductances in cardiac dysfunction during remodelling is incompletely understood. Previous studies17–20 in neonatal and adult cardiac cells suggested negligible involvement of TRPC3 in excitation–contraction coupling. TRPC3 channels display only weak Ca2+ over Na+ permeability (Ca2+:Na+ = 1.6) and a characteristic double-rectifying current to voltage feature, thus allowing for significant Na+ loading and depolarization during diastole. Hence, activation of this conductance is expected to generate significant disturbances in action potential morphology and to simultaneously shift equilibrium and operation of electrogenic Na+ transport specifically of NCX1, which has repeatedly been demonstrated as a signalling partner of TRPC channels in muscle cells.7,21,22 Analysis of acute functional consequences of TRPC3 is limited by its complex mechanism of activation, which typically involves stimulation of phospholipase C and thereby several routes linked to excitation–contraction coupling. Availability of a direct small molecule activator of TRPC3 (GSK1702934A) prompted experiments to identify the acute consequences of TRPC3 activation in a murine model of cardiac overexpression and to compare its functional effects with that of AngII , a classical mediator activating TRPC conductances. With this study, we demonstrate that TRPC3 is a determinant of cardiac contractility and excitability. This function of TRPC3 is based on its interaction with NCX1, which translates TRPC3 activity into altered cellular Ca2+ handling and action potential morphology.
2. Methods
2.1. GSK synthesis
GSK1702934A8,23 was prepared according to the method illustrated in scheme 1.
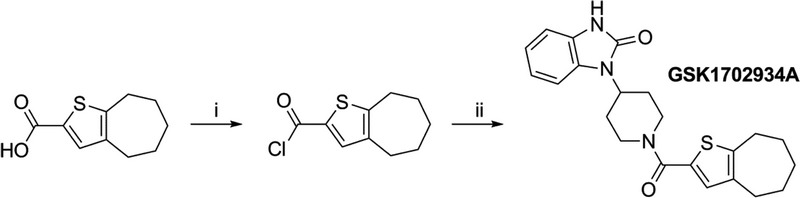
Scheme 1. Synthetic route to GSK1702934A. Reagents and conditions: (i) oxalyl chloride, DMF, DCM, 0°C to r.t., 1 h, 80%; (ii) 4-(2-keto-1-benzimidazolinyl) piperidine, DMAP, DCM, 0°C to r.t., 15 min, 82%.
2.2. Transgenic animal model, functional and biochemical experiments
A transgenic mouse model of cardiac-specific TRPC3 overexpression (low expression line 23; TRPC3-TG), generated by Prof. Molkentin,9,10,17 was compared with age-matched (2–4 months) and sex-matched wild-type (WT) littermate controls (FVB/N strain). Langendorff-perfused whole hearts,24,25 electrophysiological recordings in single isolated myocytes, fluo-4 Ca2+ imaging, western blots, immunocytochemistry, and confocal imaging were performed. Data were subjected to statistical analysis using KaleidaGraph 4.5 and Origin 6.1 software. All substances used were of the highest purity available. Experimental details are provided in an extended methods section in the Supplementary material online.
3. Results
3.1. Selective activation of a TRPC3 conductance in cardiomyocytes
A novel activator of lipid-sensitive TRPC channels, GSK1702934A (GSK), has recently been introduced.23 Availability of this compound by custom synthesis prompted us to investigate the acute functional consequences of TRPC3 activation in the heart and a scenario lacking phospholipase C stimulation and signalling pathways that hamper evaluation of the exact role of the channel. When challenged with GSK (1 µM), TRPC3-transgenic (TG) cardiomyocytes exhibited a non-selective membrane conductance with features resembling that of heterologously expressed TRPC3 channels. In contrast to control (WT) cardiomyocytes, TRPC3-TG cardiomyocytes displayed the typical double-rectifying characteristics of TRPC3 currents (Figure 1A) as previously described for the HEK293 overexpression system.26 GSK-induced membrane currents in TRPC3-overexpressing HEK293 cells are illustrated for comparison in Figure 1B.23 Time courses of GSK-sensitive currents were similar in TRPC3-TG cardiomyocytes (Figure 1A, lower panel) and transiently TRPC3-expressing HEK-293 cells (Figure 1B, lower panel). Peak currents were typically observed 10–15 s after application of the activator and declined to basal levels within 30 s. Current activation was not suppressed by elevation of the intracellular Ca2+ buffer (11 mmol/L EGTA; Figure 1A). GSK-induced currents were strictly dependent on TRPC3 expression, and GSK (1 µM) lacked any effects on major ion conductances, which determine cardiac action potential morphology and excitability such as voltage-gated K+ conductances (see Supplementary material online, Figure S1A), substantiating TRPC3 as the primary target of GSK. Applied at a concentration of 1 µM, the TRPC3 activator transiently but significantly (P < 0.05) prolonged action potential duration (APD90) in TRPC3-overexpressing myocytes from 29.5 ± 5.6 to 53.0 ± 9.0 ms as shown in Figure 1C and moderately depolarized cells from −75.2 ± 1.3 to −71.7 ± 2.2 mV (P < 0.05, tested by unpaired t-test). Our initial results on the cardiac action of GSK confirmed this compound as a suitable tool to investigate the acute consequences of enhanced TRPC3 conductances in the heart without overlapping effects on other conductances or activation of the phospholipase C signalling pathway. In parallel experiments, we characterized effects of AngII, a common arrhythmogenic mediator and stimulator of the Gq/PLC signalling pathway, and observed an AngII-induced membrane conductance that perfectly resembled the GSK-induced TRPC3 activity in terms of time course and current to voltage relation (see Supplementary material online, Figure S2). Consequently, we set out to characterize the implications of the transient TRPC3 current for electrical and mechanical function of murine hearts using the isolated perfused Langendorff preparation.
Figure 1.

Activation of TRPC3 currents in cardiomyocytes and HEK293 cells by GSK1702934A. Membrane currents and action potentials were recorded with standard whole-cell patch-clamp technique (for details, see Supplementary material online). Current–voltage (I–V) relations were obtained by applying a descending voltage ramp (+90 mV to −120 mV for 2 s) to eliminate voltage-gated Na+ and Ca2+ currents. (A) Average (upper panel) and time courses of currents at +60 mV and −80 mV (lower panel) in the absence and presence of GSK1702934A (1 µM; GSK). Voltage dependence of responses to GSK (upper panel) corresponding to peak of activation (grey) in comparison to baseline (control; black) for wild-type (WT; A, left, n = 7; N = 3), TRPC3-TG myocytes (A, centre, n = 8; N = 3), TRPC3-TG myocytes loaded with 11 mM EGTA to buffer to diastolic levels (A, right, n = 8; N = 3; EGTA buffered) and HEK293 cells overexpressing TRPC3 (B, n = 5) with buffered (3 mM EGTA; EGTA buffered). (C) Left: representative action potential (AP) recordings from a TRPC3-TG ventricular myocyte before (black line) and during administration of GSK (1 µM, grey line). Without intracellular Ca2+ buffer, GSK prolongs late repolarization up to 1.5-fold in TRPC3-TG but not in WT cardiomyocytes. Right: time course of APD90 recorded from wild-type (WT) and transgenic (TRPC3-TG) ventricular myocytes in response to 1 µM GSK [n = 10; N = 4 (WT), N = 3 (TG)].
3.2. TRPC3 activity contributes to control of cardiac contractility and arrhythmogenesis
GSK (1 µM) lacked significant effects on contractility of isolated perfused hearts from WT mice, but it generated a clear positive inotropic response in TRPC3-TG hearts (Figure 2A and see Supplementary material online, Figure S3A). The positive inotropic response was transient with left ventricular systolic pressure reaching a peak (LVSP: 119.1 ± 5.2%; dP/dt: 122 ± 4%; P < 0.05; N = 8) ∼3–4 min after start of GSK perfusion. In the majority of experiments, GSK initiated a rise in diastolic pressure, eventually leading to significant diastolic deterioration. Interestingly, we did not observe any differences in cardiac performance at baseline between WT and TRPC3-TG mouse hearts [WT, LVDevP (left ventricular developed pressure, LVSP-LVDP): 106.9 ± 1.8 mmHg vs. TRPC3-TG, LVDevP: 100.9 ± 6.9 mmHg; P > 0.1]. Thus, we report here on a novel principle of positive inotropism based on TRPC3 activation. Similar to GSK, AngII initiated transient positive inotropic effects, which amounted to 110% in WT hearts (LVSP: 114.2 ± 1.1%; dP/dt: 108.5 ± 1.3%; P < 0.01; N = 10) and was exaggerated up to 140% in TRPC3-TG mouse hearts (LVSP: 137.2 ± 14.8%; dP/dt: 138.5 ± 7.2%; P < 0.01; N = 15). This positive inotropic effect was followed by a sudden cardiac dysfunction with increasing diastolic pressure and transient loss of function, from which the majority of hearts recovered spontaneously within 1 min (see Supplementary material online, Figure S4A). This suggests an involvement of TRPC3 in functional effects of AngII at elevated expression levels of TRPC3.
Figure 2.

Activation of TRPC3 by GSK1702934A modulates cardiac contractility and favours rhythmic instability. (A) Representative recordings of left ventricular pressure (LVP) in Langendorff-perfused WT and TRPC3-TG hearts using the ISO-HEART perfusion system (Hugo Sachs Elektronik, March-Hugstetten, Germany) as previously described.25 GSK1702934A (GSK, 1 µM) modulates cardiac contractility in a TRPC3 expression-dependent manner as evident from the maximum rate of change in left ventricular pressure (dP/dt) over time (A, middle panel). GSK increased LVP in TRPC3-TG mouse hearts up to ∼120% (P < 0.05) and ∼103% (P < 0.05) in WT mouse hearts compared with baseline conditions (A, right panel); statistical significance analysed by paired t-test. (B) Left: representative LVP traces from WT (top) and TRPC3-TG (bottom) mouse hearts recorded by Langendorff perfusion. Right: comparison of arrhythmogenesis using an arrhythmia scoring system for WT (black, n = 6) and TRPC3-TG (grey, n = 8) hearts at baseline conditions (WT: median level = 1; mean = 0.83 ± 0.17; TRPC3-TG: median level = 1; mean = 1.13 ± 0.23) and in the presence of 1 µM GSK (WT: median level = 1; mean = 1.17 ± 0.31; TRPC3-TG: median level = 2; mean = 2.13 ± 0.35). (right panel); statistical significance analysed by paired Wilcoxon–Mann–Whitney (WT ± GSK; P = NS), (TRPC3-TG ± GSK; * indicates P < 0.05) and Kruskal–Wallis Anova test followed by Dunns test for multiple comparisons (WT ± GSK vs. TRPC3-TG ± GSK; P = NS for WT vs. TRPC3-TG; P < 0.1 for WT + GSK vs. TRPC3-TG + GSK); in brackets: total number of mice at each arrhythmia score.
Detailed inspection of LVP twitch traces revealed a higher incidence of arrhythmic events in GSK-exposed hearts from TRPC3-TG (median level 2) compared with WT mice (median level 1; P < 0.1) (Figure 2B). WT as well as TRPC3-TG hearts displayed rare isolated arrhythmic events (ventricular or atrial premature beats) in non-stimulated (basal) conditions. GSK exerted a significant pro-arrhythmic action in TRPC3-TG hearts (basal condition: median level 1 vs. median level 2 during GSK application; P < 0.05), characterized by paired ventricular beats, burst of atrial tachycardia, and cardiac alternans in a majority of experiments. GSK in contrast failed to significantly enhance the burden of arrhythmic events in WT hearts as quantified by an arrhythmia scoring system (basal condition: median level 1 vs. median level 1 during GSK application; P = NS)27 (Figure 2B). In three out of eight TRPC3-TG hearts, short, transient episodes of ventricular tachycardia were induced by GSK (see Supplementary material online, Figure S3B). Similarly, LVP traces from TRPC3-overexpressing hearts revealed a transient burden of arrhythmic activity during AngII challenge. This was characterized by sustained episodes of bigeminal beats during the positive inotropic phase and episodes of atrial or ventricular tachycardia and ventricular or atrial premature beats during recovery of cardiac function (see Supplementary material online, Figure S4). The AngII-induced disturbance of cardiac rhythm increased to Levels 3–4 on the arrhythmia scoring for TRPC3-TG hearts (basal condition: median level 1 vs. median level 4 during GSK application; P < 0.01), while WT hearts lacked significant rhythmical disturbances during AngII perfusion (basal condition: median level 1 vs. median level 2 during GSK application; P = NS) (see Supplementary material online, Figure S4A).
3.3. TRPC3 activity promotes arrhythmias by a mechanism involving cellular Ca2+ handling and NCX1
The arrhythmogenic action of GSK was clearly evident from AP recording in isolated myocytes. Analysis of 10 min continuous recordings during GSK perfusion revealed a higher incidence of arrhythmic depolarizations (early and late afterdepolarizations) in TRPC3-TG compared with WT cardiomyocytes (Figure 3A and B). It is of note that the effects of AngII in isolated TRPC3-TG cardiomyocytes resembled that of GSK in terms of changes in AP morphology (see Supplementary material online, Figure S4D).
Figure 3.

Arrhythmic activity in cardiomyocytes in response to TRPC3 activation requires rise of intracellular Ca2+ and NCX1 function. Action potentials were recorded from single myocytes upon intracellular current injection (1–2 nA amplitude, 2–4 ms duration) at a pacing cycle length of 2 s.28 (A) Continuous AP recordings displayed prominent arrhythmic afterdepolarizations (EADs and DADs) in TRPC3-TG myocytes during GSK infusion; WT (left panel), TRPC3-TG (right panel). (B) Frequency of arrhythmic events (number of EADs and DADs during 10 min application of GSK). Both EADs (4.7 ± 1.79 TG; n = 15; N = 3 vs. 0.3 ± 0.3 WT; n = 17; N = 4; P < 0.05) and DADs (10.75 ± 3.08 TG vs. 3.63 ± 1.32 WT; P < 0.05) were significantly increased in TRPC3-TG myocytes. Statistical significance analysed by Wilcoxon–Mann–Whitney test. (C) Incidence of afterdepolarizations (AD, left), delayed ADs (DAD, centre), and early ADs (EAD, right): WT compared with TRPC3-TG cells ± buffered to diastolic levels by 11 mM EGTA (n = 10; N = 3) or in the presence of the NCX1 inhibitor DCB (10 µM; n = 8; N = 3). Black bars represent population of cells displaying arrhythmic events during 10 min (n > 3). * indicates statistical significance (P < 0.05) in comparison to WT conditions, sharp (#) indicates (P < 0.05) in comparison to untreated TRPC3-TG myocytes. Statistical significance analysed by Fisher’s exact test.
Interestingly, when was efficiently buffered at diastolic levels by intracellular EGTA (11 mM), which did not prevent TRPC3 currents (Figure 1A), arrhythmic activity of GSK was blunted (Figure 3C) and prolongation of APD90 by GSK was barely detectable (see Supplementary material online, Figure S5). These results suggest that TRPC3-mediated arrhythmic events and changes in AP morphology are not simply a consequence of the TRPC conductance itself, but it involve a more complex linkage between the TRPC conductance and cellular Ca2+ handling.
As elevated is a well-recognized trigger for NCX-mediated arrhythmic events,28–30 we hypothesized a role of distorted Ca2+ homeostasis and NCX1 function in GSK-induced initiation of arrhythmic events. Detailed analysis of arrhythmic activity revealed that a majority of afterdepolarizations occurred either after complete repolarization or at negative potentials more negative than −50 mV during repolarization (see Supplementary material online, Figure 6) and may therefore well be based on NCX-mediated inward currents.31 Preincubation of TRPC3-TG cardiomyocytes with 10 µM 3′, 4′-dichlorobenzamil hydrochloride (DCB), a pharmacological tool to inhibit NCX forward-mode activity,32 indeed markedly reduced the number of arrhythmic events evoked by GSK, pointing towards TRPC3-triggered alteration of NCX1 function as the basis of the pro-arrhythmic action of GSK (Figure 3). NCX1 expression was found unchanged in TRPC3-TG and WT cardiomyocytes (see Supplementary material online, Figure S7), substantiating a functional link between TRPC3 and NCX1. Signalling crosstalk based on micro/nanodomain organization of the channel in proximity of the transporter has already been demonstrated for cardiovascular cells.22,33 We hypothesized that TRPC3 activation may promote NCX operation during action potentials.
Initial characterization of the effects of AngII on NCX1 forward-mode activity by tail current analysis revealed that AngII increased NCX-inward currents only in TRPC-TG myocytes (see Supplementary material online, Figure S2C). To test whether TRPC3 expression and activity indeed determines exchanger operation during cyclic de- and repolarization, we performed patch-clamp experiments in TRPC3-TG cardiomyocytes to delineate cyclic NCX function as the Ni2+-sensitive current component (Figure 4). These experiments were performed in the absence of functional sarcoplasmic reticulum (SR) using a voltage-clamp protocol to quantify reverse- and forward-mode NCX activity during depolarization and subsequent repolarization, respectively.34,35 SR function was disrupted with thapsigargin (Tg, 3 µmol/L), and voltage-gated Ca2+ entry was blocked with nitrendipine (10 µM). TRPC3-TG myocytes displayed elevated NCX outward as well as consecutive inward currents compared with wild-type myocytes when applying strong depolarizing voltage steps (+100 mV; Figure 4). Moreover, NCX currents were significantly amplified by TRPC3 overexpression in the presence of GSK. In WT myocytes, GSK promoted NCX currents only slightly to a level observed constitutively in TRPC3-TG myocytes. Thus, activation of TRPC3 at elevated expression levels significantly enhanced NCX currents in this voltage-clamp protocol, indicating that TRPC3 may dynamically affect ion concentration at the exchanger. Local, TRPC3-mediated Na+ loading at rest (holding potential) is expected to promote reverse-mode currents upon depolarization. The resulting Ca2+ accumulation during depolarization is, in turn, likely to facilitate Ca2+-induced Ca2+ release and drive forward-mode currents upon subsequent repolarization. We hypothesized that these alterations in Ca2+ cycling may promote spontaneous Ca2+ mobilization from the SR representing a potential trigger of arrhythmic events. We tested this concept by Ca2+ imaging experiments using confocal line scanning fluorescence microscopy. Ca2+ mobilization was visualized during a short subsequent, quiescent period in the absence of transmembrane Ca2+ and Na+ fluxes after an equilibrating train of stimuli in physiologic conditions as illustrated in Supplementary material online, Figure S8. Ca2+ spark frequency was significantly higher in TRPC3-TG than in WT myocytes already at basal levels of TRPC activity (Figure 5). Activation of TRPC3 by GSK (1 µM) clearly increased the frequency of Ca2+ sparks in TRPC3-TG and promoted Ca2+ discharge in WT myocytes to a level slightly below that observed in unstimulated TRPC3-TG myocytes. Interestingly, experiments designed to evaluate SR Ca2+ content did not indicate differences between TRPC3-TG and WT myocytes. Details of the Ca2+ transients measured in WT and TG myocytes are given in the supplemental information section (Table 1).
Figure 4.

Cardiac TRPC3 activity modulates NCX function. NCX currents were recorded by applying depolarizing voltage steps (500 ms) from a holding potential of −40 mV and quantified as the Ni+-sensitive current components at the end of the 500 ms depolarization steps (NCX reverse mode) and 20 ms after repolarization to −40 mV (forward mode).34,35 (A) Representative Ni+-sensitive outward currents recorded during depolarizing steps as well as subsequent tail currents upon repolarization to −40 mV for 4.5 s are shown for WT cardiomyocytes (left) and TG cardiomyocytes (right) in the absence [n = 8; N = 3 (WT); n = 9; N = 3 (TG)] (top) and presence (bottom) of 1 µM GSK [n = 8; N = 3 (WT); n = 12; N = 3 (TG)]; SR function was eliminated by thapsigargin (3 µM) and l-type Ca2+ channels were blocked by nitrendipine (10 µM). (B) Mean (±SEM) I/V plots of Ni+-sensitive peak currents. Left: tail inward currents representing NCX forward-mode activity in WT and TRPC3-TG cells and in the absence or presence of 1 µM GSK. Tail currents represent Ni+-sensitive peak inward currents at 20 ms after repolarization to −40 mV with steady-state holding current subtracted. Right: mean (±SEM) peak outward currents recorded from WT and TRPC3-TG cells in the presence and absence of 1 µM GSK. * indicates significant difference (P < 0.05) to WT, WT + GSK, and TRPC3-TG; sharp (#) significant difference (P < 0.05) to WT + GSK cells. Statistical significance analysed by two-way Anova followed by Tukey's post hoc tests.
Figure 5.
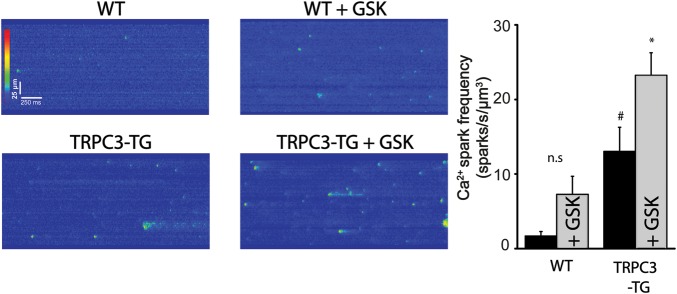
GSK enhances Ca2+ spark frequency in TRPC3-dependent manner. Confocal line scan images were recorded along the longitudinal axis of myocytes loaded with fluo-4 as described previously36 and analysed using custom-made algorithms coded in IDL.36,37 Left: time plots of Ca2+-sensitive fluorescence line scans recorded from WT cardiomyocytes (n = 14; N = 3), TRPC3-TG (n = 19; N = 7), WT + GSK (n = 19; N = 3), and TRPC3-TG + GSK (n = 19; N = 5); Right: mean calcium spark frequency (± SEM). Intracellular Ca2+ fluxes were at equilibrium and measurement was performed during a prolonged diastole. Sharp (#) indicates significant difference (P < 0.05) in comparison to WT, * indicates significance (P < 0.05) in comparison to untreated TRPC3-TG cells. Statistical significance analysed by two-way Anova followed by Tukey's post hoc tests.
Table 1.
Cardiomyocyte calcium homeostasis
| Parameter | WT untreated | WT + GSK | TRCP3 untreated | TRCP3 + GSK |
|---|---|---|---|---|
| CaT amplitude (F/F0) | 4.18 ± 0.23 (n = 28; N = 3) | 4.3 ± 0.1 (n = 14; N = 3) | 4.14 ± 0.15 (n = 37; N = 6) | 4.55 ± 0.12 (n = 26; N = 4) |
| SR Ca2+ content (F/F0) | 5.27 ± 0.11 (n = 9; N = 3) | 5.36 ± 0.14 (n = 11; N = 3) | 5.48 ± 0.08 (n = 10; N = 4) | 5.32 ± 0.12 (n = 12; N = 4) |
| TAU (s) | 2.15 ± 0.17 (n = 9; N = 3) | 1.93 ± 0.05 (n = 11; N = 3) | 2.12 ± 0.1 (n = 10; N = 4) | 1.81 ± 0.09 (n = 12; N = 4) |
| Ca2+ spark frequency (sparks/s/µm3) | 1.69 ± 0.6 (n = 14; N = 3) | 7.28 ± 2.42 (n = 19; N = 3) | 13.04 ± 3.23# (n = 19; N = 7) | 23.24 ± 3.03* (n = 19; N = 5) |
Key data of Ca2+-transient and Caffeine response parameters, (mean value ± SEM). Two-way Anova followed by the Tukey's post hoc test for multiple comparisons when an overall significance was established.
*P < 0.05, TRPC3 + GSK significantly different from TRPC3 untreated and WT + GSK.
#P < 0.05, TRPC3 significantly different from WT.
As TRPC3 activation was found to promote both NCX currents and Ca2+ mobilization from the SR with a considerably prolonged time course compared with that of current activation, we speculated about additional dynamic changes in the TRPC3-NCX crosstalk and set out to investigate localization of the proteins in cardiac myocytes.
A close spatial relation between TRPC3 and NCX1 has previously been demonstrated21,38 and is considered prerequisite for functional consequences of cardiac TRPC3 activation. We hypothesized that alterations in spatial coupling betweenTRPC3 and NCX1 may be involved in prolongation of functional consequences of TRPC3 activation. In a first set of experiments, we analysed the localization of TRPC3 and NCX1 in TRPC3-TG myocytes by immunocytochemistry (Figure 6). Our results corroborated colocalization of the two transport molecules at basal conditions. Interestingly, activation of TRPC3 channels by GSK effectively disrupted colocalization of the signalling proteins (Figure 6) and a similar effect was initiated by AngII (see Supplementary material online, Figure S9).
Figure 6.

GSK disrupts TRPC3 and NCX colocalization in adult mouse cardiomyocytes. (A) Confocal fluorescence images of freshly isolated cardiac myocytes (n = 15; N = 3) without (top) and after 5-min exposure to GSK (bottom) and stained for NCX1 (FITC, green) and TRPC3 (TRITC, red) were obtained using a Leica SP5 confocal microscope (Leica Microsystems, Mannheim, Germany). Immunostaining was performed as described previously.21 Magnified parts of the images depicting cell regions used for line scan analysis shown in B. (B) Overlap of FITC-NCX and TRITC-TRPC3 fluorescence intensities along a line scan (white line in micrograph) in the absence (top) and after 5-min exposure to GSK (bottom). Well-correlated intensity peaks are indicated by arrows, non-correlating peaks by x. (C) Mean (±SEM) pearson correlation coefficients (left) and non-linear correlation coefficients (right) of FITC-NCX and TRITC-TRPC3 fluorescence intensity as gained from line scans (n = 6) recorded in myocytes with and without exposure to 1 µM GSK as indicated. (D) Fluorescence microscopic images of HL-1 cells transfected with CFP-NCX split exchanger (CFP-NCX-265 + NCX-272, cyan), YFP-TRPC3 (green), overlay of YFP-TRPC3, and CFP-NCX split exchanger fluorescence (overlay) and net FRET after bleedthrough- and colocalization correction (nFRET) as described in Ref. 39. (E) Representative time course of rawFRET intensity recorded from a cell expressing YFP-TRPC3 and CFP-NCX-265 + NCX-272 (background subtracted). Image showing non-normalized FRET (rawFRET) recorded at the initial phase of the timeline. After 1min, 1 µM GSK was added to the chamber. Insert showing mean normalized rawFRET intensities (±SEM) recorded at indicated times from six independent experiments. Statistical significance was analysed by t-test.
To more directly monitor the disruption of TRPC3-NCX complexes by GSK in cardiac cells, we performed FRET experiments in HL-1 murine atrial myocytes transfected to express fluorescent fusions of NCX1 and TRPC3. Cells expressing equal levels of the fluorescent proteins in the plasma membrane were analysed and displayed significant FRET signals at resting conditions, indicative of direct coupling between the molecules (Figure 6D).
Upon exposure to 1 µM GSK, FRET signal abruptly declined to a significantly lower level (Figure 6E), substantiating re-localization and dissociation of the molecules after activation of the channel with GSK.
It is tempting to speculate that rapid spatial uncoupling of TRPC3 from NCX will lower local Na+ at the exchanger during repolarization, which is expected to impact differentially on reverse and forward-mode operation of NCX, promoting preferentially forward-mode Na+ entry. Our results clearly suggest activity-dependent coupling between cardiac TRPC3 and NCX1 (Figure 7) as a potential mechanism for tuning of TRPC3/NCX1-mediated control of cardiac Ca2+ homeostasis, contractility, and excitability.
Figure 7.

Hypothetical model of dynamic, functional interaction between TRPC3 and NCX1. TRPC3-mediated inotropy (upper left) requires close spatial proximity to NCX1. Cytoplasmic Ca2+ concentration is elevated by (i) suppressed intracellular Ca2+ clearance through NCX1 and (ii) by the TRPC3 Ca2+conductance itself. NCX forward-mode Ca2+ extrusion is limited to negative potentials, leading to enhanced Ca2+ loading of the SR. Excessive activation of TRPC3 results in transient Ca2+ overload followed by both channel inactivation and rapid spatial uncoupling from NCX1. While SR loading slowly declines, rapid drop in local cytoplasmic Na+ at the exchanger is expected to transiently favour arrhythmogenesis by facilitation of NCX forward mode (upper right).
4. Discussion
Using a murine model of cardiac TRPC3 overexpression and a novel pharmacological tool to directly activate TRPC3 channels, we obtained evidence for a Ca2+-dependent impact of a non-selective TRPC conductance on cardiac functions. We demonstrate that acute activation of cardiac TRPC3 increases contractility and promotes arrhythmias by a process most likely involving TRPC3/NCX1 interaction. Lipid-regulated TRPC channels are suggested to form a signalling complex with NCX1, which contribute to adaptation of cardiac contractility during remodelling processes and represent a potential player in arrhythmogenesis.
4.1. TRPC3 expression and activity determines cardiac contractility and rhythm
Although a pivotal role of lipid-regulated TRPC channels in cardiac hypertrophic remodelling has repeatedly been demonstrated,40 their impact on contractility and excitability of the heart is still elusive. Judgment of the role of cardiac TRPC channels is generally hampered by high variability of expression levels and by the fact that activation of TRPC conductances by (patho)physiological stimuli is inevitably associated with parallel changes in phospholipid and Ca2+ signalling, which make clear assignment of functional changes to a TRPC conductance difficult. Understanding of the exact link between TRPC activity and cardiac function is prerequisite for development of pharmacological strategies based on TRPC as target structure. In this study, we took advantage of a model of cardiac-specific overexpression of TRPC317 and the availability of a novel small molecule activator of TRPC3 channels.23 As TRPC3 expression is essentially low in wild-type mice, the organ-specific overexpression model allowed for evaluation of TRPC3 selectivity of the novel activator GSK. The synthetic TRPC3 agonist initiated a conductance with classical TRPC3 features in a strictly TRPC3 expression-dependent manner, indicating selective activation of TRPC3 channels. Consistently, other cardiac conductances or action potential parameters were barely affected by GSK. Although TRPC3 expression in healthy wild-type mice is low, the cation channel is typically up-regulated during maladaptive cardiac remodelling.1,15
Thus, the here deployed overexpression model allows for a specific analysis of the functional consequences of TRPC3 up-regulation in the heart. Previous studies questioned an impact of TRPC channels on cardiac excitation–contraction coupling based on a lack of TRPC localization in diade structures and proximity to l-type Ca2+ channels.18 Here we demonstrate a transient increase in contractility, associated with arrhythmogenesis as an acute response to direct TRPC3 channel activation. Elevated expression of TRPC3 has previously been reported to lack significant effects on contractility in basal conditions17 or to even reduce contractility in the absence of TRPC3-activating stimuli using post rest stimulation protocols.19 The latter effect was observed in feline myocytes and was attributed to TRPC-induced promotion of a Ca2+ leakage by TRPC3. Importantly, our results show that TRPC3 expression promotes Ca2+ turnover, as evident from enhanced release of Ca2+ from the SR at preserved filling, even in the absence of activating stimuli. This suggests that overexpression of the channel protein, which has previously been demonstrated to display constitutive activity,41 affects cardiac Ca2+ homeostasis without detectable alterations in cardiac function. Nonetheless, activation of the channel by either the synthetic agonist GSK or the (patho)physiological activator AngII resulted in a transiently increased TRPC conductance that apparently triggered positive inotropy and arrhythmias. This is in principle consistent with general biophysical considerations regarding the impact of a non-selective cation conductance on resting membrane potential, action potential shape, and Ca2+ homeostasis.1,20 The time course of TRPC3 conductance activation by either GSK or AngII was transient in both cardiac myocytes as well as in TRPC3-overexpressing HEK293 cells. Rapid decline of TRPC3 currents down to baseline during sustained stimulation of the Gq/PLC pathway has previously been reported26 and attributed in part to regulatory phosphorylation by PKC and/or a Ca2+-dependent inactivation mechanism.12 Current inactivation was also observed in the presence of GSK and was not affected by buffering of intracellular Ca2+ with EGTA. Thus, the transient nature of GSK-induced TRPC3 activity may reflect either an as yet ill-defined intrinsic inactivation process linked to phospholipid metabolism or a Ca2+-dependent inactivation process involving a Ca2+ sensor, localized closely to the inner vestibule of the TRPC3 channel. Importantly, the pro-arrhythmic actions initiated by TRPC3 activation clearly outlasted the time course of the non-selective conductance. It is of note that the effects of GSK at the organ level were even more prolonged compared with the observed current kinetics. This may be related to significantly slower kinetics of agonist concentration in the tissue or on altered kinetics of current activation in the multicellular situation. Nonetheless, the time course of TRPC3 current activation did not correlate with the observed functional effects in single cells of isolated murine hearts. Moreover, when intracellular Ca2+ was clamped by EGTA, TRPC3 channel activation by GSK or AngII remained unchanged, while effects on action potential waveform and arrhythmogenesis were profoundly suppressed. These results suggest that TRPC3 determines arrhythmogenesis not primarily by generation of a non-selective conductance but by a complex modulation of Ca2+ homeostasis and Ca2+-dependent excitability. As TRPC conductances are considered to affect sub-plasmalemmal Na+ and Ca2+ levels, we hypothesized an impact of TRPC3 on NCX operation as the basis of the observed changes in excitability.
4.2. Communication between cardiac TRPC3 and NCX1 determines action potential morphology
A signalling partnership between lipid-sensitive TRPC isoforms and NCX1 has previously been suggested for different cardiovascular cell types.21,22,33 Our results obtained in mouse ventricular myocardium corroborated this concept. Three lines of evidence suggest that TRPC3-NCX1 communication mediates functional consequences of TRPC3 activation in the heart: (i) TRPC3 and NCX1 showed significant colocalization in cardiomyocytes. (ii) Action potential prolongation and arrhythmic activity were suppressed by intracellular EGTA without significant suppression of the TRPC3 conductance. (iii) The NCX1 inhibitor DCB effectively inhibited both action potential prolongation and arrhythmias. Overall, these effects are consistent with an involvement of NCX in GSK-induced arrhythmic events. Nonetheless, it is of note that inhibition by DCB on its own is certainly insufficient to conclude NCX involvement due to limited selectivity of this pharmacological tool. TRPC conductances have been proposed to affect NCX operation by elevation of Na+ cellular levels, thereby reducing Ca2+ extrusion or even promote Ca2+ entry at positive potentials.21,33 Similar to glycoside inhibitors of K+/Na+ ATPase, elevated TRPC3 expression is expected to perturb Ca2+ extrusion by NCX1 due to elevation of local Na+ at the exchanger. We therefore hypothesized that TRPC3 channel activity increases cellular Ca2+ by mediating combined Ca2+ and Na+ entry at NCX1 to generate a cardiac glycoside-like effect,28,42 as illustrated in the hypothetical scheme shown in Figure 7. This concept was supported by our observation that TRPC3 overexpression, already at basal levels of channel activity, significantly enhanced discharge of Ca2+ from the SR. This effect was further accentuated during activation of channels with GSK and is most likely based not only on Ca2+ entering through TRPC3 channels but on reduced Ca2+ extrusion or even reverse-mode Ca2+ entry at positive potentials (Figure 7). Consistently, we observed TRPC3-dependent promotion of cyclic NCX activity in a voltage-clamp protocol (Figure 4), corroborating the principle of a rather direct modulation of NCX operation by the TRPC3 conductance. Enhanced reverse-mode currents during strong depolarizing pulses were followed by exaggerated forward-mode operation during subsequent hyperpolarization of TRPC3-overexpressing myocytes. It appears conceivable that activation of a Na+ permeable channel in proximity of NCX1 generates a gain in the dynamic function of the exchanger as previously suggested for NaV channels.43 Although reverse-mode NCX-mediated Ca2+ entry may be limited during the short murine action potential, TRPC3 is expected to profoundly elevate local Na+ as well as Ca2+ at NCX at negative potentials and thereby promote Ca2+ accumulation during depolarizations. Our results unequivocally demonstrate such tight communication between the two cardiac transport molecules. Nonetheless, local communication between NCX and TRPC3 does not explain the time course of arrhythmic events, which clearly outlasted TRPC3 current activation. However, we identified an additional mechanism that may contribute to TRPC3-mediated arrhythmogenesis. Our analysis of subcellular localization of TRPC3 and NCX1 by immunocytochemistry as well as of fluorescent fusion proteins expressed in HL-1 atrial myocytes confirmed colocalization and physical interaction. Importantly, our experiments revealed activity-dependent disruption of local coupling between the TRPC channel and the exchanger. This phenomenon was initiated by channel activation, develops within 2–10 min after agonist stimulation, and is suggested to facilitate NCX-inward currents due to a drop in local Na+ at the exchanger. Rapid reduction of Na+ entry into a sub-plasmalemmal space of restricted diffusion is expected to affect NCX function during action potentials and to promote late EADs or DADs. This effect may in part involve removal of Na+-dependent inhibition of NCX.44,45
It is thus tempting to speculate that this scenario is the basis of transient facilitation of arrhythmogenesis (see scheme in Figure 7). We suggest a model of cardiac TRPC3-NCX interaction in which NCX function is determined by proximity to TRPC3. At basal levels of TRPC3 activity and spatial coupling between the signalling molecules within a nano/microdomain, NCX forward mode is limited by local Na+ entry. Along with TRPC3-mediated Ca2+ entry, this state is proposed to account for enhanced Ca2+ mobilization from the SR and positive inotropy during channel activation. Abrupt dissociation of TRPC3 from NCX may promote arrhythmic events.
4.3. TRPC3-NCX signalplexes as potential therapeutic targets in the myocardium
Lipid-sensitive TRPC3/6/7 channels have recently emerged as potential therapeutic targets for the prevention of cardiac remodelling. Early stages of hypertrophic remodelling have been characterized by enhanced expression of lipid-sensitive TRPC channel complexes,15 and Ca2+ entry via these channels has clearly been identified as a signal that triggers Ca2+-dependent expression of prohypertrophic genes.12 Therefore, TRPC channels are proposed as elements of a critical feed-forward process in pathological remodelling, and inhibition of these channels might be considered as a strategy for prevention of heart failure development. It is important to point out that our results were obtained with a murine model of cardiac dysfunction. As this experimental model represents a specific type of cardiac muscle with Ca2+ handling being different from that in larger rodents or humans, conclusions to human pathology need to be drawn with care. Nevertheless, our current study employing a TRPC3 overexpression model unravels TRPC3 expression as a potential determinant of contractile function and susceptibility to arrhythmogenic stimuli. A recent report attributed the role of cardiac TRPC3 in arrhythmogenesis to the channel’s expression in fibroblasts.46 Here we present evidence that enhanced levels of TRPC3 in cardiomyocytes modify promote arrhythmogenic effects of AngII, which is known to evoke similar electrophysiological effects as GSK in the TRPC3-overexpressing heart. Enhanced cardiac expression of TRPC3 might be beneficial to restore contractility in early stages of remodelling. Excessive activation of the lipid-sensitive cation channels is, however, associated with enhanced arrhythmogenesis and Ca2+ overload-mediated cardiac dysfunction. This concept opens the view on novel therapeutic strategies based on targeting specific TRPC3 functions or its molecular coupling to NCX1.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by FWF grants P21925-B19 to K.G. and DK+ Metabolic and Cardiovascular Disease, grant W2126-B18. Funding to pay the Open Access publication charges for this article was provided by FWF grant W2126.
Acknowledgements
The authors thank J. Molkentin for providing the TRPC3 transgenic mouse line and K. Phillipson and J. Scott for providing NCX fusion constructs.
Conflict of interest: none declared.
References
- 1.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 2006;25:5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu W, Zou Y, Shiojima I, Kudoh S, Aikawa R, Hayashi D, Mizukami M, Toko H, Shibasaki F, Yazaki Y, Nagai R, Komuro I. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem 2000;275:15239–15245. [DOI] [PubMed] [Google Scholar]
- 3.Liu N, Gong K-Z, Cai Y-B, Li Z. Identification of proteins responding to adrenergic receptor subtype-specific hypertrophy in cardiomyocytes by proteomic approaches. Biochem Mosc 2011;76:1140–1146. [DOI] [PubMed] [Google Scholar]
- 4.Sharif-Naeini R, Folgering JHA, Bichet D, Duprat F, Delmas P, Patel A, Honoré E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol 2010;48:83–89. [DOI] [PubMed] [Google Scholar]
- 5.Goonasekera SA, Molkentin JD. Unraveling the secrets of a double life: contractile versus signaling Ca2+ in a cardiac myocyte. J Mol Cell Cardiol 2012;52:317–322. [DOI] [PubMed] [Google Scholar]
- 6.Molkentin JD. Parsing good versus bad signaling pathways in the heart: role of calcineurin-nuclear factor of activated T-cells. Circ Res 2013;113:16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest 2000;105:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muth JN, Bodi I, Lewis W, Varadi G, Schwartz A. A Ca(2+)-dependent transgenic model of cardiac hypertrophy: a role for protein kinase Calpha. Circulation 2001;103:140–147. [DOI] [PubMed] [Google Scholar]
- 9.De Windt LJ, Lim HW, Bueno OF, Liang Q, Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ, Molkentin JD. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 2001;98:3322–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998;93:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molkentin JD. Dichotomy of Ca2+ in the heart: contraction versus intracellular signaling. J Clin Invest 2006;116:623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poteser M, Schleifer H, Lichtenegger M, Schernthaner M, Stockner T, Kappe CO, Glasnov TN, Romanin C, Groschner K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc Natl Acad Sci USA. 2011;108:10556–10561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD, Houser SR. A caveolae-targeted L-Type Ca2+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res 2012;110:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escobar M, Cardenas C, Colavita K, Petrenko NB, Franzini-Armstrong C. Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J Mol Cell Cardiol 2011;50:451–459. [DOI] [PubMed] [Google Scholar]
- 15.Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 2006;281:33487–33496. [DOI] [PubMed] [Google Scholar]
- 16.Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T, Ito H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol 2007;42:498–507. [DOI] [PubMed] [Google Scholar]
- 17.Nakayama H. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. The FASEB J 2006;20:1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenner JS, Dolmetsch RE. TrpC3 regulates hypertrophy-associated gene expression without affecting myocyte beating or cell size. PLoS ONE 2007;2:e802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese D, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, Chen X, Gao E, Molkentin JD, Houser SR. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 2014;115:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao H, Wang F, Wang W, Makarewich CA, Zhang H, Kubo H, Berretta RM, Barr LA, Molkentin JD, Houser SR. Ca2+ influx through L-type Ca2+ channels and transient receptor potential channels activates pathological hypertrophy signaling. J Mol Cell Cardiol 2012;53:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eder P, Probst D, Rosker C, Poteser M, Wolinski H, Kohlwein SD, Romanin C, Groschner K. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc Res 2007;73:111–119. [DOI] [PubMed] [Google Scholar]
- 22.Poburko D, Liao C-H, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 2007;101:1030–1038. [DOI] [PubMed] [Google Scholar]
- 23.Xu X, Lozinskaya I, Costell M, Lin Z, Ball JA, Bernard R, Behm DJ, Marino JP, Schnackenberg CG. Characterization of small molecule TRPC3 and TRPC6 agonist and antagonists. Biophys J 2013;104:454a. [Google Scholar]
- 24.Wolkart G, Schrammel A, Dörffel K, Haemmerle G, Zechner R, Mayer B. Cardiac dysfunction in adipose triglyceride lipase deficiency: treatment with a PPARα agonist. Br J Pharmacol 2011;165:380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B. Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation 2001;104:3097–3102. [DOI] [PubMed] [Google Scholar]
- 26.Lichtenegger M, Stockner T, Poteser M, Schleifer H, Platzer D, Romanin C, Groschner K. A novel homology model of TRPC3 reveals allosteric coupling between gate and selectivity filter. Cell Calcium 2013;54:175–185. [DOI] [PubMed] [Google Scholar]
- 27.Wu Y. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 2002;106:1288–1293. [DOI] [PubMed] [Google Scholar]
- 28.Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskamper J, Pieske B. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res 2010;87:50–59. [DOI] [PubMed] [Google Scholar]
- 29.Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. Na/Ca exchange: regulator of intracellular calcium and source of arrhythmias in the heart. Ann NY Acad Sci 2007;1099:315–325. [DOI] [PubMed] [Google Scholar]
- 30.Homma N, Amran MS, Nagasawa Y, Hashimoto K. Topics on the Na+/Ca2+ exchanger: involvement of Na+/Ca2+ exchange system in cardiac triggered activity. J Pharmacol Sci 2006;102:17–21. [DOI] [PubMed] [Google Scholar]
- 31.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization: underlying mechanism and threshold for triggered action potentials. Circ Res 2000;87:774–780. [DOI] [PubMed] [Google Scholar]
- 32.Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol 1999;127:1846–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosker C. Ca2+ Signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem 2004;279:13696–13704. [DOI] [PubMed] [Google Scholar]
- 34.Sipido KR, Volders PGA, de Groot SHM, Verdonck F, Van de Werf F, Wellens HJJ, Vos MA. Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 2000;102:2137–2144. [DOI] [PubMed] [Google Scholar]
- 35.Hobai IA, O'Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res 2000;87:690–698. [DOI] [PubMed] [Google Scholar]
- 36.Hohendanner F, Ljubojević S, Macquaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, Antoons G, Sipido K, Pieske B, Heinzel FR. Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ Res 2013;113:527–538. [DOI] [PubMed] [Google Scholar]
- 37.Sacherer M, Sedej S, Wakuła P, Wallner M, Vos MA, Kockskamper J, Stiegler P, Sereinigg M, Lewinski von D, Antoons G, Pieske BM, Heinzel FR. CONTICA investigators. JTV519 (K201) reduces sarcoplasmic reticulum Ca2+ leak and improves diastolic function in vitro in murine and human non-failing myocardium. Br J Pharmacol 2012;167:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goel M, Zuo C-D, Sinkins WG, Schilling WP. TRPC3 channels colocalize with Na+/Ca2+ exchanger and Na+ pump in axial component of transverse-axial tubular system of rat ventricle. Am J Physiol Heart Circ Physiol 2007;292:H874–H883. [DOI] [PubMed] [Google Scholar]
- 39.Brzostowski JA, Meckel T, Hong J, Chen A, Jin T. Imaging protein-protein interactions by Förster resonance energy transfer (FRET) microscopy in live cells. Curr Protoc Protein Sci 2009;56:19.5.1–19.5.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res 2011;108:265–272. [DOI] [PubMed] [Google Scholar]
- 41.Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem 2003;278:47842–47852. [DOI] [PubMed] [Google Scholar]
- 42.Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. KB-R7943 block of Ca(2+) influx via Na(+)/Ca(2+) exchange does not alter twitches or glycoside inotropy but prevents Ca(2+) overload in rat ventricular myocytes. Circulation 2000;101:1441–1446. [DOI] [PubMed] [Google Scholar]
- 43.Lines GT, Sande JB, Louch WE, Mørk HK, Grøttum P, Sejersted OM. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys J 2006;91:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hilgemann DW, Matsuoka S, Nagel GA, Collins A. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Sodium-dependent inactivation. J Gen Physiol 1992;100:905–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuoka S, Nicoll DA, He Z, Philipson KD. Regulation of cardiac Na(+)-Ca2+ exchanger by the endogenous XIP region. J Gen Physiol 1997;109:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D, Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012;126:2051–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]