

Abstract
Mitochondrial toxin 3-nitropropionic acid (3NPA) is a neurotoxin that inhibits the activity of succinate dehydrogenase, a key enzyme of oxidative energy production, and characteristically provokes neurodegeneration in the striatum, resembling Huntington’s disease. 3NPA also affects the activity of glycogen-sinthase-kinase-3b (GSK-3b), an enzyme implicated in glycogen synthesis and in signal transduction. The aim of this study was to evaluate cardiac glycogen content and histopathological changes in the hearts of rats after subchronic treatment with 3NPA. Female adult Wistar rats were treated daily with 30mg/kg of 3NPA subcutaneously 8 days. The control group was treated with normal saline for 8 days. For the comparison of measured parameters between groups we used the Student’s t-test (p<0.05).
The stereological evaluation of glycogen content in histological sections of the heart was processed with periodic acid-Schiff (PAS). Histochemical procedure showed a significant accumulation of glycogen granules in the 3NPA group (0.028mm3/mm3±0.022), whereas the hearts of control animals were nearly devoid of glycogen granules (0.002mm3/mm3±0.001). Haematoxylin-eosin histological staining showed diffuse swelling of cardiomyocytes (3NPA=15.989μm ±1.649; saline=13.456μm ± 0.786), loss of cell cross-striations, lower myofibril volume fraction (3NPA=0.3922mm3/mm3 ± 0.0230, saline=0.4ssomm3/mm3 ± 0.0083), and mononuclear infiltration in the interstitial tissue, mostly along the blood vessels. Sirius red staining showed fibrosis of the heart (3NPA=0.0531mm3/mm3±0.0090, saline=0.013smm3/mm3 ± 0.0051). TUNEL staining showed TUNEL-positive cells in the 3NPA group (2.04cells/mm2 ± 0.92) and almost no TUNEL-positive cells in the saline group (027cells/mm2 ± 0.14).
This experiment shows that 3NPA-induced histopathological changes in the heart are accompanied by a significant accumulation of glycogen granules in cardiomyocytes.
KEY WORDS: rat, heart, 3NPA, toxicity, glycogen
INTRODUCTION
The toxin 3-nitropropionic acid (3NPA) is a well known food contaminating mycotoxin. It is naturally present in leguminous plants used to feed animals and can poison grazing livestock. Human intoxication has occurred in China when children ingested sugarcane contaminated with fungi (Arthrinium and Aspergillus) that produce 3NPA [1, 2]. Humans and experimental animals exposed to 3NPA develop severe dystonia [3] and decreased motor activity. At the cellular level, 3NPA inhibits succinate dehydrogenase [4, 5], a key enzyme of oxidative energy production that is localized in the mitochondrial inner membrane and complex II of the respiratory chain, causing ATP levels in the brain to fall. Thus, a major factor in 3NPA toxicity is cellular metabolic impairment due to mitochondrial stress. This effect develops fast and is not limited to the sites of morphological damage [6]. 3NPA induces neurodegeneration primarily in the striatum (caudate-putamen) resembling Huntington’s disease (HD) [7] and is used as a metabolic animal model of HD [7, 8]. Toxin-treated experimental animals showed degeneration also in the hippocampus and thalamus [9, 10]. As the brain, also the heart has a great dependence on mitochondrial function and ATP production [11, 12, 13]. Mirandola et al. [14] have recently shown that brain and heart mitochondria were generally more sensitive to 3NPA and Ca2+-induced mitochondrial permeability transition than mitochondria from the liver or kidneys. So far just one study has demonstrated that 3NPA in mice induces the histopathologic changes in the heart [15]. In this study caudate putamen infarction never occurred without cardiac toxicity, while lungs, liver, kidneys, pancreas, and intestines did not show significant pathology. On the other side, preconditioning with 3NPA has been shown to induce a protective effect against the consequences of brain [16, 17, 18] and heart ischemia [19]. Crespo-Biel et al. (2010) have recently shown that the treatment with 3NPA induced glycogen synthase kinase-3b (GSK-3b) truncation that augmented its kinase activity. GSK-3b is a kinase that inactivates glycogen synthase, the enzyme that catalyzes the attachment of UDP-glucose to the non-reducing end of the already formed glycogen [20]. Glycogen breakdown is mediated by glycogen phosphorylase. Glycogen is an immediate source of glucose for cardiac tissue to maintain its metabolic homeostasis [21]. In the present study we hypothesized that by affecting GSK-3b, 3NPA could affect not only the signal transduction pathways in the brain, but also the glycogen content in the hearth. Our aim was therefore to evaluate histopathological changes and the glycogen content in the heart muscle of experimental rats with 3NPA-induced striatal lesions.
MATERIALS AND METHODS
Animals, treatment, heart section staining and measurement
We used female Wistar rats weighing from 210 g to 272 g at the beginning of the experiment. The animals were handled following the guidelines of the Slovenian Law for Animal Health Protection and the Instructions for Granting Permit for Animal Experimentation for Scientific Purposes. All efforts were made to minimize animal suffering, and only the number of animals necessary to produce reliable scientific data was used. Rats were divided into two groups: the 3NPA group (n = 6): rats were treated every day with 3NPA (RBI Natick, MA, USA) in the dose 30 mg/kg for 8 days subcutaneously (s/c), and the saline group (n = 6): the group of rats that were treated every day with normal saline s/c for 8 days. The dose was chosen according to the data from the literature where the 3NPA had the effect on GSK-3b in the brain [22, 23]. Twenty-four hours after the last injection the animals were euthanized in CO2 anaesthesia. Brains and hearts were rapidly removed. For cytochrome oxidase (COX) histochemistry, the brains were quickly frozen on dry ice and stored at –80°C in a freezer until cryostat sections could be cut. The hearts were fixed in buffered 10% formalin for 24 h and embedded in paraffin. Microtome sections (4 μm) were then cut.
Visualization of striatal and hippocampal lesions by COX histochemistry
Before cutting, the brains were allowed to equilibrate at –20°C in a cryostat chamber. Coronal cryosections (10 μm) were cut through the striatum and hippocampus and thaw mounted onto microscope slides glass slides coated with a 0.01% solution of (poly)L-lysine. The slides were then vacuum-packed and stored in a freezer at –20°C until further processing. COX histochemistry was performed by following the diaminobenzidine procedure [24, 25].
Evaluation of the size of the cardiomyocytes, the volume density of glycogen, the interstitial tissue and the myofibril volume fraction
Histological sections of the heart left ventricles of each animal were stained with Hematoxylin-eosin (HE) and observed with the light microscope at an objective magnification of 40x. The diameter of 50 cardiomyocytes was measured by using Zeiss Axioscope software. For glycogen cytochemistry, the sections were stained with periodic acid-Schiff (PAS) procedure. Sirius red staining was used for the visualization of connective tissue. Stereological analysis [26] was performed using Weibel’s test system. The volume density of glycogen storage (PAS-positive granules in cytoplasm), the interstitial tissue (Sirius red staining) and the myofibril volume fraction (HE method) were estimated as described previously [13, 27, 28]. Volume density of glycogen and of interstitial tissue was estimated by counting points of grid system, which hit glycogen granules/interstitial tissue and reference space (hits on glycogen/interstitial tissue and myocytes) at an objective magnification of 40x. Volume density of interstitial tissue [29] is the quotient between hits falling on interstitial tissue and hits falling on reference space. Myofibril volume fraction was estimated by counting points of grid system, which hit myofibrils in myocytes and reference space (hits on myocytes with and without myofibrils) at an objective magnification 100x. Myofibril volume fraction is the quotient between hits falling on myofibrils and hits falling on reference space. Volume density of glycogen, interstitial tissue and myofibril volume fraction were expressed in mm3/mm3.
TUNEL assay
Detection of apoptosis was performed with the terminal deoxynucleotidyl transferase mediated-deoxyuridine triphosphate nick-end labeling (TUNEL) method (Apo Taq plus Peroxidase Kit ONCOR, Gaithersburg, MD) following the manufacturer’s instructions as described previously [27, 30]. The number of TUNEL positive cells was counted in one section and expressed as the number of cells/mm2 as described previously [31].
Statistical analysis
The average values of the measured parameters of both treatment groups were expressed as the average value ± SD. The statistical significance of the differences of the measured parameters between the 3NPA and the control group were evaluated by the Student’s t-test (p < 0.05). A statistical analysis was performed using the Microsoft Office 2003 Excel software.
RESULTS
The 3NPA-treated rats could be distinguished from the saline-treated controls by the hunched posture, a declined motor function (locomotor activity, movement pattern and vacuous chewing movements), a reduced resistance at handling and reduced body weight. One of the 3NPA rats died before the end of the experiment, so it was excluded from the study. When the body weight at the end of the experiment was compared to the body weight at the beginning of the experiment, there was a significant reduction in the 3NPA group (by -10.76% ± 3.82; paired Student’s t-test, n = 5), while the body weight of control rats did not change significantly (by + 1.50% ± 1.57; paired Student’s t-test, n = 6). COX histochemistry revealed that all 3NPA-treated animals also had striatal and occasional hippocampal lesions. Striatal injury, visualized as the % of the hypointensive COX staining area within dorsal striatum measured on average 30.6% ± 18.5, (n = 5). Neurotoxic effects of 3NPA were, however, not the focus of this study and will be published separately. In the 3NPA group we found the significant accumulation of glycogen granules in cardiomyocytes (0.028mm3/mm3 ± 0.022, n = 5) (Figure 1 A), compared with the control group in which only few glycogen granules in the cytoplasm of some cardiomyocytes were visible (0.002mm3/mm3 ± 0.001, n = 6) (Figure 1 B). The histological analysis of the hearts revealed that the myocardial fibers in the 3NPA group were interrupted by connective tissue (Figure 1 C), compared to the control group (Figure 1 D). In the 3NPA group we found an enlargement of cardiomyocytes (3NPA = 15.9891μm ± 1.649; saline = 13.4561μm ± 0.786, n = 5-6) due to cellular swelling, loss of cell cross-striations with a lower myofibril volume fraction (3NPA = 0.3922mm3/mm3±0.0230, saline = 0.4550mm3/mm3 ± 0.0083, n = 5-6) and an increased volume density of interstitial tissue (3NPA = 0.0531 mm3/mm3 ± 0.0090, saline = 0.0135mm3/mm3 ± 0.0051, n = 5-6). In the interstitial tissue, mostly along blood vessels, mononuclear infiltration was observed. We found TUNEL positive cardiomyocytes (Figure 1 E) in the 3NPA group (2.04 cells/mm2 ± 0.92, n = 5), and almost no TUNEL positive cells (0.27 cells/mm2 ± 0.14, n = 6) in the saline group (Figure 1 F).
FIGURE 1.
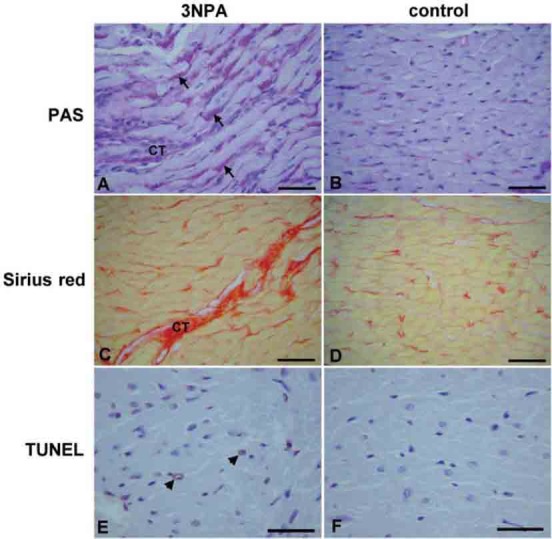
Histopathological changes in hearts of rats treated with 3NPA (A, C, E) as compared with control rats treated with normal saline (B, D, F). In 3NPA-treated animals PAS staining (A, B) showed increased amount of glycogen granules in the cytoplasm of cardiomyocytes (arrows), Sirius red staining (C, D) showed myocardial fibers interrupted with increased amount of connective tissue (CT), while TUNEL staining (E, F) showed sparse TUNEL positive cells (arrow-heads). bar (A, B, C, D) = 50 μm; bar (E, F) = 30 μm.
DISCUSSION
For the first time, this study has shown that 3NPA-induced cardiomyopathy is accompanied by a significant accumulation of glycogen granules in cardiomyocytes compared with the control group. In the 3NPA group we found the diffuse swelling of cardiomyocytes, loss of cell cross-striations with a lower myofibril volume fraction and the fibrosis with mononuclear infiltration, mostly along blood vessels. The injuries observed in our study were similar to the changes described in mice as cell loss, fibrosis, cardiomyocyte swelling and necrosis [15]. In addition we found an increased number of TUNEL positive cardiomyocites in the 3NPA group compared to the controls. It was reported that treatment with 3NPA in vivo and in vitro activates calpain [22, 23, 32], which mediates myofibrillar degeneration [33]. Calpain also plays a role in apoptosis by cleaving the caspases [32, 34]. The stereological evaluation of glycogen content in histological sections of the heart showed a significant accumulation of glycogen granules in the 3NPA group, whereas the hearts of control animals were nearly devoid of glycogen granules. Glycogen functions as the secondary long-term energy storage in animal cells. The synthesis and breakdown of glycogen is controlled by glycogen synthase and glycogen phosphorylase and the regulation of both is characterized by great complexity with many factors [35, 36, 37, 38]. It was shown that treatment with 3NPA activates calpain [22, 23, 32]. It was also shown that treatment with 3NPA induced calpain-mediated GSK-3b activation by truncation that removes its N-terminal inhibitory domain [39]. The activation of GSK-3b inactivates glycogen synthase, and this would consequently inhibit glycogen formation. Contrary to the above rationale, our experiment showed that chronic 3NPA treatment induced glycogen accumulation in cardiomyocytes. In in-vitro assays it was shown that calpain was able to partially digest glycogen phosphorylase and that glycogen phosphorylase was a natural calpain substrate [40]. We speculate that digestion by calpain reduced the activity of glycogen phosphorylase that disabled the degradation of glycogen. It is also known that myocardial glycogen levels are increased with fasting through inhibition of the glycolytic pathway by enhanced oxidation of fatty acids [41,42,43]. In our experiment the food consumption was not measured, but the reduction of the body weight in rats treated with 3NPA indicates a reduced food consumption. Taegtmeyer [43] speculates that the normal cardiac myocyte possesses mechanisms by which the cell senses exogenous fuel deprivation and stores up endogenous fuel to maintain vital processes of cell homeostasis and survival. On the other side, glycogen excess has been suggested to bring about structural and physiological impairments including ionic imbalance, change in pH, and a stimulation of pathways leading to hypertrophic signaling [44].
CONCLUSION
In conclusion, this experiment revealed that, in addition to striatal and hippocampal injuries, 3NPA-induced cardiomyopathy, that is accompanied by a significant accumulation of glycogen granules in cardiomyocytes. However, the mechanism and the pathophysiological significance of this finding for cardiotoxicity of 3NPA at present are not clear.
ACKNOWLEDGMENTS
This work was supported by the program grants from the Ministry of Higher Education, Science and Technology of Slovenia P3-0019.
DECLARATION OF INTEREST
There is no conflict of interest.
REFERENCES
- [1].Liu X, Luo X, Hu W. Studies on the epidemiology and etiology of moldy sugarcane poisoning in China. Biomed Environ Sci. 1992;5(2):161–177. [PubMed] [Google Scholar]
- [2].Ming L. Moldy sugarcane poisoning-a case report with a brief review. J Toxicol Clin Toxicol. 1995;33(4):363–367. doi: 10.3109/15563659509028924. [DOI] [PubMed] [Google Scholar]
- [3].Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitro-propionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 1991;18(4):492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- [4].Alston TA, Mela L, Bright HJ. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci USA. 1977;74(9):3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Coles CJ, Edmondson DE, Singer TP. Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem. 1979;254(12):5161–5167. [PubMed] [Google Scholar]
- [6].Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, et al. Partial inhibition of brain succinate dehydrogenase by 3-nitro-propionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70(2):794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- [7].Borlongan CV, Koutouzis TK, Freeman TB, Hauser RA, Cahill DW, Sanberg PR. Hyperactivity and hypoactivity in a rat model of Huntington's disease: The systemic 3-nitropropionic acid model. Brain Res Brain Res Protoc. 1997;1(3):253–257. doi: 10.1016/s1385-299x(96)00037-2. [DOI] [PubMed] [Google Scholar]
- [8].Ramaswamy S, McBride JL, Kordower JH. Animal models of Huntington's disease. ILAR J. 2007;48(4):356–373. doi: 10.1093/ilar.48.4.356. [DOI] [PubMed] [Google Scholar]
- [9].McCracken E, Dewar D, Hunter AJ. White matter damage following systemic injection of the mitochondrial inhibitor 3-nitropropionic acid in rat. Brain Res. 2001;892(2):329–335. doi: 10.1016/s0006-8993(00)03266-2. [DOI] [PubMed] [Google Scholar]
- [10].Szabó A, Papp A, Nagymajtényi L. Effects of 3-nitropropionic acid in rats: general toxicity and functional neurotoxicity. Arh Hig Rada Toksikol. 2005;56(4):297–302. [PubMed] [Google Scholar]
- [11].Letonja M, Petrovic D. Complete atrioventricular block induced by alcohol abuse. Pacing Clin Electrophysiol. 2003;26(11):2192–2193. doi: 10.1046/j.1460-9592.2003.00344.x. [DOI] [PubMed] [Google Scholar]
- [12].Sinkovec M, Petrovic D, Volk M, Peterlin B. Familial progressive sinoatrial and atrioventricular conduction disease of adult onset with sudden death, dilated cardiomyopathy, and brachydactyly. A new type of heart-hand syndrome? Clin Genet. 2005;68(2):155–160. doi: 10.1111/j.1399-0004.2005.00476.x. [DOI] [PubMed] [Google Scholar]
- [13].Vukojevic K, Petrovic D, Saraga-Babic M. Nestin expression in glial and neuronal progenitors of the developing human spinal ganglia. Gene Expr Patterns. 2010;10(2-3):144–151. doi: 10.1016/j.gep.2009.12.001. [DOI] [PubMed] [Google Scholar]
- [14].Mirandola SR, Melo DR, Saito A, Castilho RF. 3-nitropropionic acid-induced mitochondrial permeability transition: comparative study of mitochondria from different tissues and brain regions. J Neurosci Res. 2010;88(3):630–639. doi: 10.1002/jnr.22239. [DOI] [PubMed] [Google Scholar]
- [15].Gabrielson KL, Hogue BA, Bohr VA, Cardounel AJ, Nakajima W, Kofler J, et al. Mitochondrial toxin 3-nitropropionic acid induces cardiac and neurotoxicity differentially in mice. Am J Pathol. 2001;159(4):1507–1520. doi: 10.1016/S0002-9440(10)62536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, et al. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning”. J Cereb Blood Flow Metab. 1997;17(3):257–264. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- [17].Nakase H, Heimann A, Kempski M, Riepe MW, Dirnagl U. Ischemic tolerance induction by a neurotoxin, 3-nitropropionic acid, in a rat model of global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:S211. [Google Scholar]
- [18].Nakase H, Heimann A, Uranishi R, Riepe MW, Kempski O. Early onset tolerance in rat global cerebral ischemia induced by a mitochondrial inhibitor. Neurosci Lett. 2000;290(2):105, 108. doi: 10.1016/s0304-3940(00)01345-8. [DOI] [PubMed] [Google Scholar]
- [19].Turan NN, Basgut B, Aypar E, Ark M, Iskit AB, Cakici I. Chemical preconditioning effect of 3-nitropropionic acid in anesthetized rat heart. Life Sci. 2008;82(17-18):928–933. doi: 10.1016/j.lfs.2008.02.011. [DOI] [PubMed] [Google Scholar]
- [20].Nielsen JN, Derave W, Kristiansen S, Ralston E, Ploug T, Richter EA. Glycogen synthase localization and activity in rat skeletal muscle is strongly dependent on glycogen content. J Physiol. 2001;531:757–769. doi: 10.1111/j.1469-7793.2001.0757h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Arkinstall MJ, Bruce CR, Clark SA, Rickards CA, Burke LM, Hawley JA. Regulation of fuel metabolism by preexercise muscle glycogen content and exercise intensity. J Appl Physiol. 2004;97(6):2275–2283. doi: 10.1152/japplphysiol.00421.2004. [DOI] [PubMed] [Google Scholar]
- [22].Crespo-Biel N, Camins A, Pelegrí C, Vilaplana J, Pallàs M, Canudas AM. 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci Lett. 2007;421(1):77–81. doi: 10.1016/j.neulet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- [23].Crespo-Biel N, Camins A, Gutiérrez-Cuesta J, Melchiorri D, Nicoletti F, Pallàs M, et al. Regulation of GSK-3beta by calpain in the 3-nitropropionic acid model. Hippocampus. 2010;20(8):962–970. doi: 10.1002/hipo.20691. [DOI] [PubMed] [Google Scholar]
- [24].Wong-Riley M. Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res. 1979;171(1):11–28. doi: 10.1016/0006-8993(79)90728-5. [DOI] [PubMed] [Google Scholar]
- [25].Milatovic D, Zivin M, Gupta RC, Dettbarn WD. Alterations in cytochrome c oxidase activity and energy metabolites in response to kainic acid-induced status epilepticus. Brain Res. 2001;912(1):67–78. doi: 10.1016/s0006-8993(01)02657-9. [DOI] [PubMed] [Google Scholar]
- [26].Weibel ER. Practical methods for biological Morphometry. London: Academic press; 1979. Stereological Methods. [Google Scholar]
- [27].Zorc M, Vraspir-Porenta O, Zorc-Pleskovic R, Radovanovic N, Petrovic D. Apoptosis of myocytes and proliferation markers as prognostic factors in end-stage dilated cardiomyopathy. Cardiovasc Pathol. 2003;12(1):36–39. doi: 10.1016/s1054-8807(02)00134-5. [DOI] [PubMed] [Google Scholar]
- [28].Petrovic D. Cytopathological basis of heart failure--cardiomyocyte apoptosis, interstitial fibrosis and inflammatory cell response. Folia Biol (Praha) 2004;50(2):58–62. [PubMed] [Google Scholar]
- [29].Hasanovic A, Mornjakovic Z, Pikula B, Dilberovic F. Morphologic findings of the ischemic myocardium. Bosn J Basic Med Sci. 2006;6(1):82–85. doi: 10.17305/bjbms.2006.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vukojevic K, Carev D, Sapunar D, Petrovic D, Saraga-Babic M. Developmental patterns of caspase-3, bax and bcl-2 proteins expression in the human spinal ganglia. J Mol Histol. 2008;39(3):339–349. doi: 10.1007/s10735-008-9171-4. [DOI] [PubMed] [Google Scholar]
- [31].Suput D, Zorc-Pleskovic R, Petrovic D, Milutinovic A. Cardiotoxic injury caused by chronic administration of microcystin-YR. Folia Biol (Praha) 2010;56(1):14–18. [PubMed] [Google Scholar]
- [32].Bizat N, Hermel JM, Boyer F, Jacquard C, Cre’minon C, Ouary S, et al. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: Implications for Huntington's disease. J Neurosci. 2003;23(12):5020–5030. doi: 10.1523/JNEUROSCI.23-12-05020.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Festuccia WT, Laplante M, Brulé S, Houde VP, Achouba A, Lachance D, et al. Rosiglitazone-induced heart remodelling is associated with enhanced turnover of myofibrillar protein and mTOR activation. J Mol Cell Cardiol. 2009;47(1):85–95. doi: 10.1016/j.yjmcc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- [34].Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275(7):5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- [35].Johnson LN. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. FASEB J. 1992;6(6):2274–2282. doi: 10.1096/fasebj.6.6.1544539. [DOI] [PubMed] [Google Scholar]
- [36].Lees SJ, Franks PD, Spangenburg EE, Williams JH. Glycogen and glycogen phosphorylase associated with sarcoplasmic reticulum: effects of fatiguing activity. J Appl Physiol. 2001;91(4):1638–1644. doi: 10.1152/jappl.2001.91.4.1638. [DOI] [PubMed] [Google Scholar]
- [37].Nielsen JN, Wojtaszewski JFP. Regulation of glycogen synthase activity and phosphorylation by exercise. Proc Nutr Soc. 2004;63(2):233–237. doi: 10.1079/PNS2004348. [DOI] [PubMed] [Google Scholar]
- [38].Greenberg CC, Jurczak MJ, Danos AM, Brady MJ. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab. 2006;291(1):E1–8. doi: 10.1152/ajpendo.00652.2005. [DOI] [PubMed] [Google Scholar]
- [39].Goni-Oliver P, Lucas JJ, Avila J, Hernández F. N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J Biol Chem. 2007;282(31):22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- [40].Purintrapiban J, Wang M, Forsberg NE. Identification of glycogen phosphorylase and creatine kinase as calpain substrates in skeletal muscle. Int J Biochem Cell Biol. 2001;33(5):531–540. doi: 10.1016/s1357-2725(01)00012-7. [DOI] [PubMed] [Google Scholar]
- [41].Evans G. The glycogen content of the rat heart. J Physiol. 1934;82(4):468–480. doi: 10.1113/jphysiol.1934.sp003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schneider CA, Taegtmeyer H. Fasting in vivo delays myocardial cell damage after brief periods of ischemia in the isolated working rat heart. Circ Res. 1991;68(4):1045–1050. doi: 10.1161/01.res.68.4.1045. [DOI] [PubMed] [Google Scholar]
- [43].Taegtmeyer H. Glycogen in the heart - An expanded view. J Mol Cell Cardiol. 2004;37(1):7–10. doi: 10.1016/j.yjmcc.2004.05.001. [DOI] [PubMed] [Google Scholar]
- [44].Puthanveetil P, Wang F, Kewalramani G, Kim MS, Hosseini-Beheshti E, Ng N, et al. Cardiac glycogen accumulation after dexamethasone is regulated by AMPK. Am J Physiol Heart Circ Physiol. 2008;295(4):H1753–H1762. doi: 10.1152/ajpheart.518.2008. [DOI] [PubMed] [Google Scholar]