

NORE1A is a Ras senescence effector that modulates HIPK2-dependent posttranslational modifications of p53.
Abstract
The Ras oncoprotein is a key driver of cancer. However, Ras also provokes senescence, which serves as a major barrier to Ras-driven transformation. Ras senescence pathways remain poorly characterized. NORE1A is a novel Ras effector that serves as a tumor suppressor. It is frequently inactivated in tumors. We show that NORE1A is a powerful Ras senescence effector and that down-regulation of NORE1A suppresses senescence induction by Ras and enhances Ras transformation. We show that Ras induces the formation of a complex between NORE1A and the kinase HIPK2, enhancing HIPK2 association with p53. HIPK2 is a tumor suppressor that can induce either proapoptotic or prosenescent posttranslational modifications of p53. NORE1A acts to suppress its proapoptotic phosphorylation of p53 but enhance its prosenescent acetylation of p53. Thus, we identify a major new Ras signaling pathway that links Ras to the control of specific protein acetylation and show how NORE1A allows Ras to qualitatively modify p53 function to promote senescence.
Introduction
Activation of Ras oncogenes is a driving event in many human cancers (Malumbres and Barbacid, 2003; Campbell and Der, 2004; Schubbert et al., 2007). Activated forms of Ras stimulate multiple mitogenic signaling pathways, including the Raf–MAPK and PI3 kinase pathways to promote transformation (Pylayeva-Gupta et al., 2011). However, activated forms of Ras also promote oncogene induced senescence (Serrano et al., 1997; Cox and Der, 2003; Lowe et al., 2004). The detection of Ras-induced senescence in multiple cell culture systems (Serrano et al., 1997; Ferbeyre et al., 2002), in vivo mouse models (Collado et al., 2005; Morton et al., 2010; Kennedy et al., 2011), human rasopathies (Courtois-Cox et al., 2006), and premalignant activated Ras-containing human pancreatic tumors (Caldwell et al., 2012) confirms that the process is physiological (Dimauro and David, 2010; Kuilman et al., 2010). However, the mechanisms by which Ras drives senescence and how these mechanisms are subverted during the development of malignancy (Chen et al., 2005; Collado et al., 2005; Kuilman et al., 2010) remains poorly understood (Bianchi-Smiraglia and Nikiforov, 2012). One factor that is clearly important is the activation of the p53 tumor suppressor by Ras (Serrano et al., 1997).
p53 is a key mediator of the cellular response to a broad variety of stress stimuli (Levine, 1997). It is thought to play a major role in restricting cancer development, as its function is impaired in almost half of human tumors (Velculescu and El-Deiry, 1996). p53 primarily serves as a transcription factor that can modulate multiple genes involved in apoptosis, cell cycle control, and senescence (Vogelstein et al., 2000; Beckerman and Prives, 2010). The regulation of p53 is extremely intricate. p53 protein levels are under control of the mdm2 ubiquitin ligase (Ringshausen et al., 2006). However, posttranslational modifications of p53 such as phosphorylation and acetylation on multiple sites can have a powerful effect on the qualitative output of p53 (Carter and Vousden, 2009; Gu and Zhu, 2012), biasing it toward apoptosis or senescence. The mechanisms underlying the regulation of p53 by Ras remain unclear, although Ras has been shown to promote p53 acetylation during senescence induction (Pearson et al., 2000).
NORE1A (RASSF5) is a member of the RASSF family of tumor suppressors (van der Weyden and Adams, 2007). NORE1A binds directly to Ras oncoproteins with all the characteristics of an effector that promotes apoptosis (Khokhlatchev et al., 2002; Vos et al., 2003) or cell cycle arrest (Aoyama et al., 2004; Calvisi et al., 2009). NORE1A lacks enzymatic activity and is thought to act as a scaffolding molecule. Like other RASSF family proteins, it can bind the proapoptotic kinases MST1 and MST2 and feeds into the Hippo signaling pathway (Khokhlatchev et al., 2002). However, NORE1A tumor suppressor activity does not require the interaction with MST kinases (Aoyama et al., 2004), suggesting other effectors are the key to its function. NORE1A is frequently down-regulated during tumor development (Donninger et al., 2007), and its dysregulation is implicated in a rare familial cancer syndrome (Chen et al., 2003). In primary tumors, inactivation of NORE1A expression often correlates with up-regulation of Ras activity (Calvisi et al., 2008) and exogenous expression of NORE1A suppresses the tumorigenic phenotype (Vos et al., 2003). Moreover, NORE1A−/− mouse embryo fibroblasts (MEFs) are susceptible to one-step transformation by activated Ras, which wild-type MEFs are not (Park et al., 2010). Thus, NORE1A serves as a Ras effector/tumor suppressor that likely plays a key role in restraining the transforming effects of mutant Ras.
Our recent studies have suggested that although NORE1A can induce apoptosis, a more physiological role may be in the regulation of the cell cycle by inducing a p53-dependent activation of the CDK inhibitor p21CIP1 (Calvisi et al., 2009). Moreover, in primary human tumors, NORE1A down-regulation and p53 inactivation are usually mutually exclusive, suggesting they lie in the same pathway (Calvisi et al., 2009). As Ras senescence pathways are known to operate, in part, via p53 (Serrano et al., 1997), we examined the role of NORE1A in Ras-mediated senescence and p53 activation.
We found that NORE1A is a potent inducer of p53-dependent senescence in multiple cell systems. Moreover, knockdown of NORE1A impaired Ras-induced senescence and enhanced the transforming activity of activated Ras. However, deletion mutagenesis of NORE1A showed that these biological effects were independent of canonical Hippo pathway signaling.
Further studies revealed that NORE1A forms an endogenous, Ras-regulated complex with the kinase HIPK2 and that this interaction is essential for full Ras/NORE1A-induced senescence. HIPK2 can positively regulate total p53 levels by down-regulating the p53-negative regulator mdm2 (Di Stefano et al., 2005a). It can also modulate the apoptotic or the senescence-promoting activity of p53 by promoting discrete posttranslational modifications (Puca et al., 2010). It stimulates the proapoptotic activity of p53 by direct phosphorylation of p53 residue S46 (D’Orazi et al., 2002; Di Stefano et al., 2004). However, HIPK2 can also indirectly promote the acetylation of p53 by recruiting the acetyl transferases PCAF and CBP/p300 into a complex (Hofmann et al., 2002; Di Stefano et al., 2005b). Acetylation of p53 can enhance its affinity for the promoters of prosenescence genes such as p21CIP1 and is associated with senescence (Pearson et al., 2000; Di Stefano et al., 2005b; Brooks and Gu, 2011). Our studies show that NORE1A inhibits the proapoptotic phosphorylation of p53 on S46 and inhibits p53 proapoptotic signaling. However, NORE1A stimulates the acetylation of p53 and activates prosenescent signaling in a HIPK2-dependent manner. This appears to be due to a Ras-regulated scaffolding of p53 and HIPK2 by NORE1A.
Thus, we identify a novel NORE1A–HIPK2–p53 pathway that allows Ras to qualitatively control the posttranslational modification code of p53 to favor senescence over apoptosis. This pathway explains mechanistically the previous observations that Ras can promote p53 acetylation (Pearson et al., 2000), p21CIP1 activation (Di Stefano et al., 2005b), and senescence (Serrano et al., 1997). Inactivation of this pathway is frequent in primary tumors and its loss may play a key role in the ability of Ras-driven tumor cells to breach the senescence barrier and progress to malignancy.
Results
NORE1A is a Ras senescence effector
NORE1A is a Ras effector/tumor suppressor with a poorly characterized mode of action. We have recently shown that NORE1A activates p21CIP1 via p53 (Calvisi et al., 2009). As p53 activation of p21CIP1 is associated with the induction of senescence, we hypothesized that NORE1A might be part of the elusive senescence signaling machinery of Ras. To determine if this is the case, we examined the ability of NORE1A to induce senescence. A549 cells are a human lung tumor cell line that contains an activated Ras gene and is wild-type for p53 but does not express NORE1A (Aoyama et al., 2004). A549 cells were transiently transfected with a NORE1A expression vector and senescence assayed by β-galactosidase activity. NORE1A was found to be a potent inducer of senescence in A549 cells (Fig. 1 a). To confirm that NORE1A was acting via p53, we used siRNA knockdown of p53 in the cells to show that the senescence inducing activity of NORE1A is p53 dependent (Fig. 1 a). Additional studies showed NORE1A also induces senescence in primary MEFs (Fig. 1 b). This ability was impaired in a NORE1A mutant that is defective for binding to Ras (NORE1A(RA*); Aoyama et al., 2004). Moreover, knockdown of NORE1A using two previously validated short hairpin RNAs (shRNAs) against mouse NORE1A (Calvisi et al., 2009) inhibited the ability of Ras to induce senescence in MEFs (Fig. 1 b). Thus, NORE1A appears to serve as a key component of a Ras–p53 senescence pathway.
Figure 1.

NORE1A is a Ras senescence effector. (A) NORE1A induces p53-dependent senescence in human cells. A549 human lung tumor cells (p53 wild type) were transfected with 1ug of pcDNA-HA-NORE1A and 100 nM of scrambled siRNA or two different p53 siRNAs. Cells were incubated for 72 h before assaying for β-galactosidase activity. p53 and NORE1A levels are shown in the inset. (B) NORE1A is a Ras senescence effector. Primary MEFs were transfected with 1 µg of wild type NORE1A, a Ras binding defective (RA*) mutant of NORE1A, 100 ng of activated H-Ras with vector control shRNA or 100 ng of activated H-Ras with two different shRNAs against murine NORE1A. After 72 h, cells were assayed for β-galactosidase activity. *, P < 0.05 compared with vector transfected cells. (C) NORE1A is essential for senescence in human bronchial epithelial cells. HBEC-3KT cells are a nonvirally immortalized human bronchial epithelial cell line. Stable transduction of shRNA against NORE1A into the cells promoted a dramatic reduction of the basal levels of senescence measured by β-galactosidase activity. (left) Quantification of two separate assays. (right) A representative image of the cells and the relative knockdown of NORE1A protein. Bar, 1,000 µM. (D) The HBEC-3KT+/− NORE cells were scored for senescence-associated heterochromatin foci (SAHF) by analyzing fifty randomly selected cells. The process was repeated twice. *, P < 0.05 compared with cells stably expressing the scrambled shRNA. (E) Western blot analysis of Dec1 expression from the HBEC-3KT cells stably knocked down for NORE1A. The density of the bands was quantitated using ImageJ software and relative Dec1 expression was calculated after normalizing to β-actin expression.
To confirm the importance of NORE1A to Ras-induced senescence and to examine the role of the pathway in suppressing Ras-driven transformation, we used HBEC-3KT cells. These cells are a human lung epithelial cell line that has been immortalized in the absence of viral oncoproteins by telomerase and cdk4 (Ramirez et al., 2004). The cells retain NORE1A expression and exhibit a high background of senescence, as measured by β-galactosidase activity, under normal growth conditions (Fig. 1 c). However, after stable transduction of NORE1A shRNA constructs and knockdown of NORE1A expression, the senescence is repressed (Fig. 1 c). Fig. 1 c (left) shows quantification of senescence in the cell system. Fig. 1 c (right) shows a representative image of the cells and the degree of NORE1A protein knockdown. To substantiate that the high level of β-galactosidase staining observed in these cells was indeed senescence related, we measured two additional well-established markers of senescence, senescence-associated heterochromatin foci (SAHF) and Dec1 (Collado and Serrano, 2010). We observed that loss of NORE1A reduced both the levels of SAHF (Fig. 1 d) and Dec1 expression (Fig. 1 e), confirming that NORE1A is an important effector of senescence. Trypan blue staining showed no difference in the levels of cell viability (unpublished data).
Suppression of NORE1A enhances Ras-mediated transformation
To determine the effects of NORE1A knockdown on Ras-mediated senescence and transformation in the HBEC-3KT cells, we stably transfected a doxycycline-inducible expression vector for H-Ras12v (Ellis et al., 2002) into a matched pair of HBEC-3KT cells containing either the scrambled shRNA control or two different NORE1A shRNAs. Doxycycline was added to the cells and, after 72 h, the cells were assayed for senescence. The control cells showed a 30% increase over the high background, whereas the NORE1A knockdown cells showed only an ∼5 and ∼10% increase, respectively (Fig. 2 a). The levels of Ras protein induced in all three cell systems were approximately the same (Fig. 2 b). We then plated the cells in soft agar in the presence or absence of doxycycline and assayed for the growth of colonies after 3 wk. Colony formation was enhanced in the NORE1A knockdown cells (Fig. 2 c).
Figure 2.
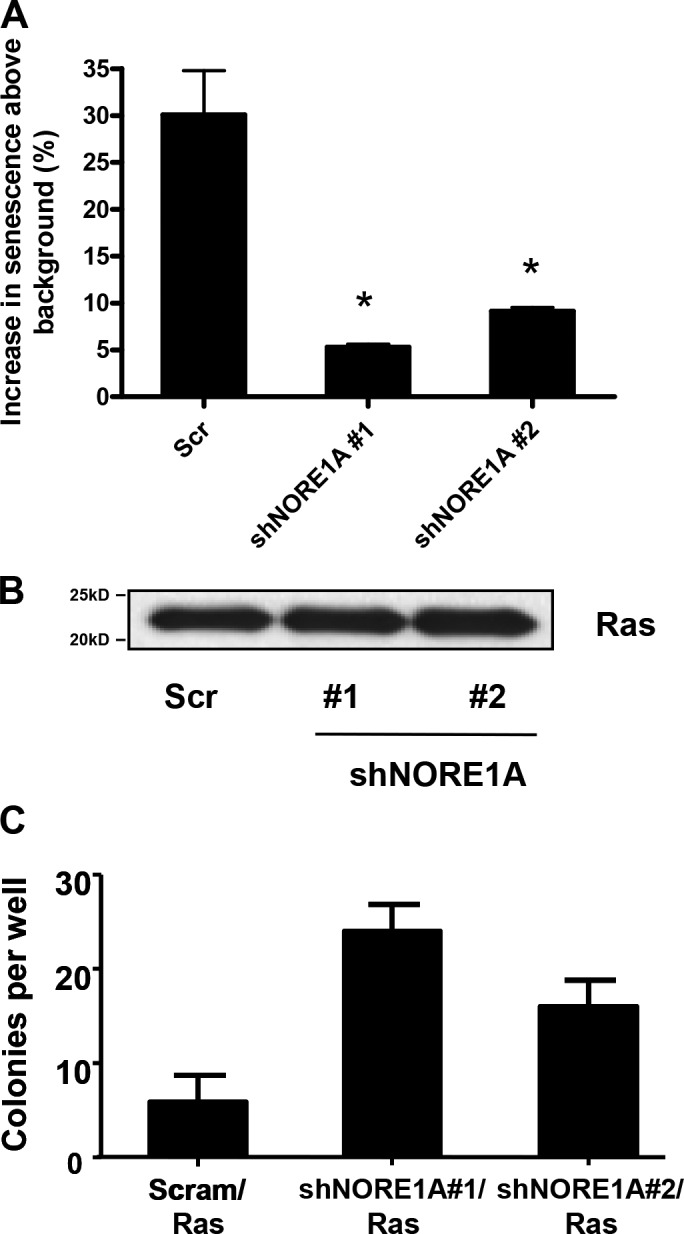
NORE1A suppression impairs Ras-mediated senescence induction and enhances Ras-mediated transformation. (A) An inducible form of activated H-Ras was transfected into the scrambled shRNA or the two NORE1A shRNA transduced HBEC-3KT stable cell lines described in Fig. 1. Ras was induced with doxycycline and, after 72 h, β-galactosidase activity was quantified. The percent increase over the uninduced control cells was measured. Data represent the mean ± SD of triplicate experiments. *, P < 0.05 compared with scrambled control. (B) Duplicate dishes were induced, and lysed, and equal amounts of protein were loaded on a gel and immunoblotted for the expression of H-Ras to confirm equivalent expression. (B) The Ras-inducible HBEC-3KT cell lines were then plated in soft agar in the presence of doxycycline. Frequency of colony formation was quantified. Experiments were done in duplicate and the error bars show SD.
NORE1A senescence signaling does not require the Hippo pathway
NORE1A can modulate apoptosis by binding to the MST kinases that regulate the Hippo pathway using its C-terminal SARAH motif (Khokhlatchev et al., 2002). Yet it can modulate cell cycle arrest in an MST kinase-independent manner (Aoyama et al., 2004). To determine if the senescence-activating effects of NORE1A were dependent on the Hippo signaling pathway, we generated a deletion mutant of NORE1A (NORE1A-delSARAH) that lacks the region containing the SARAH motif essential for its interaction with MST kinases (Khokhlatchev et al., 2002). We then repeated the senescence assays in A549 cells including the mutant NORE1A. The deletion mutant retained almost WT levels of senescence induction in A549 cells (Fig. 3).
Figure 3.
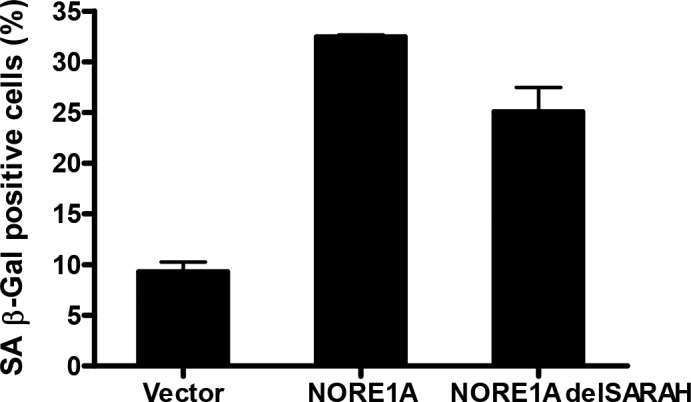
NORE1A-mediated senescence is independent of the canonical Hippo pathway. A549 cells were transfected with WT or a C-terminal deletion mutant of NORE1A lacking the SARAH motif connecting NORE1A to the Hippo pathway (delSARAH). Senescence was scored by β-galactosidase assay. In each case, data are represented as mean ± SD of triplicate experiments. P < 0.05 compared with control.
NORE1A forms an endogenous, Ras-regulated complex with the p53 regulator HIPK2
The aforementioned experiments implicate NORE1A as a key Ras senescence effector that acts via p53 but does not use the canonical Hippo pathway. While attempting to determine the mechanism behind the effect, we examined the subcellular localization of endogenous NORE1A and found that it is primarily localized to the nucleus (Fig. 4 a). Transient transfections of NORE1A tagged with GFP confirmed a mainly nuclear localization but also revealed that some cells demonstrated NORE1A in nuclear speckles (Fig. 4 b, top). Systematic analysis of a series of fluorescently tagged known nuclear speckle proteins showed that NORE1A specifically co-localized with the kinase HIPK2, a known regulator of p53 (Puca et al., 2010; Fig. 4 b, bottom). Further analysis confirmed that endogenous NORE1A could be co-immunoprecipitated with endogenous HIPK2 from HuH6 and HepG2 liver cancer cell lines (Fig. 4 c).
Figure 4.

NORE1A forms a Ras regulated complex with HIPK2. (A) NORE1A is primarily nuclear. NCI-H1792 cells (that retain NORE1A expression) were fractionated into nuclear and cytoplasmic fractions to determine the sub-cellular localization of endogenous NORE1A. The distribution of TFIIH and Actin served as controls for effective fractionation. (B) Exogenously expressed NORE1A forms nuclear speckles with HIPK2. (top) COS-7 cells were transfected with GFP-NORE1A which localized to the nucleus and in some cells to nuclear speckles. (bottom) RFP-NORE1A co-localized with GFP-HIPK2 speckles in the nuclei of COS-7 cells. A representative nucleus is shown. Bar, 40 µM. (C) NORE1A and HIPK2 can be detected in endogenous complex. HepG2 cells and HuH6 cells were immunoprecipitated (IP) for NORE1A and immunoblotted (IB) for HIPK2. (D) Activated Ras enhances the association of NORE1A and HIPK2. HEK-293T cells were cotransfected with expression constructs for HIPK2, NORE1A and activated H-Ras or activated K-Ras for 24 h, lysed and equal amounts of protein immunoprecipitated with anti-GFP. The immunoprecipitates were analyzed by Western blotting with anti-HA and anti-GFP antibodies. (E) Activated Ras enhances the interaction between endogenous NORE1A and HIPK2. HBEC-3KT cells expressing inducible activated H-Ras (Fig. 2) were left untreated (−) or treated (+) with 2 µg/ml doxycycline for 72 h to induce Ras expression and lysed, and equal amounts of protein were immunoprecipitated with an anti-NORE1A antibody. The immunoprecipitates were analyzed by Western blotting with anti-NORE1A, anti-HIPK2, and anti–H-Ras antibodies. (left) Immunoprecipitation; (right) inputs.
To determine the effects of Ras on the interaction of NORE1A and HIPK2, we performed co-transfection experiments in HEK-293T cells and measured the degree of complex formation. In the presence of activated Ras, the degree of complex formation between NORE1A and HIPK2 was dramatically increased (Fig. 4 d). To confirm the effects of Ras on the endogenous interaction between NORE1A and HIPK2, we used the HBEC-3KT cells stably transfected with an inducible Ras construct (Fig. 2). We found that after doxycycline treatment to induce Ras expression, there was a strong increase in the complex formation between endogenous NORE1A and endogenous HIPK2 (Fig. 4 e).
Ras/NORE1A act to stabilize HIPK2
During these experiments, we often observed an apparent increase in the levels of HIPK2 in the lysates of cells transfected with Ras and NORE1A (Fig. 4 d). This was particularly apparent when we examined the effects of activated Ras induction on endogenous HIPK2 levels (Fig. 4 e). HIPK2 is known to be an unstable protein (Calzado et al., 2009). By using cyclohexamide treatment to block translation after transient transfection of HEK-293 cells, we were able to analyze the effects of Ras and NORE1A on HIPK2 protein stability. We found that Ras acts to stabilize NORE1A and that both NORE1A and Ras stabilize HIPK2. The most effective stabilization was a combination of Ras and NORE1A. A representative experiment is shown in Fig. 5 a. Quantification of three experiments is shown below as relative percentage decay (Fig. 5 b).
Figure 5.

Ras/NORE1A stabilizes HIPK2 protein. (A) HEK-293 cells were transfected in triplicate with HIPK2, NORE1A, and activated H-Ras expression constructs. The cells were treated with cyclohexamide and one dish lysed at each time point. Levels of protein were determined by immunoblot (IB). A representative blot is shown. Relative decay of HIPK2 levels over 4 h was determined by quantification of the band intensity from multiple experiments (n = 3) and is shown below in B. Error bars show SD.
Interaction with HIPK2 is required for NORE1A-mediated senescence
HIPK2 is a tumor suppressor that binds p53 and modulates its activity both directly and indirectly (Puca et al., 2010). Thus, HIPK2 is a good candidate for a downstream component of NORE1A senescence signaling. To determine the importance of HIPK2 for NORE1A-mediated senescence, we generated stable clones of A549 cells transfected with shRNAs against HIPK2. Compared with the scrambled shRNA control, the HIPK2 knockdown cells were resistant to NORE1A induced senescence (Fig. 6 a). Suppression of HIPK2 similarly reduced baseline senescence in HBEC-3KT cells (Fig. 6 b). To further confirm the biological importance of the interaction between NORE1A and HIPK2, we used deletion mutagenesis followed by point mutagenesis to identify the minimal domain required for the interaction of NORE1A with HIPK2. Ultimately, Arginines 92–94 of NORE1A were mutated to Alanines. The resultant NORE1AR92-94A mutant (NORE1AR3A) was defective for the interaction with HIPK2 and defective for the induction of senescence in A549 cells (Fig. 6 c) and showed a reduced ability to stabilize HIPK2 (Fig. S1 a). However, it retained WT binding to MST1 and retained WT apoptosis capacity (Fig. 6 d).
Figure 6.

The interaction with HIPK2 is essential for NORE1A-mediated senescence. (A) HIPK2 knockdown blocks NORE1A induced senescence. A549 cells were stably transfected with shRNA constructs against HIPK2 or a scrambled control. The cells were then transfected with NORE1A and the induction of senescence measured by β-galactosidase assay. HIPK2 and NORE1A expression was assessed by Western blot (inset). (B) Loss of HIPK2 reduces baseline senescence in HBEC-3KT cells. HBEC-3KT cells were transfected with the same HIPK2 shRNA constructs and 72 h later senescence measured by β-galactosidase assay. (C) A HIPK2 binding mutant of NORE1A is defective for senescence induction. A mutant form of NORE1A designated NORE1AR3A was generated by mutating the Arginines at residues 92–94 to Alanines. The mutant was co-transfected with HIPK2 in HEK-293T cells and its ability to co-immunoprecipitate with HIPK2 compared with the WT protein (left). The NORE1AR3A mutant was then assayed for the relative ability to induce senescence in A549 cells (right). (D) NORE1AR3A retains the ability to bind MST1 and induce apoptosis. The NORE1AR3A mutant was then examined to determine if it retained WT MST1 binding activity (left) and apoptosis inducing capacity, using a pCaspase-3-sensor fluorescent protein indicator assay (right). Indicator cells were COS-7 and the assay was scored 48 h after transfection. Data represent the mean ± SD of triplicate experiments. P < 0.05 compared with vector transfected cells.
NORE1A acts as a Ras-regulated scaffold for p53 and HIPK2
NORE1A has no apparent enzymatic activity and is thought to act as a scaffolding molecule to assemble and regulate signaling complexes. If so, we would anticipate that it would enhance the association of p53 and HIPK2. To determine if this is the case, we first determined if we could detect a complex between NORE1A and endogenous p53 in HEK-293 cells. We found that NORE1A can be co-precipitated with p53 and that the interaction of NORE1A and p53 is enhanced in the presence of activated Ras (Fig. 7 a). We then examined the effects of NORE1A on the complex formation between HIPK2 and p53. Fig. 7 b shows that the association of HIPK2 with endogenous p53 is enhanced by the presence of NORE1A and this effect is strongly enhanced by the additional presence of activated Ras.
Figure 7.

NORE1A scaffolds p53 and HIPK2. (A) NORE1A forms a Ras-regulated complex with endogenous p53. HEK-293 cells were transfected with NORE1A in the presence or absence of activated Ras. Cells were lysed, immunoprecipitated (IP) for NORE1A, and immunoblotted (IB) for p53. (B) NORE1A promotes the formation of a p53–HIPK2 complex. HEK-293 cells were transfected with HIPK2, NORE1A, and activated Ras. Cells were lysed and immunoprecipitated (IP) for GFP-HIPK2. The IP was then examined for the presence of endogenous p53.
NORE1A promotes prosenescence but inhibits proapoptotic posttranslational modifications of p53 via HIPK2
HIPK2 activates p53 via several different mechanisms. It binds and promotes the degradation of the p53-negative regulator mdm2 stabilizing p53 (Di Stefano et al., 2005a). It also directly phosphorylates p53 on serine 46, and this activates the proapoptotic action of p53 by enhancing its affinity for the promoters of proapoptotic genes such as Bax (Hofmann et al., 2002). However, HIPK2 also has indirect effects on p53, as it can recruit the acetyltransferases PCAF and CBP/p300 to acetylate p53 on specific lysine residues (Puca et al., 2010). This acetylation can switch the activity of p53 from apoptosis to senescence by enhancing its affinity for specific promoters such as p21CIP1 (Di Stefano et al., 2005b). Ras has previously been shown to promote the acetylation of p53 on residue K382 by an unknown mechanism (Pearson et al., 2000).
As we had observed induction of senescence by NORE1A, we examined the effects of NORE1A on p53 acetylation at residue K382 using a specific antibody. In transient transfections of A549 cells, we observed that NORE1A, but not the HIPK2 binding defective mutant of NORE1A, induced p53 acetylation at residue 382 (Fig. 8 a). We then examined the relationship between NORE1A and acetylated p53 in a panel of WT p53 primary human tumor samples. We found that the expression levels of NORE1A closely correlated with the acetylation of p53 (correlation coefficient [r] = 0.811; r2 = 0.6578; P < 0.0001; Fig. 8 b).
Figure 8.

NORE1A mediates specific posttranslational modifications of p53 via HIPK2. (A) NORE1A promotes acetylation of p53 on residue K382. A549 cells were transfected with NORE1A WT or the HIPK2-defective NORE1AR3A mutant. The cells were lysed and immunoblotted for p53 acetylated at K382 using a K382-specific antibody. (B) NORE1A expression correlates with acetylated p53 expression in primary human tumor samples. The relationship between the protein levels (as assessed by Western blotting and quantification of the chemiluminescence signal) of NORE1A and those of acetylated p53 (K382) was measured in a collection of human HCC (n = 40) harboring WT p53. Axes are shown as relative luminescent units from samples with equal total protein loading. A significant, direct correlation was found between NORE1A and acetylated p53 levels. GraphPad Prism 5.01 was used to evaluate statistical significance by Tukey–Kramer and linear regression analyses (correlation coefficient [r] = 0.811, r2 = 0.6578, P < 0.0001). (C) Ras requires NORE1A to modulate p53 acetylation. HEK-293 cells were transiently transfected with the NORE1A shRNAs and activated H-Ras. Cells were lysed and assayed for endogenous p53 K382 acetylation after 24 h. Immunoreactive bands were quantified by densitometry and the results of 3 experiments plotted as a bar graph with data normalized to the scrambled control, *, P < 0.05 compared with cells transfected with the scrambled control. (D) NORE1A requires HIPK2 to modulate p53 acetylation. HEK-293 cells were transfected with NORE1A in the presence or absence of the HIPK2 shRNAs. After 24 h cells were lysed and assayed for endogenous p53 K382 acetylation. Immunoreactive bands were quantified by densitometry and the results of three experiments plotted as a bar graph, with data normalized to control cells transfected with the scrambled shRNA. *, P < 0.05 compared with cell transfected with the scrambled shRNA. (E) NORE1A suppresses S46 phosphorylation of p53. HEK-293 cells were transfected with expression constructs for NORE1A or the NORE1AR3A mutant. The cells were lysed and immunoblotted for endogenous p53 phosphorylated at S46 (left) or acetylated at K382 (right). (F) Ras suppresses p53 S46 phosphorylation. HEK-293 cells were transfected with expression constructs for NORE1A, activated H-Ras, or both. The level of S46 phosphorylation of endogenous p53 was then assayed by Western blot using a phospho S46–specific antibody. The Western blot shown is representative of three experiments. Total levels of endogenous p53 and transfected NORE1A/Ras are shown in the lower panels.
Further studies using transient transfections in HEK-293 with the validated NORE1A shRNAs showed that Ras requires NORE1A to acetylate p53 (Fig. 8 c) and that NORE1A requires HIPK2 to acetylate p53 (Fig. 8 d). Thus, NORE1A/HIPK2 appears to be the link between Ras and p53 acetylation.
We then examined the effects of NORE1A on the HIPK2-dependent proapoptotic posttranslational modification of p53. Using transient transfections in HEK-293 cells and a phospho S46-specific antibody, we found that NORE1A does not activate phosphorylation of p53 on S46 (Fig. 8 e, left). Indeed, it appears to suppress it. This contrasts with the effects on p53 K382 acetylation which is up-regulated by WT NORE1A in the HEK-293 cells (Fig. 8 e, right). Thus, in the same cells, NORE1A is modulating the posttranslational modifications of p53, activating it toward a prosenescent function. Interestingly, the NORE1A point mutant R3A that is defective for HIPK2 binding, and does not enhance p53 K382 acetylation, also failed to activate p53 S46 phosphorylation (Fig. 8 e). Co-transfection experiments with activated Ras also showed that it fails to stimulate phosphorylation of p53 S46. Indeed, S46 phosphorylation is inhibited by Ras/NORE1A (Fig. 8 f).
Upon phosphorylation by HIPK2, p53 preferentially binds and activates the promoters of proapoptotic genes (Hofmann et al., 2002). As we observed the inhibition of p53 phosphorylation by NORE1A we examined the effects of NORE1A on a Bax luciferase reporter. Fig. 9 a shows that NORE1A suppresses the activation of Bax. Because NORE1A induces senescence, we also examined its effects on p21, a p53-responsive gene that is associated with senescence (Pearson et al., 2000; Di Stefano et al., 2005b; Brooks and Gu, 2011) and found that NORE1A strongly increases p21 expression (Fig. 9 b). Thus, NORE1A appears to repress p53-responsive genes associated with apoptosis and enhance expression of p53-responsive genes associated with senescence, suggesting that NORE1A is a key part of the signaling machinery that determines if p53 drives apoptosis or senescence.
Figure 9.
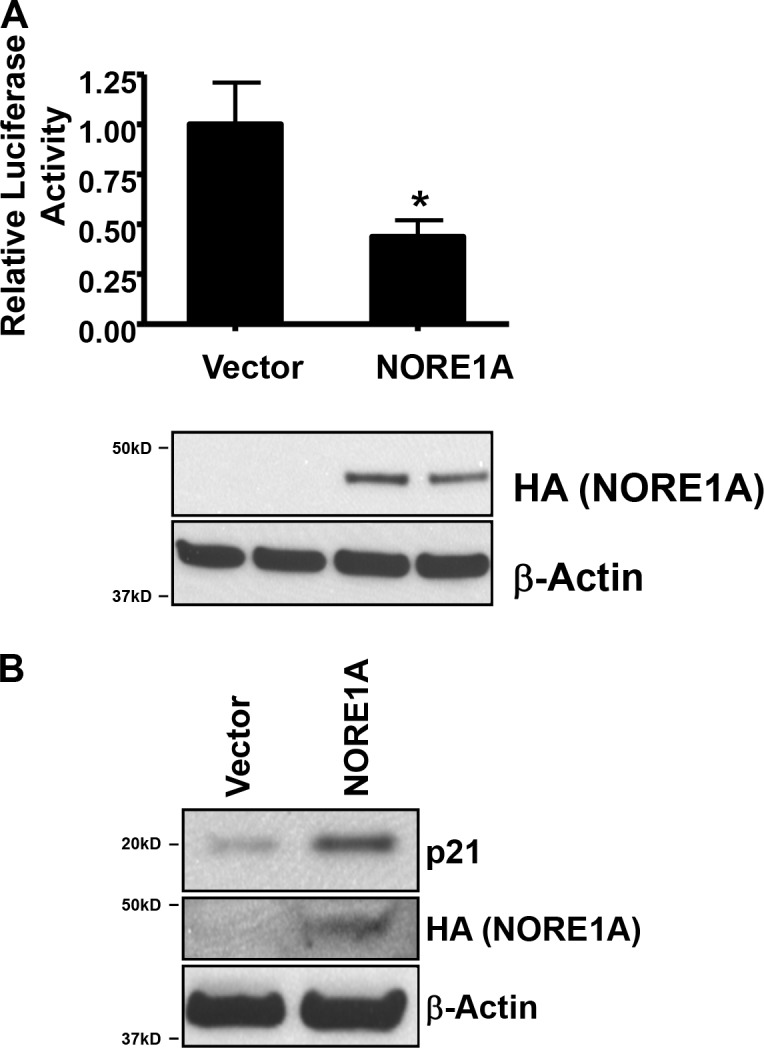
NORE1A differentially regulates the expression of p53-responsive genes. (A) NORE1A suppresses Bax promoter activation. A549 cells were transfected with NORE1A and a Bax-luciferase reporter, and activation of the reporter assessed by luciferase assay. (bottom) Expression levels of NORE1A are shown in the Western blot. Assays were performed twice in duplicate. Data are represented as mean ± SD. *, P < 0.05 compared with vector. (B) NORE1A enhances p21 expression. A549 cells were transfected with an expression construct for NORE1A or empty vector, the cells lysed and equal amounts of protein analyzed by Western blotting for p21 expression. β-actin was used as a loading control.
Discussion
Ras oncoproteins are powerfully transforming, but aberrant Ras signaling can also activate potent cell death pathways, including both apoptosis and senescence (Serrano et al., 1997; Feig and Buchsbaum, 2002; Cox and Der, 2003; Lowe et al., 2004). It has been hypothesized that Ras-induced senescence pathways serve to impede the development of cancer in cells that acquire activation of Ras (Prieur and Peeper, 2008; Collado and Serrano, 2010; Overmeyer and Maltese, 2011). Their loss should facilitate Ras-driven transformation. Although Ras-induced senescence under physiological conditions now appears a more complex process than originally thought, depending on both signal intensity and cell type (Studebaker et al., 2008; Kuilman et al., 2010; Bianchi-Smiraglia and Nikiforov, 2012), in vivo studies have now confirmed the occurrence and importance of Ras-driven senescence in tumor systems (Chen et al., 2009; Collado and Serrano, 2010; Dimauro and David, 2010; Morton et al., 2010).
The mechanisms underlying Ras induced senescence pathways and how they are subverted to facilitate human tumor development remain poorly understood. One component that has been shown to play a clear role in Ras-mediated senescence is the p53 tumor suppressor (Serrano et al., 1997). Ras has been shown to induce senescence-associated posttranslational modifications of p53 (Pearson et al., 2000), but the connection between Ras and p53 remains unclear.
NORE1A is a member of the RASSF family of proteins (RASSF5). RASSF family members are tumor suppressors that can serve as Ras death effectors (Donninger et al., 2007). Both RASSF1A and NORE1A (RASSF5) can serve as proapoptotic Ras effectors connecting Ras to the Hippo pathway (Khokhlatchev et al., 2002; Matallanas et al., 2011). Thus, the frequent loss of RASSF1A and NORE1A expression in human tumors due to epigenetic inactivation may be a major permissive factor for Ras transformation by uncoupling Ras from apoptosis.
Although initial studies identified an apoptotic function for NORE1A (Khokhlatchev et al., 2002; Vos et al., 2003), we previously identified a more likely physiological role in the regulation of cell cycle arrest by p53 (Calvisi et al., 2009). In following through on these studies, we have identified NORE1A as a key component of Ras-driven senescence signaling to p53. Thus, the frequent loss of NORE1A in human tumors will partially uncouple p53-dependent senescence from upstream regulating events such as Ras. This may explain why MEFs derived from NORE1A knockout mice can be transformed in a single step by oncogenic Ras (Park et al., 2010), instead of undergoing senescence (Serrano et al., 1997).
The link between Ras/NORE1A and p53 appears to be primarily the HIPK2 protein. NORE1A forms an endogenous, Ras-regulated complex with HIPK2 that stabilizes the protein. As NORE1A has been shown to directly bind the related HIPK1 (Lee et al., 2012), it seems likely that the interaction with HIPK2 is also direct. HIPK2 is itself a tumor suppressor (D’Orazi et al., 2012) with a broad range of functions. It has been shown to regulate apoptosis, in large part by phosphorylating p53 on residue S46 (D’Orazi et al., 2002; Hofmann et al., 2002). This enhances the affinity for p53 for the promoters of proapoptotic genes (Puca et al., 2010). However, HIPK2 can also recruit the acetyltransferases CBP/p300 (Hofmann et al., 2002) and PCAF (Di Stefano et al., 2005b) to acetylate p53. The former induces acetylation of p53 at residue K382, whereas the latter acetylates K320. Acetylation at K382 in combination with S46 phosphorylation appears to promote p53 apoptotic functions (Puca et al., 2010). Acetylation of K382 alone has been associated with senescence (Pearson et al., 2000). Moreover, acetylation at K320 has been reported to enhance p53 association with the p21CIP1 promoter, enhancing expression, which can lead to senescence (Taira and Yoshida, 2012). Here, we show that in the presence of Ras/NORE1A signaling, HIPK2 promotes the acetylation of p53 at K382 but suppresses the phosphorylation of p53 at S46. We have also detected NORE1A-induced, prosenescent acetylation of p53 at K320 (Fig. S1 b). Thus, NORE1A is specifically inducing prosenescent posttranslational modifications of p53 via HIPK2 and suppressing proapoptotic posttranslational modifications. The complex balance of factors that control the net levels of the various posttranslational modifications of p53 remain poorly understood. NORE1A appears to be able to scaffold HIPK2 to p53 under the influence of Ras in such a way that acetylation but not phosphorylation effects are enhanced. Thus, the NORE1A levels of a cell may play an important role in determining how the cells modulate p53 in response to stress stimuli, such as aberrant oncogene activity. The NORE1A–HIPK2 interaction now provides a mechanistic explanation for the previously reported induction of p53 K382 acetylation during Ras-driven senescence (Pearson et al., 2000). Although we have been unable to confirm a direct interaction between NORE1A and p53 as yet, the stoichiometry of the interaction of the overexpressed proteins suggests it is possible.
NORE1A has recently been shown to bind the main negative regulator of p53 protein levels, the ubiquitin ligase mdm2 (Lee et al., 2012). Both p53 and HIPK2 regulate and are regulated by mdm2 (Di Stefano et al., 2005a; Rinaldo et al., 2009). As we have previously shown that NORE1A stabilizes p53 in the nucleus (Calvisi et al., 2009), it may be interesting to examine the action of NORE1A on mdm2, which could allow both quantitative and qualitative (via HIPK2) regulation of p53.
Currently, the main RASSF family activated anti-growth pathway that has been described is the Hippo pathway (Fausti et al., 2012). In its simplest form the Hippo pathway consists of an MST kinase, which phosphorylates and activates a LATS kinase that then phosphorylates the related transcriptional co-activators YAP and TAZ to modulate their activity. RASSF proteins such as NORE1A can bind and activate MST kinases to contribute to the Hippo pathway (Avruch et al., 2012). In these studies, we included a NORE1A mutant that does not bind MST kinases to determine that the p53 pathway/senescence-inducing effects of NORE1A appear to be largely independent of the canonical Hippo pathway. However, recent studies have identified a potential role for HIPK2 in modulating the Hippo pathway by interacting with YAP (Poon et al., 2012), and YAP has now been implicated in p53-mediated senescence (Xie et al., 2013). Therefore, NORE1A-induced senescence may be more complex, and there may be a role for NORE1A and other RASSF proteins in the induction of the Hippo pathway by noncanonical activity at the level of YAP/TAZ, independently of MST kinases.
Thus, we identify a novel Ras–NORE1A–HIPK2–p53 signaling pathway that is critical and specific for Ras-mediated senescence. The pathway promotes the induction of specific prosenescence posttranslational modifications of p53. Its inactivation uncouples Ras from p53, allowing Ras to drive transformation without the restriction of senescence, even in the presence of WT p53. NORE1A is down-regulated in many human tumors and we found that hepatocellular carcinomas that lose NORE1A expression usually demonstrate reduced p53 acetylation. This observation supports the concept that senescence pathways must be inactivated for malignant progression of cancer and that NORE1A is a critical component of such a pathway. This work also identifies a novel mechanism of action for a RASSF family protein: the regulation of protein acetylation. We suspect this function may be common to other RASSF family members, and may involve targets in addition to p53.
Materials and methods
Plasmids
Human NORE1A cDNA was obtained from Origene and confirmed by sequencing. HA-, KATE-, and GFP-NORE1A expression plasmids were generated by cloning the full-length NORE1A cDNA as a BglII–EcoRI fragment into a pcDNA vector with an HA epitope tag, pmKATE2 (Evrogen) and pEGFP-C1 (Takara Bio). pmKATE2 encodes the far-red fluorescent protein mKATE2 and pEGFP-C1 a red-shifted variant of WT GFP. All three fusion proteins were under the control of a CMV promoter. pCGN-HA-H-Ras12v was created by cloning the full-length H-Ras12v cDNA as a BamHI fragment into pCGN vector under the control of a CMV promoter (Ellis et al., 2002) and pLRT-HRas12v by cloning the full-length H-Ras12v cDNA as a XhoI–NotI fragment into the pLRT retroviral vector (Watsuji et al., 1997). The GFP-tagged HIPK2 expression plasmid was generated by cloning the HIPK2 cDNA into the pEGFP-C2 expression vector and was a gift of Y. Kim (National Institutes of Health, Bethesda, MD). shRNAs for human NORE1A (#1: 5′-TATATATAGCTATATGCCT-3′; #2: 5′-AGCTTTGTGCTAAAGGAGA-3′; Scrambled: 5′-ATCTCGCTTGGGCGAGAGTAAG-3′; RHS4531-EG83593) were obtained from Open Biosystems and HIPK2 shRNAs (#1: 5′-CTCGCCAGCCTCCACCGTCTACACTGGAT-3′; #2: 5′-CCTTTGTGGAGACTGAAGACACCAGATGA-3′; TR304106) were obtained from Origene. p53 and control siRNAs were from Applied Biosystems (ID 2714) and mouse NORE1A shRNAs (#1: 5′-GGCTGCTCAAGAAGTTCATGGTTGTGGAC-3′; #2: 5′-GCGACGTGAGGAGCATCTTCGAGCAGCCG-3′) from Origene and have been described previously (Calvisi et al., 2009). The NORE1AR3A mutant (92/94: RRR:AAA) was generated using a QuikChange kit (Stratagene) as described by the manufacturer. The NORE1A RA mutant was also generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene) in a similar manner based on the mutant described in Aoyama et al. (2004). The NORE1A-sarah-del mutant was generated by PCR and lacks the C-terminal 168 bases pairs. All mutants were sequenced to confirm fidelity before use. MST1 constructs were a generous gift (J. Chernoff, Fox Chase Cancer Center, Philadelphia, PA).
Tissue culture and transfections
HEK-293, A549, NCI-H1792, HepG2, and COS-7 cells were obtained from the American Type Culture Collection. MEFs were a gift from D. Dean (University of Louisville, Louisville, KY). Cells were grown in DMEM/10% FBS. HBEC-3KT cells were provided courtesy of Jerry Shay (UT South Western, Dallas, TX) and cultured in keratinocyte serum-free medium containing bovine pituitary extract and recombinant epidermal growth factor (Invitrogen). A summary of the cell lines used in this study with respect to their Ras and p53 status is provided in Tables S1 and S2. Transient transfections were performed using Jet-Prime (Polyplus) as described by the manufacturer. GFP and RFP proteins were visualized using an Olympus IX50 fluorescent microscope at room temperature in growth medium with a 100×/1.30 Olympus UPlanFl oil immersion objective. β-galactosidase assays were performed using a BioVison kit (BioVision). Samples were imaged using an IX50 inverted system microscope (Olympus) with a UPlanFl 4×/0.13 PhL or LCPlanFl 20×/0.40 Ph1 objective (Olympus) and a SPOT camera (Diagnostic Instruments Inc.). Senescence-associated heterochromatin foci (SAHF) were detected essentially as previously described (Aird and Zhang, 2013). In brief, cells were plated in glass-bottom microwell dishes (MatTek Corp). The cells were fixed with 4% paraformaldehyde for 10 min at room temperature and then washed with PBS. They were then permeabilized with 0.2% (vol/vol) Triton X-100 for 5 min at room temperature, blocked with 3% BSA in PBS for 5 min, and stained with 0.15 µg/ml DAPI diluted in 3% BSA in PBS for 3 min at room temperature. SAHF were observed using an Olympus IX50 fluorescent microscope with a 100×/1.30 Olympus UPlanFl oil immersion objective. Apoptosis assays were performed using the pCaspase-3-Sensor indicator plasmid, as described previously (Vos et al., 2003). In brief, COS-7 cells were co-transfected with either red fluorescent expression vector pmKATE-NORE1A or NORE1AR3A and yellow fluorescent protein-pSensor caspase activity reporter plasmid (Takara Bio). Localization of the pSensor indicator protein in cotransfected cells was determined 24 h later by fluorescent microscopy. Cyclohexamide (Sigma-Aldrich) treatments were performed 24 h after transfection at a concentration of 20 µg/ml. Soft agar assays were performed as described previously (Ellis et al., 2002). Cells (104) were resuspended in 0.35% agar and plated on a prehardened 0.7% agar base and incubated at 37°C for 14 d before scoring by microscopic evaluation. pLRT-H-Ras12v transfected cells were selected in 5 µg/ml Blasticidin (Invitrogen) and induced with 1 µg/ml doxycycline (Sigma-Aldrich).
Human tissue samples
A collection of 40 p53 WT HCCs were used. Liver tissues were provided by S.S. Thorgeirsson (Laboratory of Experimental Carcinogenesis, National Cancer Institute, Bethesda, MD). Institutional Review Board approval was obtained at participating hospitals in the Liver Tissue Procurement and Distribution System (University of Minnesota, Minneapolis, MN, and University of Pittsburgh, Pittsburgh, PA) and the National Institutes of Health. The clinicopathological features of HCC patients are summarized in Table S2.
Western blots and immunoprecipitation
Monoclonal NORE1A antibodies were a gift from A. Khokhlatchev (University of Virginia, Richmond, VA), and were raised against human NORE1A amino acid residues 119–416. Rabbit polyclonal NORE1A antibodies were raised against the synthetic human NORE1A peptide: KYDKFRQKLEEALRESQGKPG by ProSci (Poway, CA). Rabbit monoclonal HIPK2 antibodies were from Epitomics (Burlingame, CA). Rabbit polyclonal Dec1 was from Imgenex (Littleton, CO), and rabbit polyclonal RFP and mouse monoclonal GFP were from Santa Cruz Biotechnology, Inc. Mouse monoclonal anti-H-Ras antibody 146 was from the NCI antibody repository. Mouse monoclonal FLAG and β-actin antibodies were obtained from Sigma-Aldrich and mouse monoclonal HA antibodies were from Covance. Rabbit polyclonal p53 Ac382 and Phospho-S46 and p38 were obtained from Cell Signaling Technology. Immunoprecipitations were performed using llama anti-GFP, Allele Biotech, or mouse monoclonal HA beads (Sigma-Aldrich). Goat anti–mouse and anti–rabbit secondary antibodies conjugated to peroxidase were from Thermo Fisher Scientific. Western blots were developed using a West-Pico kit (Thermo Fisher Scientific). Protein acetylation was detected by immunoprecipitation with rabbit anti–acetyl-lysine beads (ImmuneChem) followed by Western blot with the appropriate antibody. Alternatively, the protein was immunoprecipitated and then Western blotted with rabbit anti-acetyl lysine (Cell Signaling Technology).
Image acquisition and processing
Images were scanned and quantified using a Pharos FX plus Molecular Imager (Bio-Rad Laboratories) and Quantity One software (Bio-Rad Laboratories). Images were compiled using Photoshop (Adobe).
Online supplemental material
Fig. S1shows that the Ras binding defective mutant of NORE1A (NORE1AR3A) fails to stabilize HIPK2 protein and that NORE1A also promotes acetylation of p53 at K320. Table S1 provides a description of the cell lines used. Table S2 provides a description of the clinicopathological features of HCC patient samples. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201408087/DC1.
Supplementary Material
Acknowledgments
We would like to thank Dr. Etorre Appella (National Cancer Insitute; NCI) for thoughtful discussions and encouragement.
This work was funded in part by National Institutes of Health grants RR18733 and CA133171-01 and NCI intramural funds (G.J. Clark).
The authors declare no competing financial interests.
Footnotes
Abbreviations:
- MEF
- mouse embryon fibroblast
- shRNA
- short hairpin RNA
References
- Aird K.M., and Zhang R.. 2013. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 965:185–196 10.1007/978-1-62703-239-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama Y., Avruch J., and Zhang X.F.. 2004. Nore1 inhibits tumor cell growth independent of Ras or the MST1/2 kinases. Oncogene. 23:3426–3433 10.1038/sj.onc.1207486 [DOI] [PubMed] [Google Scholar]
- Avruch J., Zhou D., Fitamant J., Bardeesy N., Mou F., and Barrufet L.R.. 2012. Protein kinases of the Hippo pathway: regulation and substrates. Semin. Cell Dev. Biol. 23:770–784 10.1016/j.semcdb.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman R., and Prives C.. 2010. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2:a000935 10.1101/cshperspect.a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi-Smiraglia A., and Nikiforov M.A.. 2012. Controversial aspects of oncogene-induced senescence. Cell Cycle. 11:4147–4151 10.4161/cc.22589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C.L., and Gu W.. 2011. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2:456–462 10.1007/s13238-011-1063-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell M.E., DeNicola G.M., Martins C.P., Jacobetz M.A., Maitra A., Hruban R.H., and Tuveson D.A.. 2012. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene. 31:1599–1608 10.1038/onc.2011.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi D.F., Pinna F., Pellegrino R., Sanna V., Sini M., Daino L., Simile M.M., De Miglio M.R., Frau M., Tomasi M.L., et al. 2008. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int. J. Cancer. 123:2057–2064 10.1002/ijc.23720 [DOI] [PubMed] [Google Scholar]
- Calvisi D.F., Donninger H., Vos M.D., Birrer M.J., Gordon L., Leaner V., and Clark G.J.. 2009. NORE1A tumor suppressor candidate modulates p21CIP1 via p53. Cancer Res. 69:4629–4637 10.1158/0008-5472.CAN-08-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzado M.A., De La Vega L., Munoz E., and Schmitz M.L.. 2009. From top to bottom: the two faces of HIPK2 for regulation of the hypoxic response. Cell Cycle. 8:1659–1664 10.4161/cc.8.11.8597 [DOI] [PubMed] [Google Scholar]
- Campbell P.M., and Der C.J.. 2004. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 14:105–114 10.1016/j.semcancer.2003.09.015 [DOI] [PubMed] [Google Scholar]
- Carter S., and Vousden K.H.. 2009. Modifications of p53: competing for the lysines. Curr. Opin. Genet. Dev. 19:18–24 10.1016/j.gde.2008.11.010 [DOI] [PubMed] [Google Scholar]
- Chen J., Lui W.O., Vos M.D., Clark G.J., Takahashi M., Schoumans J., Khoo S.K., Petillo D., Lavery T., Sugimura J., et al. 2003. The t(1;3) breakpoint-spanning genes LSAMP and NORE1 are involved in clear cell renal cell carcinomas. Cancer Cell. 4:405–413 10.1016/S1535-6108(03)00269-1 [DOI] [PubMed] [Google Scholar]
- Chen X., Mitsutake N., LaPerle K., Akeno N., Zanzonico P., Longo V.A., Mitsutake S., Kimura E.T., Geiger H., Santos E., et al. 2009. Endogenous expression of Hras(G12V) induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc. Natl. Acad. Sci. USA. 106:7979–7984 10.1073/pnas.0900343106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Trotman L.C., Shaffer D., Lin H.K., Dotan Z.A., Niki M., Koutcher J.A., Scher H.I., Ludwig T., Gerald W., et al. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 436:725–730 10.1038/nature03918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M., and Serrano M.. 2010. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer. 10:51–57 10.1038/nrc2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M., Gil J., Efeyan A., Guerra C., Schuhmacher A.J., Barradas M., Benguría A., Zaballos A., Flores J.M., Barbacid M., et al. 2005. Tumour biology: senescence in premalignant tumours. Nature. 436:642 10.1038/436642a [DOI] [PubMed] [Google Scholar]
- Courtois-Cox S., Genther Williams S.M., Reczek E.E., Johnson B.W., McGillicuddy L.T., Johannessen C.M., Hollstein P.E., MacCollin M., and Cichowski K.. 2006. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 10:459–472 10.1016/j.ccr.2006.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A.D., and Der C.J.. 2003. The dark side of Ras: regulation of apoptosis. Oncogene. 22:8999–9006 10.1038/sj.onc.1207111 [DOI] [PubMed] [Google Scholar]
- Dimauro T., and David G.. 2010. Ras-induced senescence and its physiological relevance in cancer. Curr. Cancer Drug Targets. 10:869–876 10.2174/156800910793357998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano V., Rinaldo C., Sacchi A., Soddu S., and D’Orazi G.. 2004. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Exp. Cell Res. 293:311–320 10.1016/j.yexcr.2003.09.032 [DOI] [PubMed] [Google Scholar]
- Di Stefano V., Mattiussi M., Sacchi A., and D’Orazi G.. 2005a. HIPK2 inhibits both MDM2 gene and protein by, respectively, p53-dependent and independent regulations. FEBS Lett. 579:5473–5480 10.1016/j.febslet.2005.09.008 [DOI] [PubMed] [Google Scholar]
- Di Stefano V., Soddu S., Sacchi A., and D’Orazi G.. 2005b. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21Waf1 after nonapoptotic DNA damage. Oncogene. 24:5431–5442 10.1038/sj.onc.1208717 [DOI] [PubMed] [Google Scholar]
- Donninger H., Vos M.D., and Clark G.J.. 2007. The RASSF1A tumor suppressor. J. Cell Sci. 120:3163–3172 10.1242/jcs.010389 [DOI] [PubMed] [Google Scholar]
- D’Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., et al. 2002. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 4:11–19 10.1038/ncb714 [DOI] [PubMed] [Google Scholar]
- D’Orazi G., Rinaldo C., and Soddu S.. 2012. Updates on HIPK2: a resourceful oncosuppressor for clearing cancer. J. Exp. Clin. Cancer Res. 31:63 10.1186/1756-9966-31-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis C.A., Vos M.D., Howell H., Vallecorsa T., Fults D.W., and Clark G.J.. 2002. Rig is a novel Ras-related protein and potential neural tumor suppressor. Proc. Natl. Acad. Sci. USA. 99:9876–9881 10.1073/pnas.142193799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausti F., Di Agostino S., Sacconi A., Strano S., and Blandino G.. 2012. Hippo and rassf1a Pathways: A Growing Affair. Mol. Biol. Int. 2012:307628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig L.A., and Buchsbaum R.J.. 2002. Cell signaling: life or death decisions of ras proteins. Curr. Biol. 12:R259–R261 10.1016/S0960-9822(02)00787-X [DOI] [PubMed] [Google Scholar]
- Ferbeyre G., de Stanchina E., Lin A.W., Querido E., McCurrach M.E., Hannon G.J., and Lowe S.W.. 2002. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 22:3497–3508 10.1128/MCB.22.10.3497-3508.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B., and Zhu W.G.. 2012. Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8:672–684 10.7150/ijbs.4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T.G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., and Schmitz M.L.. 2002. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4:1–10 10.1038/ncb715 [DOI] [PubMed] [Google Scholar]
- Kennedy A.L., Morton J.P., Manoharan I., Nelson D.M., Jamieson N.B., Pawlikowski J.S., McBryan T., Doyle B., McKay C., Oien K.A., et al. 2011. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell. 42:36–49 10.1016/j.molcel.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A., Rabizadeh S., Xavier R., Nedwidek M., Chen T., Zhang X.F., Seed B., and Avruch J.. 2002. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 12:253–265 10.1016/S0960-9822(02)00683-8 [DOI] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Mooi W.J., and Peeper D.S.. 2010. The essence of senescence. Genes Dev. 24:2463–2479 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D., Park S.J., Sung K.S., Park J., Lee S.B., Park S.Y., Lee H.J., Ahn J.W., Choi S.J., Lee S.G., et al. 2012. Mdm2 associates with Ras effector NORE1 to induce the degradation of oncoprotein HIPK1. EMBO Rep. 13:163–169 10.1038/embor.2011.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A.J.1997. p53, the cellular gatekeeper for growth and division. Cell. 88:323–331 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- Lowe S.W., Cepero E., and Evan G.. 2004. Intrinsic tumour suppression. Nature. 432:307–315 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- Malumbres M., and Barbacid M.. 2003. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 3:459–465 10.1038/nrc1097 [DOI] [PubMed] [Google Scholar]
- Matallanas D., Romano D., Al-Mulla F., O’Neill E., Al-Ali W., Crespo P., Doyle B., Nixon C., Sansom O., Drosten M., et al. 2011. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell. 44:893–906 10.1016/j.molcel.2011.10.016 [DOI] [PubMed] [Google Scholar]
- Morton J.P., Timpson P., Karim S.A., Ridgway R.A., Athineos D., Doyle B., Jamieson N.B., Oien K.A., Lowy A.M., Brunton V.G., et al. 2010. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA. 107:246–251 10.1073/pnas.0908428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmeyer J.H., and Maltese W.A.. 2011. Death pathways triggered by activated Ras in cancer cells. Front Biosci (Landmark Ed). 16:1693–1713(Landmark Ed) 10.2741/3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Kang S.I., Lee S.Y., Zhang X.F., Kim M.S., Beers L.F., Lim D.S., Avruch J., Kim H.S., and Lee S.B.. 2010. Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 285:35029–35038 10.1074/jbc.M110.165506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P.P., and Pelicci P.G.. 2000. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 406:207–210 10.1038/35021000 [DOI] [PubMed] [Google Scholar]
- Poon C.L., Zhang X., Lin J.I., Manning S.A., and Harvey K.F.. 2012. Homeodomain-interacting protein kinase regulates Hippo pathway-dependent tissue growth. Curr. Biol. 22:1587–1594 10.1016/j.cub.2012.06.075 [DOI] [PubMed] [Google Scholar]
- Prieur A., and Peeper D.S.. 2008. Cellular senescence in vivo: a barrier to tumorigenesis. Curr. Opin. Cell Biol. 20:150–155 10.1016/j.ceb.2008.01.007 [DOI] [PubMed] [Google Scholar]
- Puca R., Nardinocchi L., Givol D., and D’Orazi G.. 2010. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene. 29:4378–4387 10.1038/onc.2010.183 [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y., Grabocka E., and Bar-Sagi D.. 2011. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 11:761–774 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez R.D., Sheridan S., Girard L., Sato M., Kim Y., Pollack J., Peyton M., Zou Y., Kurie J.M., Dimaio J.M., et al. 2004. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 64:9027–9034 10.1158/0008-5472.CAN-04-3703 [DOI] [PubMed] [Google Scholar]
- Rinaldo C., Prodosmo A., Siepi F., Moncada A., Sacchi A., Selivanova G., and Soddu S.. 2009. HIPK2 regulation by MDM2 determines tumor cell response to the p53-reactivating drugs nutlin-3 and RITA. Cancer Res. 69:6241–6248 10.1158/0008-5472.CAN-09-0337 [DOI] [PubMed] [Google Scholar]
- Ringshausen I., O’Shea C.C., Finch A.J., Swigart L.B., and Evan G.I.. 2006. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 10:501–514 10.1016/j.ccr.2006.10.010 [DOI] [PubMed] [Google Scholar]
- Schubbert S., Shannon K., and Bollag G.. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 7:295–308 10.1038/nrc2109 [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., and Lowe S.W.. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 88:593–602 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- Studebaker A.W., Storci G., Werbeck J.L., Sansone P., Sasser A.K., Tavolari S., Huang T., Chan M.W., Marini F.C., Rosol T.J., et al. 2008. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 68:9087–9095 10.1158/0008-5472.CAN-08-0400 [DOI] [PubMed] [Google Scholar]
- Taira N., and Yoshida K.. 2012. Post-translational modifications of p53 tumor suppressor: determinants of its functional targets. Histol. Histopathol. 27:437–443. [DOI] [PubMed] [Google Scholar]
- van der Weyden L., and Adams D.J.. 2007. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta. 1776:58–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu V.E., and El-Deiry W.S.. 1996. Biological and clinical importance of the p53 tumor suppressor gene. Clin. Chem. 42:858–868. [PubMed] [Google Scholar]
- Vogelstein B., Lane D., and Levine A.J.. 2000. Surfing the p53 network. Nature. 408:307–310 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- Vos M.D., Martinez A., Ellis C.A., Vallecorsa T., and Clark G.J.. 2003. The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J. Biol. Chem. 278:21938–21943 10.1074/jbc.M211019200 [DOI] [PubMed] [Google Scholar]
- Watsuji T., Okamoto Y., Emi N., Katsuoka Y., and Hagiwara M.. 1997. Controlled gene expression with a reverse tetracycline-regulated retroviral vector (RTRV) system. Biochem. Biophys. Res. Commun. 234:769–773 10.1006/bbrc.1997.6705 [DOI] [PubMed] [Google Scholar]
- Xie Q., Chen J., Feng H., Peng S., Adams U., Bai Y., Huang L., Li J., Huang J., Meng S., and Yuan Z.. 2013. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 73:3615–3624 10.1158/0008-5472.CAN-12-3793 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.