

Supplemental digital content is available in the text.
Key Words: ALS, C9orf72, Dipeptide-repeat protein, Drosha, FTLD, TDP-43
Abstract
Frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) are 2 neurodegenerative disorders that share clinical, genetic, and neuropathologic features. The presence of abnormal expansions of GGGGCC repeats (G4C2 repeats) in a noncoding region of the Chromosome 9 open reading frame 72 (C9orf72) gene is the major genetic cause of both FTLD and ALS. Transcribed G4C2 repeats can form nuclear RNA foci and recruit RNA-binding proteins, thereby inhibiting their normal function. Moreover, through a repeat-associated non-ATG translation mechanism, G4C2 repeats translation leads to dipeptide-repeat protein aggregation in the cytoplasm of neurons. Here, we identify Drosha protein as a new component of these dipeptide-repeat aggregates. In C9orf72 mutation cases of FTLD-TDP (c9FTLD-TDP) and ALS (c9ALS), but not in FTLD or ALS cases without C9orf72 mutation, Drosha is mislocalized to form neuronal cytoplasmic inclusions in the hippocampus, frontal cortex, and cerebellum. Further characterization of Drosha-positive neuronal cytoplasmic inclusions in the hippocampus, frontal cortex, and cerebellum revealed colocalization with p62 and ubiquilin-2, 2 pathognomonic signatures of c9FTLD-TDP and c9ALS cases; however, Drosha inclusions rarely colocalized with TDP-43 pathology. We conclude that Drosha may play a unique pathogenic role in the onset or progression of FTLD-TDP/ALS in patients with the C9orf72 mutation.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) are 2 devastating neurodegenerative disorders with overlapping clinical, genetic, and neuropathologic features (1). The presence of abnormal GGGGCC repeats expansion (G4C2 repeats) in an intronic region of the Chromosome 9 open reading frame 72 (C9orf72) gene is found in patients with familial FTLD (3%–48%) and familial ALS (3%–46%), those with sporadic forms of FTLD (2%–23%) and sporadic ALS (0.4%–21%), and those with the combination of both syndromes (10%–88%) (2). Dominant mutations in TARDBP and FUS/TLS were identified as causative of FTLD and ALS associated with TDP-43 (FTLD-TDP) and FUS (FTLD-FUS) inclusions, respectively (3, 4). Similarly, mutations in the VCP (5, 6), GRN (7, 8), and UBQLN2 (9) genes were associated with both familial FTLD and ALS cases, providing genetic evidence of common pathologic mechanisms linked to accumulations of TDP-43 inclusions and neurodegeneration (10). Recently, the presence of abnormal G4C2 repeats expansion in C9orf72 were identified as the most common genetic abnormality in FTLD/ALS spectrum disorders characterized by TDP-43 pathology, which we refer to here as c9FTLD-TDP and c9ALS, respectively (11, 12). The number of G4C2 repeats in the normal population ranges from 2 to 24 (11–15), whereas up to several thousand repeats have been described in the pathologically expanded allele (11, 13, 16), associated with approximately 10% of sporadic cases of FTLD-TDP and ALS and 25% to 40% familial cases (17).
Since the identification of C9orf72 mutation as the major genetic factor linked to c9FTLD-TDP/c9ALS in 2011, enormous efforts have been made to elucidate the pathogenic mechanisms of these G4C2 repeats. It has been proposed that these mechanisms involve haploinsufficiency, protein toxicity of dipeptide repeat (DPR) aggregates produced from a repeat-associated non-ATG translation of G4C2 expanded sequences (18), and RNA-gain of toxic function (19–21). Moreover, several RNA-binding proteins including hnRNPA3, Pur α, ASF/S2, ADARB2, or nucleolin (22–26) bind specifically to G4C2 repeated sequences, thereby affecting their ability to bind their natural RNA targets. The consequences of the RNA-binding protein recruitment could lead to disturbances in RNA processing, changes in expression levels of mRNA and/or microRNAs (miRNAs). In this regard, in other repeat expansions diseases, such as Fragile X–associated tremor/ataxia syndrome, it was shown that nuclear RNA foci containing CGG-repeats expansions recruit DGCR8 and partially its partner Drosha protein, 2 key players in miRNA biogenesis (27). Consequently, the processing of primary miRNAs is reduced in cells expressing CGG-repeats and in postmortem brain samples from Fragile X–associated tremor/ataxia syndrome patients, resulting in decreased levels of mature miRNAs.
The emerging importance of miRNAs as key players in mechanisms of neurodegeneration may in part be caused by the complexity of miRNA-based regulatory networks that influence gene expression. Indeed, a growing number of studies point to the differential expression of miRNAs in postmortem brain samples from patients with neurodegenerative disease such as Alzheimer disease (AD), Parkinson disease, and Huntington disease, among others, as potential mediators of the diverse disease processes in these different disorders (28–33). Here, we show that Drosha protein, but not its cofactor DGCR8, is mislocalized and forms neuronal cytoplasmic inclusions (NCIs) in the hippocampus, frontal cortex, and cerebellum of autopsy-confirmed c9FTLD-TDP and c9ALS cases, but not in FTLD-TDP and ALS cases without C9orf72 mutation, other neurodegenerative diseases, or control individuals. Interestingly, these cytoplasmic Drosha inclusions colocalize with DPR aggregates and with p62 and ubiquilin-2, 2 important factors involved in degradation of proteins via the ubiquitin/proteasome pathway.
MATERIALS AND METHODS
Autopsy Cohort
Human postmortem brain samples were obtained from the University of Pennsylvania, Center for Neurodegenerative Disease Brain Bank, under institutional review board approval, as recently reviewed (34). Regions sampled included midfrontal cortex, hippocampus, and cerebellum from c9FTLD-TDP and c9ALS patients and age-matched FTLD-TDP, ALS, and control individuals (Table, Supplemental Digital Content 1, http://links.lww.com/NEN/A714). Also included were age-matched AD, hippocampal sclerosis, dementia with Lewy bodies, and FTLD non-TDP43 (FTLD-FUS and FTLD-Tau) cases (Table, Supplemental Digital Content 1, http://links.lww.com/NEN/A714). Histopathologic subtyping of our FTLD-TDP cohort was done according to established guidelines (35) (Table, Supplemental Digital Content 1, http://links.lww.com/NEN/A714). Genetic testing for C9orf72 expansions was performed as previously described (36, 37). All necessary written informed consent forms were obtained from the patients or their next of kin and confirmed at the time of death.
Immunohistochemistry and Immunofluorescence
For immunohistochemistry (IHC) studies, paraffin-embedded 6-μm-thick sections from various brain regions were deparaffinized in xylene and rehydrated in graded alcohol concentrations. Endogenous peroxidases were quenched by incubating sections in a solution of 5% H2O2/methanol for 30 minutes at room temperature. After washing in water for 10 minutes, antigen retrieval was performed in 1% Antigen Unmasking Solution (Vector) by microwaving for 15 minutes at 99°C. Slides were allowed to cool at room temperature and then washed with 0.1 mol/L Tris buffer, pH 7.6 for 5 minutes. To reduce nonspecific signals, sections were immersed in blocking buffer (0.1 mol/L Tris–2% fetal bovine serum, pH 7.6) for 1 hour at room temperature and then incubated overnight at 4°C in a humidified chamber, with the primary antibody diluted in blocking buffer; anti-Drosha (rabbit, 1:500, Ab12286; Abcam, Cambridge, MA), anti–Gly-Pro (GP, no. 2325, rabbit, 1:20,000, generated in CNDR), anti–Gly-Ala (GA, no. 2328, rabbit, 1:20,000, generated in CNDR), and anti-GA (no. 5 F2, mouse, 1:1000, kindly provided by Dr. Edbauer [38]). After 3 washes in 0.1 mol/L Tris buffer for 5 minutes, sections were incubated with a biotin-conjugated secondary antibody diluted in blocking buffer for 1 hour at room temperature. Antigen-antibody reactions were visualized using VECTASTAIN AB solution (Vector Laboratories, Inc., Burlingame, CA) and ImmPACT DAB solution (Vector Laboratories, Inc.). Hematoxylin-counterstained slides were dehydrated through graded alcohol concentrations and xylene and mounted with Cytoseal (Thermo Scientific, Rockford, IL). Bright-field images were acquired using a Nikon Eclipse TE2000 microscope using NIS-Elements software.
For double immunofluorescence (IF), paraffin sections were processed as previously described but omitting the endogenous peroxidase–blocking step. Incubation with primary antibodies was done overnight at 4°C in a humidified chamber: anti-Drosha (rabbit, 1:250, Ab12296, Abcam, Cambridge, MA), p62 (mouse, 1:500, H00008878-M01; Abnova, Walnut, CA), anti–ubiquilin-2 (clone 5 F5, mouse, 1:10,000, H00029978-M03; Abnova), anti-GA (no. 5 F2, mouse, 1:500), anti-p409/410 (TAR5P-1D3, rat, 1:200 [39]). After several washes in 0.1 mol/L Tris buffer, samples were incubated in the dark with the corresponding secondary antibodies at 1:1000 dilution in blocking buffer for 1 hour at room temperature in a humidified chamber: goat anti-rabbit Alexa Fluor 594–conjugated, goat anti-mouse Alexa Fluor 488–conjugated, and goat anti-mouse Alexa Fluor 488–conjugated (Molecular Probes, Eugene, OR). A final step to reduce endogenous autofluorescence was performed by immersing sections in a 0.3% Sudan black/70% ethanol solution for 5 minutes, followed by a vigorous wash in water for 10 minutes after which the sections were mounted in Vectashield medium containing DAPI. Images were obtained in a high-resolution Leica DMI6000 microscope using the Leica LAS-AF software. A total of 10 images (40×) per section of each brain region containing Drosha NCIs were analyzed, and the percentages of Drosha NCIs that colocalized with DPR, p62, or ubiquilin-2 proteins in the c9FTLD-TDP (n = 4) and c9ALS cases (n = 4) were determined. The analysis and quantification were performed manually using ImageJ software. Results were expressed as the mean ± SD (n = 4). Statistical differences of colocalization between brain regions were evaluated with a 2-tailed Student t-test.
RESULTS
Drosha Mislocalization in c9FTLD-TDP and c9ALS Cases
To elucidate if the presence of aberrant G4C2 repeats could alter the normal nuclear distribution of the RNA-binding proteins Drosha and DGCR8, their subcellular localization was analyzed in postmortem brain samples of FTLD-TDP and ALS cases with and without C9orf72 mutations and in age-matched control individuals (Table, Supplemental Digital Content 1, http://links.lww.com/NEN/A714). Interestingly, IHC of Drosha in the hippocampus, frontal cortex, and cerebellum revealed the presence of frequent Drosha-positive NCIs in c9FTLD-TDP and c9ALS cases but not in FTLD and ALS cases without G4C2 repeats expansion or in control individuals (Figs. 1, 2). We confirmed Drosha antibody specificity by competition experiments with the immunizing peptide using Western blot (Figure, Supplemental Digital Content 2, part a, http://links.lww.com/NEN/A715), double IF (Figure, Supplemental Digital Content 2, part b, http://links.lww.com/NEN/A715), and IHC (Figure, Supplemental Digital Content 2, part c, http://links.lww.com/NEN/A715, Materials and Methods, Supplemental Digital Content 3, http://links.lww.com/NEN/A716).
FIGURE 1.
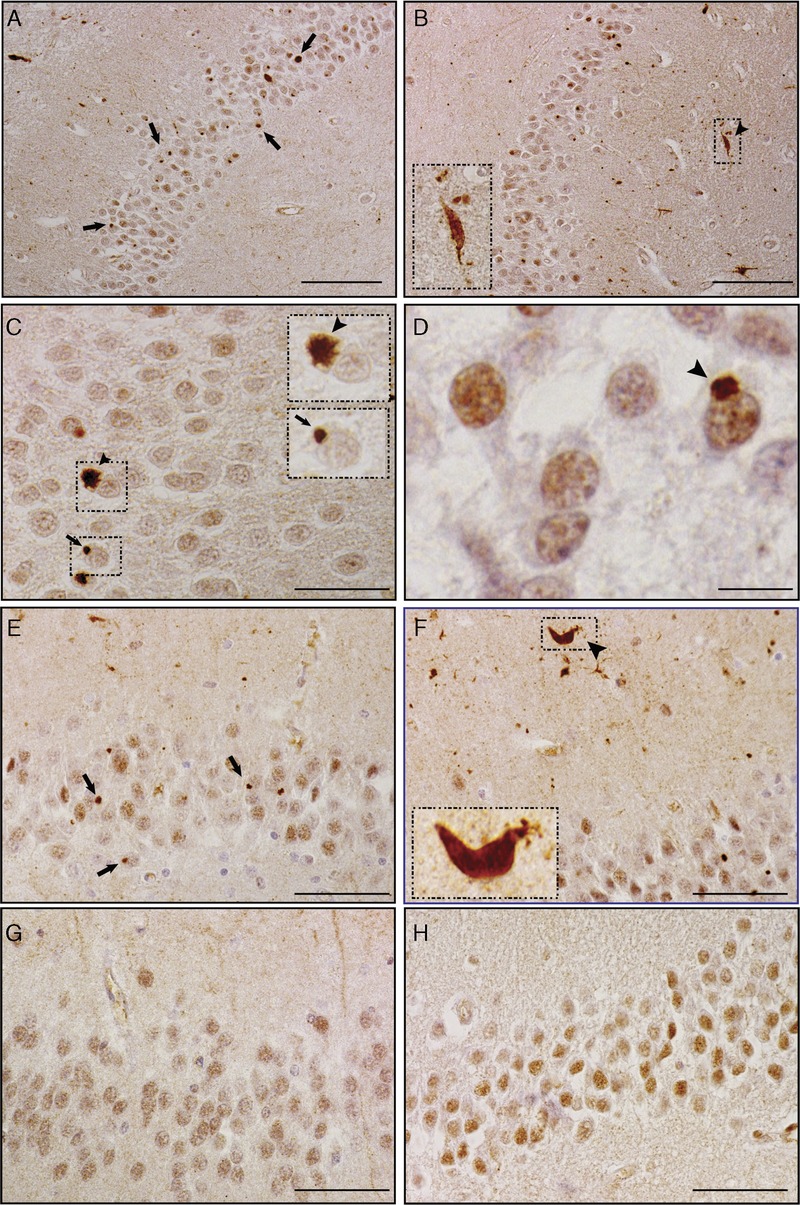
Mislocalized Drosha in the hippocampus of c9FTLD-TDP and c9ALS patients. Drosha immunohistochemistry in the hippocampus of c9FTLD-TDP (A–D), c9ALS (E, F), FTLD-TDP (G), and ALS (H) cases. Drosha-positive neuronal cytoplasmic inclusions (NCIs) were exclusively observed in the dentate gyrus in C9orf72 mutation carriers (see arrows in A, C, E). Drosha NCIs show starlike (C, arrowhead and inset) or dotlike shapes (C, arrow and inset) in the cytoplasm of dentate granule neurons. Drosha accumulation into NCIs does not result in nuclear clearance of Drosha protein (D, arrowhead). Drosha-positive dystrophic neurites (DNs) also were frequently found in c9FTLD-TDP and c9ALS cases (B, F, arrowhead and insets), but, unlike the NCIs, Drosha-positive DNs were not exclusively associated with the presence of C9orf72 mutations. Nuclear Drosha was observed in dentate granular cells of FTLD-TDP (G) and ALS (H) cases, but no NCIs were seen. The insets are higher-magnification images of the boxed areas in (B), (C), and (F). Scale bars = (A, B) 100 μm; (C, E–H) 50 μm; (D) 10 μm.
FIGURE 2.
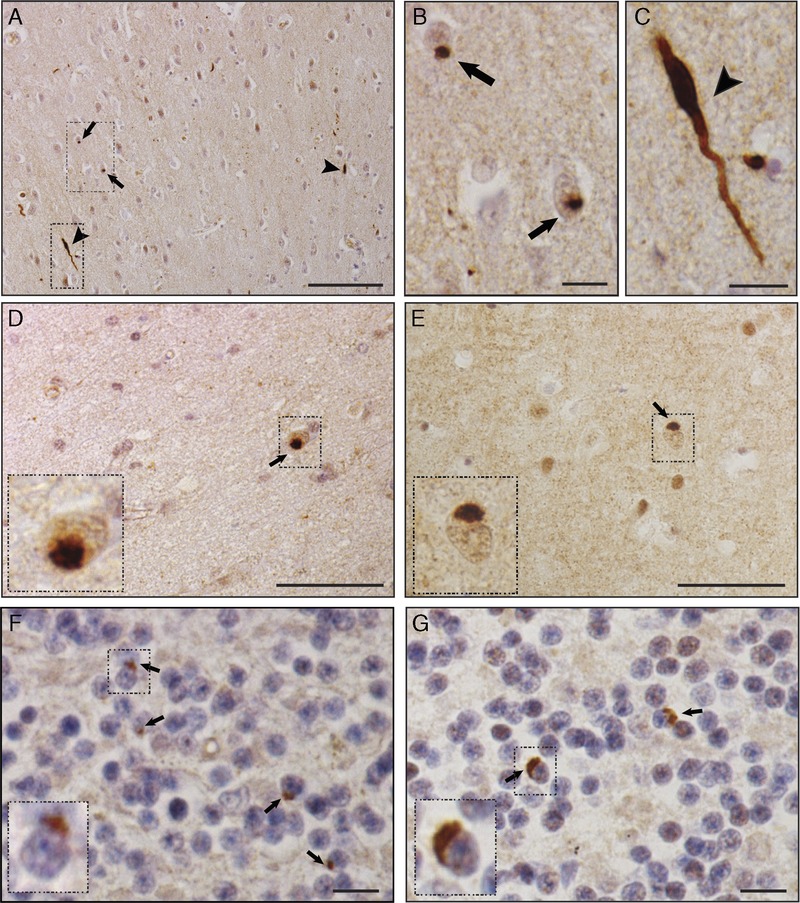
Mislocalized Drosha protein in the frontal cortex and cerebellum of c9FTLD-TDP and c9ALS patients. Drosha immunohistochemistry in the frontal cortex (A–E) and cerebellum (F, G) in c9FTLD-TDP (A–D, F) and c9ALS (E, G) cases. In the frontal cortex, Drosha-positive neuronal cytoplasmic inclusions (NCIs) were observed in scattered cells both in superficial (A, arrows) and deep layers (D, E, arrows, insets). A few Drosha-immunoreactive dystrophic neurites (DNs) were observed in cortical areas (A and C, arrowhead). The boxed areas in A are shown in B and C at higher magnification. Drosha-positive NCIs were found in the granular cell layer of the cerebellum in c9FTLD-TDP (F) and c9ALS (G) cases (arrows and insets). The insets are higher-magnification images of the boxed areas in D to G. Scale bars = (A) 100 μm; (D, E) 50 μm; (B, C, F, G) 10 μm.
In the hippocampus, all c9FTLD-TDP (8 of 8) and c9ALS (15 of 15) cases analyzed showed Drosha-positive punctate (Fig. 1A, E) and star-shaped (Fig. 1C) NCIs exclusively in granule cell neurons of the dentate gyrus. By contrast, no Drosha-positive NCIs were detected in the FTLD-TDP (0 of 9) and ALS (0 of 12) cases analyzed (Fig. 1 G, H); the same was true for all the age-matched control individuals wherein Drosha was mainly in the nuclear compartment (Figure, Supplemental Digital Content 5, http://links.lww.com/NEN/A718). Moreover, no Drosha NCIs were detected in the hippocampi of patients with other neurodegenerative diseases, including AD, dementia with Lewy bodies, hippocampal sclerosis, or non-TDP43 FTLD (Figure, Supplemental Digital Content 4, http://links.lww.com/NEN/A717). Remarkably, Drosha accumulation as NCIs in c9FTLD-TDP and c9ALS cases did not result in the clearance of Drosha from the nucleus (Fig. 1D). Furthermore, Drosha-immunoreactive dystrophic neurites (DNs) were observed in the molecular layer of the hippocampus in c9FTLD-TDP and c9ALS (Fig. 1B, F). The amount and frequency of Drosha-immunoreactive DNs were similar in all c9FTLD-TDP and c9ALS cases; however, Drosha-immunoreactive DNs were observed in a minority of FTLD-TDP cases (2 of 9) (data not shown) but none in the age-matched controls. Thus, our results suggest that Drosha-positive NCIs but not DNs are specific for cases with C9orf72 mutations.
In the midfrontal cortex, Drosha-immunopositive NCIs were found in c9FTLD-TDP (5 of 5) and c9ALS (4 of 4) cases; however, these NCIs were restricted to a few scattered cells in superficial and deep layers (Fig. 2). None of the FTLD-TDP (0 of 7) or ALS (0 of 4) cases without G4C2 expansion or age-matched control individuals showed Drosha-positive NCIs in that region. Drosha-immunoreactive DNs were also detected in the frontal cortex of all the c9FTLD-TDP (5 of 5) and c9ALS (4 of 4) cases analyzed (Fig. 2A, C, arrowhead) and in a few FTLD-TDP cases (2 of 7) (data not shown). Similar to the finding in the hippocampi, none of the age-matched controls showed Drosha-positive DNs.
In the cerebellum, IHC revealed Drosha-immunopositive NCIs in the granular cell layer of c9FTLD-TDP and c9ALS cases (Fig. 2F, G, arrows). However, unlike the hippocampi and midfrontal cortex, Drosha inclusions in the cerebellum were found in only subsets of c9FTLD (4 of 6) and c9ALS (2 of 8) cases (Table, Supplemental Digital Content 1, http://links.lww.com/NEN/A714). The FTLD and ALS cases without C9orf72 mutation and age-matched controls showed no significant changes in Drosha distribution. Finally, analysis of Drosha distribution in spinal cord samples did not reveal any significant differences between c9ALS, ALS, and control age-matched individuals (data not shown), and no Drosha-positive NCIs were detected in the spinal cord region. Moreover, none of these cases or those without C9orf72 mutations or any of the controls showed changes in DGCR8 protein distribution, which was mainly nuclear (Figure, Supplemental Digital Content 5, http://links.lww.com/NEN/A718).
Drosha Is a New Component of DPR
Next, we compared the distribution of Drosha inclusions and GA and GP DPRs in hippocampus and cerebellum of our autopsy cohort. As reported by others (18), GA- and GP-immunoreactive protein aggregates were only found in c9FTLD-TDP and c9ALS cases (data not shown). Furthermore, side-by-side comparison of Drosha-positive NCI and GA or GP DPRs in consecutive brain sections showed fewer Drosha-positive inclusions compared with GA- and GP-positive DPR aggregates (Fig. 3A–F), suggesting that Drosha mislocalization does not occur in all cells with DPR aggregates.
FIGURE 3.
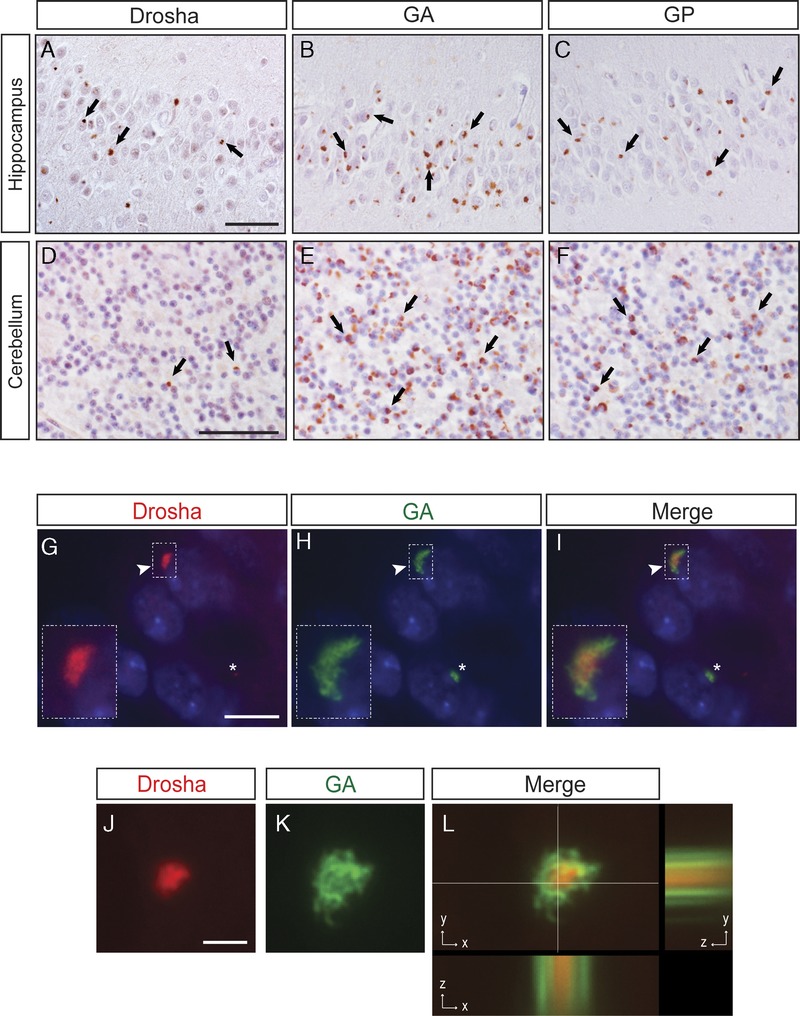
Drosha is a component of dipeptide-repeat (DPR) aggregates. (A–F) Immunohistochemistry (IHC) results for Drosha (A, D), polyGly-Ala (GA) (B, E), and polyGly-Pro (GP) (C, F) proteins in the hippocampus and cerebellum of patients with C9orf72 expansion mutations. Comparative analyses of Drosha, GA, and GP IHC in consecutive brain sections show fewer Drosha-positive inclusions compared with the numbers of GA- and GP-positive aggregates (see examples identified by arrows). (G–L) Double immunofluorescence staining with anti-Drosha (red, G, J, insets) and anti-GA (green, H, K, insets) antibodies in dentate granule neurons shows that Drosha-positive neuronal cytoplasmic inclusions (NCIs) colocalize with GA-immunoreactive aggregates albeit with Drosha in the center of these inclusions that are surrounded by GA positivity (I, arrowhead and inset). Asterisks indicate cell wherein there are GA-positive aggregates that do not contain mislocalized Drosha (G–I). Cell nuclei stained with Dapi (blue). xyz cut of a Drosha-positive NCI (red, J and L) surrounded by GA-immunoreactive material (green, K and L). Scale bars = (G–I) 10 μm; (J–L) 2 μm.
We then analyzed the percentages of Drosha NCIs that contained GA DPR and found that, although not all the DPR aggregates contained mislocalized Drosha, almost all Drosha NCIs are immunopositive for GA DPR in c9FTLD-TDP and c9ALS cases: hippocampus (94.47% ± 11.1%), frontal cortex (94.9% ± 11.67%) and cerebellum (99.1% ± 1.75%). Nonsignificant differences were observed in the percentage of colocalization between the 3 brain areas analyzed. Images in Figure 3 show Drosha-positive NCIs forming the core of inclusions that were surrounded by GA protein immunoreactivity (Fig. 3G–L).
Drosha Inclusions Are Immunopositive for p62 and Ubiquilin-2
The presence of p62-positive/TDP-43–negative NCIs in the hippocampus and cerebellum has been described as a key pathologic feature in c9FTLD-TDP and c9ALS cases (36, 40–47). In addition, NCIs and DNs immunoreactive for the ubiquilin-2 have been found in several brain regions of c9FTLD-TDP and c9ALS cases and are diagnostic signatures of the presence of C9orf72 mutations in these disorders (36). To determine if Drosha, TDP-43, p62, and/or ubiquilin-2 colocalize in the inclusions found in C9orf72 mutation cases, we examined pathologic NCIs in the hippocampus, frontal cortex, and cerebellum for the presence of these proteins. Analysis of double IF of Drosha-positive NCIs in the hippocampus, frontal cortex, and cerebellum of both c9FTLD-TDP and c9ALS cases showed that high percentages of them were also immunopositive for p62: 89.3% ± 11.9%, 97.1% ± 9.2%, and 92.8% ± 6.2%, respectively (Fig. 4A–C). Similar colocalization of Drosha and ubiquilin-2 was also found: 89.1% ± 14.1% in the hippocampus, 94.9% ±18.8% in the frontal cortex, and 99.5% ±1.43% in the cerebellum (Fig. 4D–F). Nonsignificant differences were observed in the percentage of colocalization between the 3 brain areas analyzed. Interestingly, only rare Drosha NCIs were found to colocalize with phosphorylated TDP-43, a hallmark of FTLD-TDP and ALS pathology (data not shown). Moreover, we noted that not all p62- and ubiquilin-2–immunoreactive inclusions contained mislocalized Drosha.
FIGURE 4.
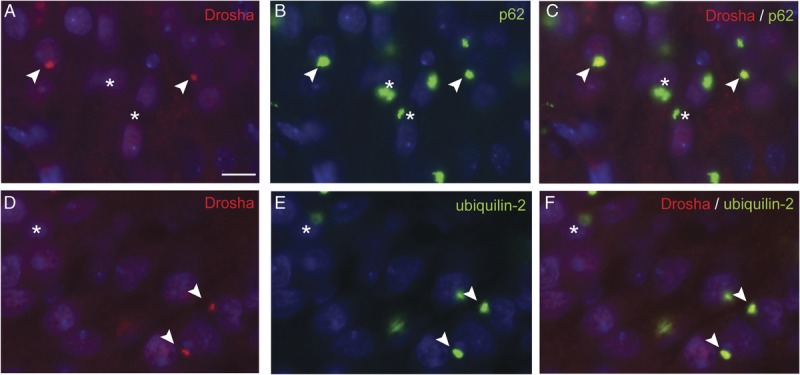
Colocalization of Drosha-positive inclusions with p62 and ubiquilin-2. Double immunofluorescence (IF) results of Drosha, p62, and ubiquilin-2 in the hippocampus of c9FTLD-TDP cases. Double labeling of Drosha (A, D, red) and p62 (B) or ubiquilin-2 (E) in dentate granule neurons shows colocalization of Drosha-positive inclusions with p62 protein (C, arrowhead) and ubiquilin-2 pathology (F, arrowhead). Asterisks show some cytoplasmic aggregates positive for p62 (B, C) or ubiquilin-2 (E, F) protein that did not contain mislocalized Drosha protein (A, D). Cell nuclei are lightly stained with DAPI (blue) to enable visualization of the double label IF results. Scale bar = 10 μm.
DISCUSSION
Here, we describe the presence of mislocalized Drosha protein in NCIs in c9FTLD-TDP and c9ALS cases. Drosha is a Class II RNase III endonuclease and together with its cofactor DGCR8 are the main components of the microprocessor complex involved in the first steps of miRNA biogenesis (48–51). The primary miRNA, which may contain sequences encoding multiple miRNAs, is cleaved in the nucleus by Drosha at the base of the stem-loop into shorter precursor miRNA (52–55). Because the presence of secondary RNA structures forming stem-loops is important to DGCR8 recognition and Drosha processing and because structural studies have shown that G4C2 repeats can form secondary G-quadruplex structures (23, 56–58), we hypothesized a potential RNA-toxic effect of G4C2 expansions recruiting these 2 microprocessor components. Interestingly, Sellier et al (27) showed earlier that, in Fragile X–associated tremor/ataxia syndrome, a trinucleotide repeat expansion-associated disease, the expression of transcripts containing pathologic CGG repeats was found to sequester DGCR8 and Drosha proteins, thereby supporting the idea of an RNA-gain of function.
Initially, the distribution of Drosha and DGCR8 was analyzed in affected brain regions of c9FTLD and c9ALS cases with p62 pathology and abundant DPR-immunoreactive inclusions, including the hippocampus, frontal cortex, and cerebellum and spinal cord (18, 45, 59). Consistent with a canonical role of the microprocessor complex in the nucleus, our IHC analyses in control cases showed that Drosha and DGCR8 proteins were mainly localized in the nuclear subcellular compartment. No significant differences in DGCR8 protein distribution were observed between c9FTLD-TDP or c9ALS cases and age-matched FTLD-TDP, ALS, or control individuals. Interestingly, despite the fact that we hypothesized that Drosha could bind G4C2-rich sequences as potential targets and be sequestered into the nuclear RNA-foci, Drosha protein was mainly mislocalized to the cytoplasm of some neurons in the dentate gyrus of the hippocampus, frontal cortex, and granular layer of the cerebellum in c9FTLD-TDP and c9ALS patients. It is important to point out that Drosha NCIs are only observed in specific neuronal populations affected by DPR inclusions and not in hippocampal pyramidal neurons or cerebellar Purkinje cells, among others, indicating a specific neuronal vulnerability of Drosha mislocalization to the cytoplasm.
The mechanism that drives Drosha protein to accumulate in the cytoplasm of a subset of neurons remains unknown. Similar findings have been reported by Mori et al (25) for hnRNPA3, another nuclear RNA-binding protein, which was also found to form NCIs in patients with C9orf72 mutations. The fact that, in vitro, hnRNPA3 can bind to G4C2-expanded sequences together with its function in mRNA export suggests that hnRNPA3 plays a role in the G4C2 transcript nuclear export. In vitro, other RNA-binding proteins are able to recognize and bind specifically to G4C2 repeats (22–26), but only a few of them, Pur-α, hnRNPH, hnRNPA1, and ADARB2 colocalize with G4C2 repeats RNA-foci in cellular models or in human postmortem tissue (22–26). Although none of the in vitro studies enumerated above identified Drosha as a candidate protein that directly binds to G4C2 repeat sequences, we cannot exclude the possibility that Drosha could bind to the G4C2 repeats transcripts through an unknown cofactor and be transported to the cytoplasm.
Furthermore, redistribution of Drosha protein into the cytoplasm has been associated with the processing of virus-derived cytoplasmic miRNAs, a noncanonical Drosha function independent of DGCR8 protein (50–52). Because virus-derived cytoplasmic miRNAs are able to drive changes in phosphatase-kinase balance in mammalian cells, Drosha was shown to be actively transported between the nucleus and the cytoplasm by a CRM1-dependent manner (60–62). Drosha protein also has been proposed as a vestigial remnant of the small RNA–mediated defense mechanism that is evolutionarily conserved and is involved in the cleavage of mRNA during times of cellular stress (52). In this regard, whether the expression of aberrant G4C2 repeats transcripts could activate analogous signaling pathways linked to cellular stress and defense against foreign RNA remains to be investigated.
Although Drosha accumulates in NCIs in c9FTLD-TDP and c9ALS cases, the complete clearance of Drosha from the nucleus was not observed; however, we cannot rule out the possibility that changes in protein homeostasis could affect the processing of miRNAs and/or RNAs. Indeed, it has been reported that changes in Drosha expression levels are sufficient to modify a variety of miRNA that are processed in cells (63). Recently, it has been reported that TDP-43 protein interacts with Drosha in vitro and its downregulation affects the biogenesis of miRNAs (64). Although it is plausible that Drosha could be sequestered by pathologic TDP-43 species in the cytoplasm, our present results do not support this hypothesis because Drosha-positive NCIs were rarely positive for phosphorylated TDP-43. Furthermore, Drosha-positive NCIs were only found in C9orf72 mutation cases. Although the mechanism that drives both proteins to aggregate in c9FTLD-TDP and c9ALS patients is not known, the disturbance of these 2 functional related proteins involved in RNA/miRNA processing could contribute to abnormal neuronal functions.
Further characterization of Drosha NCIs in the hippocampus, frontal cortex, and cerebellum of c9FTLD-TDP and c9ALS cases revealed that Drosha protein was a component in some GA-positive DPR aggregates. The sequestration of Drosha protein into some DPR aggregates again indicates a potential vulnerability of neuronal population (also described for Unc119 protein [65]) and that could contribute specifically to FTLD and ALS pathology in patients with C9orf72 mutations.
Previous reports showed that cytoplasmic DPR aggregates were surrounded by p62 protein (38, 66), a ubiquitin-binding scaffold protein (Sequestosome 1) that colocalizes with ubiquitinated protein aggregates in many neurodegenerative diseases (41, 47, 67–70). Here we showed by double IF that Drosha-positive inclusions are p62 and ubiquilin-2 positive but TDP-43 negative in the hippocampus, frontal cortex, and cerebellum, although the TDP-43 inclusions are characteristic signatures in c9FTLD-TDP and c9ALS cases. In addition, the fact that Drosha NCIs colocalized with the ubiquitin-like protein ubiquilin-2 could indicate that both DPRs and Drosha are misfolded proteins that are likely to be degraded via the ubiquitin proteasome system; however, the presence of p62 and ubiquilin-2 into Drosha-positive NCIs could be caused by the coaggregation with DPR aggregates.
In summary, in the present study, we show that the mislocalization of Drosha to NCIs is specific for both FTLD-TDP and ALS cases with C9orf72 mutation. The Drosha NCIs also contain DPR proteins, p62 and ubiquilin-2, and rarely pTDP-43. However, not all DPR NCIs are immunoreactive for Drosha. This mislocalization of Drosha to NCIs did not appear to be random or solely as function of pathologic protein misfolding because it is not observed in FTLD-TDP and ALS cases without C9orf72 mutation or other neurodegenerative diseases. Although the mechanism of Drosha aggregation in the cytoplasm is still unknown, our data support the hypothesis that this is directly related to DPR aggregation. Thus, although the presence of Drosha NCIs might not be a diagnostic tool compared with DPR NCIs, it may increase our understanding on the contribution of DPR proteins and their aggregations in c9FTLD and c9ALS. Our finding of Drosha sequestration in DPR aggregates in c9FTLD and c9ALS supports the idea that disturbance of RNA/miRNA processing may play an important role in these 2 neurodegenerative diseases.
ACKNOWLEDGMENTS
The authors thank the patients and their families for their contributions. The authors thank Colin Bredenberg for his helpful assistance. The authors also thank Dr. Edbauer for kindly providing the anti-GA 5 F2 antibody, Drs. Manuela Neumann and Elizabeth Kremmer for providing the phosphorylation-specific TDP-43 rat monoclonal antibody TAR5P-1D3, and Dr. Tuschl for supplying the Myc-Drosha plasmid. VM-YL is the John H. Ware, 3rd, Professor of Alzheimer’s Disease Research. JQT is the William Maul Measey-Truman G. Schnabel, Jr, Professor of Geriatric Medicine and Gerontology.
Footnotes
This study was supported by the National Institutes of Health (grants AG017586, AG010124, and AG032953) and the Koller Family Foundation.
The authors declare no conflict of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.jneuropath.com).
REFERENCES
- 1. Ferrari R, Kapogiannis D, Huey ED, et al. FTD and ALS: A tale of two diseases. Curr Alzheimer Res 2011; 8: 273– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Woollacott IO, Mead S. The C9ORF72 expansion mutation: Gene structure, phenotypic and diagnostic issues. Acta Neuropathol 2014; 127: 319– 32 [DOI] [PubMed] [Google Scholar]
- 3. Lagier-Tourenne C, Polymenidou M, Hutt KR, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 2012; 15: 1488– 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9: 995– 1007 [DOI] [PubMed] [Google Scholar]
- 5. Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857– 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004; 36: 377– 81 [DOI] [PubMed] [Google Scholar]
- 7. Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916– 19 [DOI] [PubMed] [Google Scholar]
- 8. Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442: 920– 24 [DOI] [PubMed] [Google Scholar]
- 9. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477: 211– 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morris HR, Waite AJ, Williams NM, et al. Recent advances in the genetics of the ALS-FTLD complex. Curr Neurol Neurosci Rep 2012; 12: 243– 50 [DOI] [PubMed] [Google Scholar]
- 11. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245– 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011; 72: 257– 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cruts M, Gijselinck I, Van Langenhove T, et al. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci 2013; 36: 450– 59 [DOI] [PubMed] [Google Scholar]
- 14. Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol 2012; 11: 54– 65 [DOI] [PubMed] [Google Scholar]
- 15. van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: Geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013; 34: 363– 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013; 92: 345– 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol 2012; 11: 323– 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013; 77: 639– 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gendron TF, Belzil VV, Zhang YJ, et al. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol 2014; 127: 359– 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 2014; 127: 347– 57 [DOI] [PubMed] [Google Scholar]
- 21. Neumann M. Frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Molecular similarities and differences. Rev Neurol (Paris) 2013; 169: 793– 98 [DOI] [PubMed] [Google Scholar]
- 22. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013; 80: 415– 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014; 507: 195– 200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 2013; 5: 1178– 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mori K, Lammich S, Mackenzie IR, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 2013; 125: 413– 23 [DOI] [PubMed] [Google Scholar]
- 26. Xu Z, Poidevin M, Li X, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 2013; 110: 7778– 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sellier C, Freyermuth F, Tabet R, et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 2013; 3: 869– 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hebert SS, Horre K, Nicolai L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A 2008; 105: 6415– 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J, Inoue K, Ishii J, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007; 317: 1220– 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martí E, Pantano L, Bañez-Coronel M, et al. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res 2010; 38: 7219– 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miñones-Moyano E, Porta S, Escaramis G, et al. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 2011; 20: 3067– 78 [DOI] [PubMed] [Google Scholar]
- 32. Nunez-Iglesias J, Liu CC, Morgan TE, et al. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One 2010; 5: e8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shioya M, Obayashi S, Tabunoki H, et al. Aberrant microRNA expression in the brains of neurodegenerative diseases: MiR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol 2010; 36: 320– 30 [DOI] [PubMed] [Google Scholar]
- 34. Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014; 10: 477– 84e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011; 122: 111– 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brettschneider J, Van Deerlin VM, Robinson JL, et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol 2012; 123: 825– 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Deerlin VM, Gill LH, Farmer JM, et al. Familial frontotemporal dementia: From gene discovery to clinical molecular diagnostics. Clin Chem 2003; 49: 1717– 25 [DOI] [PubMed] [Google Scholar]
- 38. Mackenzie IR, Arzberger T, Kremmer E, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol 2013; 126: 859– 79 [DOI] [PubMed] [Google Scholar]
- 39. Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009; 117: 137– 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol 2013; 126: 385– 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011; 122: 691– 702 [DOI] [PubMed] [Google Scholar]
- 42. Cooper-Knock J, Hewitt C, Highley JR, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012; 135: 751– 64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mahoney CJ, Beck J, Rohrer JD, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: Clinical, neuroanatomical and neuropathological features. Brain 2012; 135: 736– 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 2011; 122: 673– 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pikkarainen M, Hartikainen P, Alafuzoff I. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol 2008; 67: 280– 98 [DOI] [PubMed] [Google Scholar]
- 46. Stewart H, Rutherford NJ, Briemberg H, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012; 123: 409– 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Troakes C, Maekawa S, Wijesekera L, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 2012; 32: 505– 14 [DOI] [PubMed] [Google Scholar]
- 48. Denli AM, Tops BB, Plasterk RH, et al. Processing of primary microRNAs by the Microprocessor complex. Nature 2004; 432: 231– 35 [DOI] [PubMed] [Google Scholar]
- 49. Gregory RI, Yan KP, Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004; 432: 235– 40 [DOI] [PubMed] [Google Scholar]
- 50. Han J, Lee Y, Yeom KH, et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 2004; 18: 3016– 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 2004; 14: 2162– 67 [DOI] [PubMed] [Google Scholar]
- 52. Bohnsack MT, Czaplinski K, Gorlich D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004; 10: 185– 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lund E, Guttinger S, Calado A, et al. Nuclear export of microRNA precursors. Science 2004; 303: 95– 98 [DOI] [PubMed] [Google Scholar]
- 54. Yi R, Qin Y, Macara IG, et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003; 17: 3011– 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zeng Y, Cullen BR. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res 2004; 32: 4776– 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fratta P, Mizielinska S, Nicoll AJ, et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2012; 2: 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reddy K, Zamiri B, Stanley SY, et al. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem 2013; 288: 9860– 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zamiri B, Reddy K, Macgregor RB, Jr, et al. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem 2014; 289: 4653– 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pikkarainen M, Hartikainen P, Alafuzoff I. Ubiquitinated p62-positive, TDP-43–negative inclusions in cerebellum in frontotemporal lobar degeneration with TAR DNA binding protein 43. Neuropathology 2010; 30: 197– 99 [DOI] [PubMed] [Google Scholar]
- 60. Shapiro JS. Processing of virus-derived cytoplasmic primary-microRNAs. Wiley Interdiscip Rev RNA 2013; 4: 463– 71 [DOI] [PubMed] [Google Scholar]
- 61. Shapiro JS, Langlois RA, Pham AM, et al. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA 2012; 18: 1338– 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shapiro JS, Schmid S, Aguado LC, et al. Drosha as an interferon-independent antiviral factor. Proc Natl Acad Sci U S A 2014; 111: 7108– 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sperber H, Beem A, Shannon S, et al. miRNA sensitivity to Drosha levels correlates with pre-miRNA secondary structure. RNA 2014; 20: 621– 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A 2012; 109: 3347– 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS–associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 2014; 128: 485– 503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mann DM, Rollinson S, Robinson A, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2013; 1: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. King A, Maekawa S, Bodi I, et al. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology 2011; 31: 239– 49 [DOI] [PubMed] [Google Scholar]
- 68. Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012; 135: 723– 35 [DOI] [PubMed] [Google Scholar]
- 69. Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012; 135: 693– 708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wooten MW, Hu X, Babu JR, et al. Signaling, polyubiquitination, trafficking, and inclusions: Sequestosome 1/p62’s role in neurodegenerative disease. J Biomed Biotechnol 2006; 2006: 62079. [DOI] [PMC free article] [PubMed] [Google Scholar]