

Abstract
Inhibition of the functions of L1 cell adhesion molecule (L1) by ethanol has been implicated in the pathogenesis of the neurodevelopmental aspects of the fetal alcohol syndrome (FAS). Ethanol at pharmacological concentrations has been shown to inhibit L1-mediated neurite outgrowth of rat post-natal day 6 cerebellar granule cells (CGN). Extracellular signal-related kinases (ERK) 1/2 activation occurs following L1 clustering. Reduction in phosphoERK1/2 by inhibition of mitogen-activated protein kinase kinase (MEK) reduces neurite outgrowth of cerebellar neurons. Here, we examine the effects of ethanol on L1 activation of ERK1/2, and whether this activation occurs via activation of fibroblast growth factor receptor 1 (FGFR1). Ethanol at 25 mm markedly inhibited ERK1/2 activation by both clustering L1 with cross-linked monoclonal antibodies, or by L1-Fc chimeric proteins. Clustering L1 with subsequent ERK1/2 activation did not result in tyrosine phosphorylation of the FGFR1. In addition, inhibition of FGFR1 tyrosine kinase blocked basic fibroblast growth factor (bFGF) activation of ERK1/2, but did not affect activation of ERK1/2 by clustered L1. We conclude that ethanol disrupts the signaling pathway between L1 clustering and ERK1/2 activation, and that this occurs independently of the FGFR1 pathway in cerebellar granule cells.
Keywords: ethanol, fetal alcohol syndrome, L1 cell adhesion molecule, mitogen-activated protein kinases, signal transduction
Heavy drinking during pregnancy is the cause of fetal alcohol syndrome (FAS), the leading known cause of mental retardation (Abel and Sokol 1991). Drinking during pregnancy can also result in a spectrum of effects known as alcohol-related developmental disorder, which range from severe cognitive and behavioral impairment without the classic facial dysmorphology to relatively subtle neurobehavioral deficits (Stratton et al. 1996). It is estimated that 1% of all newborns are affected by prenatal ethanol exposure (Sampson et al. 1997). The L1 cell adhesion molecule (L1) has been implicated in the pathogenesis of FAS as a result of an extensive overlap of neuroanatomic features (Bearer 2001). In vitro studies have suggested that ethanol decreases L1-mediated cell adhesion (Ramanathan et al. 1996; Wilkemeyer et al. 2003). In other systems, L1 adhesivity is unaffected by ethanol (Vallejo et al. 1997; Bearer et al. 1999b). Ethanol has been shown to inhibit L1-mediated neurite outgrowth of cerebellar granule cells (Bearer et al. 1999b; Watanabe et al. 2004). We hypothesize that one mechanism of alcohol teratogenesis is the inhibition of L1 function via inhibition of L1 signaling cascades.
L1 is a 220-kDa member of the immunoglobulin (Ig) superfamily of cell adhesion molecules (Lindner et al. 1983), whose functions include cell adhesion, neurite outgrowth, axon fascicle formation, and neural migration (Lindner et al. 1983; Wong et al. 1995; Kamiguchi 2003). L1 is a cell surface glycoprotein and is expressed on post-mitotic neurons. L1 binds homophilically or heterophilically to itself or a number of other binding partners, both in cis and in trans (Lemmon et al. 1989; Kuhn et al. 1991; Brummendorf et al. 1993; Felsenfeld et al. 1994; Milev et al. 1996; Malhotra et al. 1998; Castellani et al. 2000). Initially, adhesion molecules were thought to influence axonal growth, based on their ability to mediate adhesive interactions. However, a number of experiments have shown that the response of a neurite to naturally occurring substrates such as fibronectin, laminin, or L1 cannot be predicted based on the adhesiveness of the neurite–substrate interactions (Lemmon et al. 1992; Zheng et al. 1994). Instead, the neurite responses are because of the activation of signaling cascades and endocytosis following binding of the adhesion molecule (Schuch et al. 1989; Von Bohlen Und Halbach et al. 1992; Williams et al. 1992; Kamiguchi and Yoshihara 2001; Long et al. 2001).
There is evidence for two signaling pathways for L1. In one signaling pathway, L1 is endocytosed (Schaefer et al. 1999), leading to the activation of P13 kinase, Rac1, mitogen-activated protein kinase kinase (MEK) and extracellular signal-regulated kinases (ERK1/2) (Schmid et al. 2000). Inhibition of this pathway by inhibition of ERK1/2 activation reduced mean neurite length of mouse cerebellar granule cells (CGN) by 56% (Schmid et al. 2000). The second pathway proposes a cascade common to the neurite promoting activity of several cell adhesion molecules (for review, see Doherty and Walsh (1996). The fibroblast growth factor receptor 1 (FGFR1) tyrosine kinase has been reported to be activated by L1, neural cell adhesion molecule (NCAM) and N-cadherin as well as basic fibroblast growth factor (bFGF). Binding of L1 increases the phosphorylation of FGFR1. Phosphorylation activates the FGFR1 tyrosine kinase which initiates a cascade of events leading to arachidonic acid release and resulting in neurite outgrowth.
In this study, we investigate the effect of ethanol on L1-mediated activation of ERK1/2, and whether L1 activates ERK1/2 via the FGFR1. Here, we have identified the ERK1/2 pathway as a target for ethanol inhibition of neurite outgrowth, and provided evidence that one of the two proposed L1 pathways mediates this inhibition in a simple, well-characterized cell system.
Materials and methods
Antibodies
Antibodies used in this research were the following: rabbit polyclonal antibody against the cytoplasmic domain of L1 (Schaefer et al. 1999) (anti-L1CD), rabbit polyclonal antibody against dually phosphorylated, activated ERK1/2 (New England Biolabs, Ipswich, MA, USA; anti-phosphoERK1/2), rabbit polyclonal antibodies against ERK2 (anti-ERK2) and a peptide of FGFR1 (anti-flg sc-121), as well as the peptide itself which is used to block the binding of the antibody to FGFR1 (anti-flg blocking peptide sc-121 P) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal antibodies to FGFR1 (Upstate Biotechnology, Lake Placid, NY, USA), mouse monoclonal antibody to phosphotyrosine (PY20) (Sigma, St Louis, MO, USA), goat anti-mouse IgG conjugated to Oregon Green (Molecular Probes, Eugene, OR, USA), human IgG (Fc), rabbit polyclonal antibody to mouse IgG, rabbit polyclonal antibody to mouse IgG (H+L) conjugated to hydrogen peroxidase, and goat polyclonal antibodies to rabbit IgG (H+L) conjugated to hydrogen peroxidase (all from Jackson Immuno-Research, West Grove, PA, USA).
Mouse monoclonal antibody to rat L1 (ASCS4) was produced from a hybridoma cell line developed by P. H. Patterson and obtained from the Developmental Studies Hybridoma Bank which was developed under the auspices of the National Institute of Child Health and Development and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA, USA. The cells were grown in Iscove’s modified Dulbecco’s medium, 20% fetal bovine serum (FBS) with penicillin/streptomycin, then seeded into the CELLMAX system (Cellco Inc., Rancho Dominguez, CA, USA). The serum content of the media was gradually decreased. Media was harvested weekly, and antibody was affinity purified using Protein-G sepharose. ASCS4 has not been previously characterized for its binding site on L1.
Preparation of L1-Fc
L1-Fc containing plasmid was prepared as previously described (Bearer et al. 1999b). The plasmid was stably expressed in NIH 3T3 cells using lipofectamine. Cells were co-transfected with the neomycin-resistant plasmid, pMAMneo (ratio 1 : 9) (a gift from Susann Brady-Kalnay, Case Western Reserve University, Cleveland, OH, USA). Transfected cells were selected with G418 (Gibco, Rockville, MD, USA) at 0.9 mg/mL. Clones were selected and subcloned by limiting dilution. Supernatants were assayed for L1-Fc. A stably transfected clone expressing L1-Fc was propagated using the CELLMAX system (Cellco Inc.) using the manufacturer’s instructions. Serum in the media in the CELLMAX system was gradually removed. L1-Fc was purified from the harvested serum-free media by modification of the method of Sakurai et al. (1998). Briefly, proteins were ammonium sulfate precipitated, then re-suspended in 17 mm NaH2PO4, pH 6.3 (Buffer A), and dialyzed overnight at 4°C. Twenty milliliters of 50% DE52 slurry in Buffer A was added to the dialysate, and rocked at 4°C overnight. The DE52 slurry was centrifuged, and washed three times with Buffer A. L1-Fc was eluted with Buffer A containing 0.4 m KCl and the protein concentration of the eluant was determined. Protein A-conjugated sepharose beads at 0.1 mL/mg protein were added, the pH adjusted with 0.1 mL 1 m HEPES, pH 8.0 per ml of eluant and Na azide added to give a final concentration of 0.01%. The slurry is rocked overnight and stored at 4°C until use. To determine purity and L1-Fc content, an aliquot of the L1-Fc-bound sepharose beads was washed with phosphate-buffered saline, and L1-Fc was eluted with 50 mm glycine, pH 2.80 into test tubes containing 1 m HEPES, pH 8.0 to neutralize the pH. Protein concentration was determined by the bicinchoninic acid assay (Pierce, Rockford, IL, USA). The purity and immunoreactivity of remaining eluted L1-Fc was determined by polyacrylamide electrophoresis and, either silver stained for protein, or by western blot using anti-Fc antibodies for immunoreactivity. For each experiment, the appropriate amount of L1-Fc-containing protein A-sepharose slurry was eluted for L1-Fc as described. Following elution, an aliquot of L1-Fc was taken for protein determination, and 1% bovine serum albumin (BSA) was added to each fraction. Soluble L1-Fc prepared in this manner was added to cell cultures within 1 h of elution.
Preparation of CGN cells
Rat CGN were obtained from post-natal day 6 Sprague–Dawley rat pups (Zivic–Miller) as previously described (Bearer et al. 1999b). To minimize pain and discomfort, pups were rapidly decapitated as approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and guidelines from the US Public Health Service. Viability was assessed with trypan blue and was > 90%. These cells have been extensively characterized as > 90% CGN (Hockberger et al. 1987; Beattie and Siegel 1993; Nakai and Kamiguchi 2002).
Immunofluorescence
CGN grown overnight on poly l-lysine coated 8-well Lab-Tek slides in DMEM/10% FBS (HyClone, Logan, UT, USA) were placed on ice. Affinity-purified ASCS4 (30 μg) was added to the wells, and incubated for 1 h. Cells were fixed with 4% paraformaldehyde at 4°C, then stained with fluorescent secondary antibodies, equilibrated in Slow-Fade, and observed using a Nikon Optiphot-2 fluorescent microscope at × 20.
ERK1/2 activation
ERK1/2 was activated by addition of a cross-linked anti-L1 monoclonal antibody as described by Schmid et al. (2000). To cluster L1 with ASCS4, 6 × 105 cells were plated on poly l-lysine-coated 60-mm tissue culture dishes in DMEM with 10% FBS and 20 mm HEPES, pH 7.2 and grown overnight. Two hours prior to the addition of clustering antibody, the media was removed and replaced with DMEM, 20 mm HEPES, pH 7.2 to reduce background phosphoERK1/2. To form multimeric complexes, ASCS4 was cross-linked by mixing with rabbit anti-mouse IgG (1 : 2.5 g/g). The mixture was incubated for 1 h at 4°C prior to addition to cells. Mouse IgG was used as control as previously described (Schmid et al. 2000). Ten minutes after addition of the cross-linked ASCS4 (clASCS4) or mouse IgG, the cells were placed on ice, the media was removed, the cells were washed with ice-cold Hank’s balanced salt solution (HBSS), and cell lysates were prepared. For activation of ERK1/2, 50 ng/mL bFGF was added to the cells. For experiments to inhibit bFGF activation of ERK1/2, 3-(4-dimethylamino-benzylidenyl)-2-indolinone (DMBI) was added 20 min prior to the addition of bFGF or clASCS4.
To cluster L1 using L1-Fc, 2 × 105 CGN were plated on poly l-lysine-coated 35-mm tissue culture dishes in serum-free defined media consisting of neurobasal media (Gibco) with the following additions: B27 supplement (Gibco), 20 mm l-glutamine, 6 g/L glucose, 20 mm HEPES, pH 7.2, penicillin/streptomycin. Serum-free defined media was used in these experiments to duplicate the assay conditions where we found the greatest inhibition of ethanol on L1-mediated neurite outgrowth (Bearer et al. 1999a). Cells were grown overnight in a humidified 37°C with 10% CO2. Freshly eluted L1-Fc in 1% BSA or elution buffer with 1% BSA alone was added to the appropriate dishes. At the indicated times, tissue culture dishes were placed on ice, the media was removed, the cells were washed with ice-cold HBSS, and cell lysates were prepared.
Determination of tyrosine-phosphorylated FGFR1
For determination of phosphorylated FGFR1, 7 × 106 CGN were plated on poly l-lysine-coated 10-cm tissue culture dishes in serum-free defined media. ClASCS4 prepared as described above, or bFGF 50 ng/mL, were added to the media. At the indicated times, cells were removed from the incubator, placed on ice, washed with ice-cold HBSS, and cell lysates were prepared.
Preparation of cell lysates
All procedures were at 4°C. Cells were extracted in lysis buffer consisting of: 20 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 10% glycerol, 10 mm Na-vanadate, 10 mm NaF, 2 mm aprotinin, 0.1 mm phenylmethylsulfonyl fluoride, 1 μm leupeptin, 1 μg/mL pepstatin, 10 μg/mL turkey trypsin inhibitor, phosphatase inhibitor cocktail I (Sigma) and phosphatase inhibitor cocktail II (Sigma). Cell extracts were incubated for 30 min and then centrifuged in a microfuge at maximum speed for 10 min.
Immunoprecipitation of proteins
The cell lysate supernatants were transferred to clean microfuge tubes and pre-cleared with 1 μg rabbit IgG or mouse IgG for 1 h, then with 30 μL Protein A–agarose for 1 h, followed by low-speed centrifugation. Supernatants were collected, and 2 μg anti-phosphotyrosine antibody was added. After 1 h incubation, 30 μL of protein A–agarose was added and lysates were incubated overnight. The tubes were centrifuged in a microfuge at 1000 g for 4 min. In some cases, the supernatants were saved for phosphoERK1/2 determination. The pellets were washed with lysis buffer, and boiled for 5 min in sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer. The samples were separated by SDS–PAGE (4–15% gradient gel), transferred to a polyvinylidene difluoride membrane, and the membrane was blocked in Tris-buffered saline containing 2% BSA and 0.1% Tween-20. The membrane was incubated with antibodies to FGFR1, washed, probed with horseraddish peroxidase (HRP)-goat anti-rabbit IgG, and reactive proteins were visualized by chemiluminescence. In some cases, the blocking peptide sc-121 P was added to the anti-FGFR1 immunoblotting antibody as per manufacturer’s instructions prior to addition to the immunoblots.
SDS–PAGE/western blotting for phosphoERK1/2 and ERK2
Proteins were precipitated from cell lysate and immunoprecipitation supernatants by addition of methanol–chloroform. Precipitants were dried, washed with lysis buffer, and boiled for 5 min in SDS–PAGE sample buffer. The samples were separated by SDS–PAGE (12% gel) and transferred to a PDVF membrane. The membrane was blocked in Tris-buffered saline containing 2% BSA and 0.1% Tween-20. The membrane was incubated with antibodies to dually phosphorylated ERK1/2, washed, probed with HRP-goat anti-rabbit IgG, and reactive proteins were visualized by chemiluminescence. Blots were stripped and re-probed with anti-ERK2 antibodies to assess protein loading. The relative intensity of the bands was quantified using transmittance densitometry using Kodak 1D software. The phosphoERK1/2 band densities were normalized for the amount of total ERK2 protein for all quantitative analyses.
Results
L1 activation of the ERK1/2 pathway was investigated in primary cultures of rat post-natal day 6 CGN. Dimerization has been shown to initiate signaling by members of the Ig superfamily such as NCAM, integrins and some receptor tyrosine kinases (Heldin 1995). Clustering of L1 in mouse CGN by addition of cross-linked monoclonal antibodies to mouse L1 resulted in significant increases in phosphoERK1/2 (Schmid et al. 2000). We first sought to characterize a monoclonal antibody ASCS4 (Yamazaki et al. 1997) to rat L1, and then to determine if ASCS4 could activate ERK1/2 in rat cerebellar granule neurons. On living CGN, uncross-linked ASCS4 bound to the cell surface (Fig. 1a), suggesting that ASCS4 recognized a site on the extracellular domain of L1.
Fig. 1.
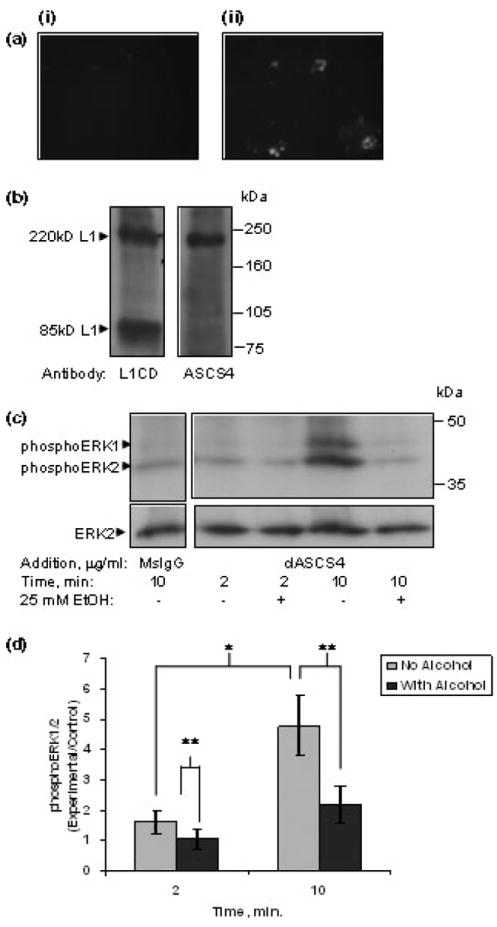
(a) ASCS4 binds to the surface of living cells. Cerebellar granule cells grown in the presence of 10% FBS were cooled to 4°C. Affinity-purified ASCS4 (not clustered, a monoclonal antibody to L1) was added to the media, and incubated for 1 h. Cells were subsequently fixed, and probed with fluorescent secondary antibodies. (i) No ASCS4 was added to cells; (ii) patchy distribution of ASCS4 staining is seen. (b) ASCS4 recognizes an extracellular epitope. Western blots of rat whole brain lysate were probed with a polyclonal antibody to the cytoplasmic domain of L1 (L1CD) or with ASCS4. The blots were probed with HRP-conjugated secondary antibodies, and reactive proteins visualized by chemiluminesence. L1CD recognizes the 220 kDa full-length L1 protein as well as the 85-kDa fragment which represents the carboxy terminal protein following cleavage in the third fibronectin domain. In contrast, ASCS4 only recognizes the 220-kDa full-length L1. (c) ERK1/2 is activated by cross-linked ASCS4 which is inhibited by ethanol. Serum-starved cerebellar granule cells were treated with 25 mm ethanol for 1 h, then either mouse IgG, or clASCS4 was added for 2 or 10 min. ERK1/2 activation was assayed by western blot analysis. Western blots showed that addition of cross-linked ASCS4 increased activation of ERK1/2 (phosphoERK1/2) at 10 min and a marked inhibition of ASCS4 mediated ERK1/2 activation in ethanol pretreated cells (one of seven representative blots shown). (d) PhosphoERK1/2 is significantly increased at 10 min following addition of cross-linked ASCS4. Pretreatment (1 h) with 25 mm ethanol significantly reduces phosphoERK1/2. Densitometric quantification of ERK1/2 phosphorylation corrected for total ERK1/2 shown in (c) is plotted as relative densitometric units relative to the MsIgG control. The values of seven separate experiments are shown. The bar indicates the mean of the values ± SE. *A statistically significant increase in ERK1/2 activation at 10 min following addition of cross-linked ASCS4 versus 2 min (p < 0.003, paired t-test); **a statistically significant decrease in ERK1/2 activation following alcohol pretreatment (p < 0.02, paired t-test).
In addition, western blot analysis of whole rat brain lysate showed distinct bands when immunoblotted with ASCS4 versus rabbit anti-L1CD, an antibody to the cytoplasmic domain of L1 (Schaefer et al. 1999) (Fig. 1b). Both antibodies recognize the full-length L1 at 220 kDa. In addition, rabbit anti-L1CD recognizes the 85-kDa fragment of L1 which contains the fourth and fifth fibronectin domains, the transmembrane domain and the cytoplasmic domain (Sadoul et al. 1988; Silletti et al. 2000). The lack of binding of ASCS4 to the 85 kDa polypeptide fragment indicates that the antibody’s binding site is on the NH2 terminal extracellular domain before the plasmin-sensitive dibasic sequences beginning at amino acid 840 (Silletti et al. 2000).
Using clASCS4 to induce clustering of L1, we found a significant increase in phosphoERK1/2 at 10 min. ASCS4 was first cross-linked by the addition of rabbit anti-mouse IgG, and then added to CGN. PhosphoERK1/2 was not significantly increased at 2 min. However, a significant activation of ERK1/2 was found at 10 min following the addition of antibody (Figs 1c and d) consistent with the findings of Schmid et al. (2000).
Next, we investigated the effect of ethanol on L1-mediated activation of ERK1/2. We have previously shown that ethanol inhibits L1-mediated neurite outgrowth with a maximal effect at a concentration of 25 mm (Bearer et al. 1999b). We have found increased inhibition of L1-mediated neurite outgrowth with a 1 h pre-incubation of neurons in 25 mm ethanol prior to addition of L1-Fc to promote neurite outgrowth (unpublished observations). Schmid et al. (2000) showed that inhibition of ERK1/2 activation using the MEK specific inhibitors PD98059 or U0125 inhibited neurite outgrowth on L1. To determine whether ethanol might inhibit L1-mediated neurite outgrowth by inhibiting L1 activation of ERK1/2, 25 mm ethanol was added to CGN for 1 h, then clASCS4 was added, and the phosphorylation status of ERK1/2 was determined at 2 and 10 min. The amount of phosphoERK1/2 was significantly reduced in the presence of ethanol both at 2 and 10 min following addition of clASCS4 (Figs 1c and d). The inhibition at 2 min in the absence of L1-related ERK1/2 activation may indicate that ethanol inhibits other factors from activating ERK1/2 which are not directly being tested in this system.
To determine if other means of clustering L1 and activating ERK1/2 are also sensitive to ethanol, we investigated the effect of the addition of L1-Fc on activation of ERK1/2. L1-Fc contains disulfide bonds within the Fc region and hence is composed of two L1 extracellular domains. In addition, multimerization of L1-Fc into trimeric and higher order complexes may occur through interactions of the third fibronectin repeat (Silletti et al. 2000). Thus, binding of L1-Fc to cell surface L1 would be expected to dimerize and/or multimerize the cell surface L1 and activate ERK1/2. L1-Fc increased the phosphorylation of ERK1/2 with a maximum attained at 10 min, corrected for protein loading by measuring total ERK2 (data not shown). This finding was consistent with findings by Schaefer et al. (1999) and Schmid et al. (2000). Both 10 and 20 μg/mL of L1-Fc added to the media increased the phosphorylation of ERK1/2 at 10 min (Figs 2a and b). As a control, human Fc was added to the cells at 20 μg/mL, and ERK1/2 was assayed after 10 min. No activation of ERK1/2 was seen above that of vehicle alone (Figs 2c and d), while L1-Fc significantly increased phosphoERK1/2 (Figs 2c and d).
Fig. 2.
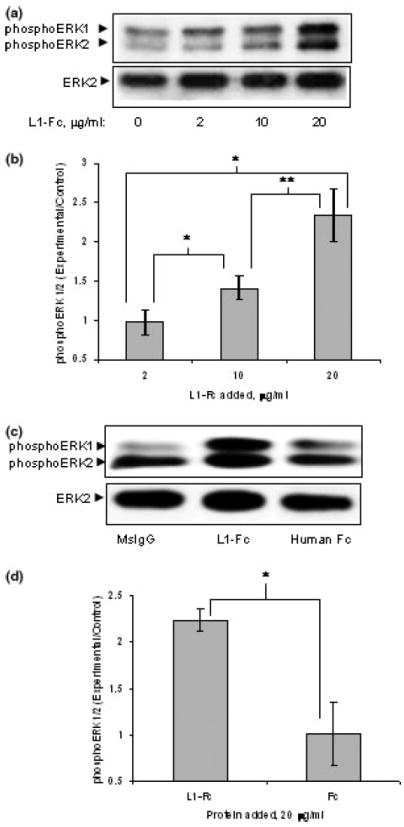
(a) L1-Fc activates ERK1/2. ERK1/2 activation (phosphoERK1/2) was seen with increasing amounts of L1-Fc added to the media, assayed at 10 min following addition. Vehicle alone is denoted as 0 μg/mL, first lane. (b) L1-Fc significantly increases phosphoERK1/2 at 10 and 20 μg/mL. Densitometric quantification of ERK1/2 phosphorylation corrected for total ERK2 shown in (a) is plotted as relative densitometric units relative to the vehicle alone control. The values of five separate experiments are shown. L1-Fc (2 μg/mL) was not significantly different than vehicle alone. However, both 10 and 20 μg/mL significantly increased phosphoERK1/2 (*p < 0.005, paired t-test), and activation with 20 μg/mL was significantly greater than that with 10 μg/mL (**p < 0.01, paired t-test). (c) As a control, 20 μg/mL human Fc was added to cells, and assayed for ERK1/2 activation at 10 min. One of three representative western blots is shown. (d) Densitometric quantification of ERK1/2 phosphorylation corrected for total ERK2 as shown in (c) is plotted as relative densitometric units relative to the vehicle alone control. The values from three separate experiments are shown. The bar indicates the mean ± SE. The asterisk indicates a statistically significant increase in ERK1/2 activation in the presence of 20 μg/mL of L1-Fc versus Fc (p < 0.02, paired t-test). There was no significant increase with Fc alone.
Next, we determined the effect of ethanol on L1-Fc activation of ERK1/2. As shown in Fig. 3(a and b), ethanol significantly reduced phosphoERK1/2 following addition of either vehicle alone or 20 μg/mL L1-Fc. Thus, both clAS-CS4 and L1-Fc dimer activation of ERK1/2 are significantly inhibited by ethanol. These results indicate that the mechanism of inhibition is most likely to involve the cell surface L1, and not be an effect of ethanol on the cross-linked antibody or L1-Fc.
Fig. 3.
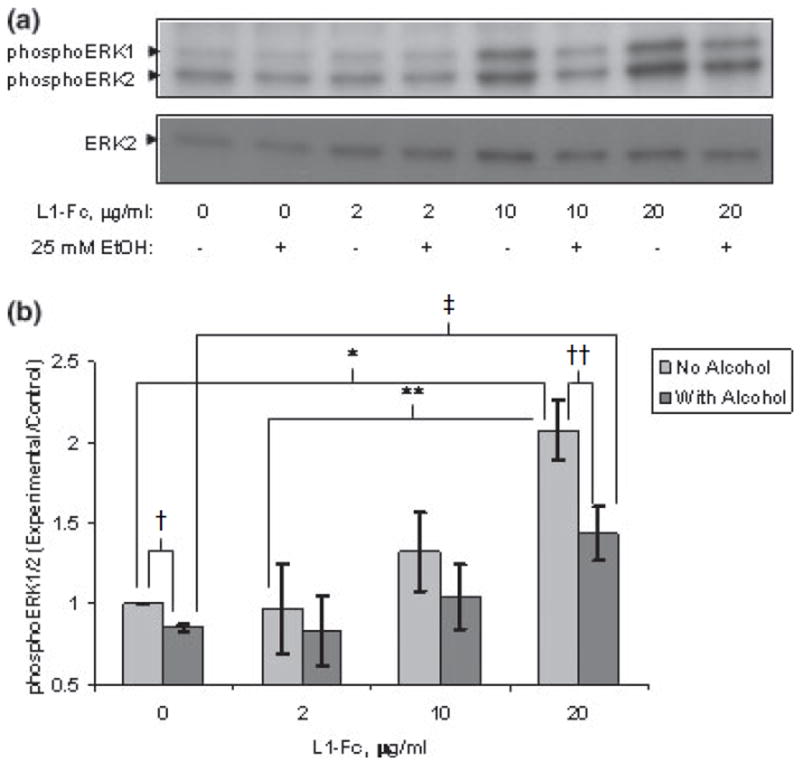
(a) Ethanol inhibits L1-Fc activation of ERK1/2. Cerebellar granule cells grown in serum-free defined media were treated with or without 25 mm ethanol for 1 h prior to addition of L1-Fc at increasing concentrations. ERK1/2 activation (phosphoERK1/2) was assayed by western blot analysis after addition of L1-Fc or vehicle alone. A representative blot of four experiments is shown. (b) Ethanol significantly inhibits L1-Fc activation of ERK1/2. Densitometric quantification of ERK1/2 phosphorylation corrected for total ERK2 shown in (a) is plotted as relative densitometric units relative to the vehicle alone control. The values of four separate experiments are shown. The bar indicates the mean of the values ± SE. Addition of 20 μg/mL of L1-Fc significantly increased phosphoERK1/2 above both addition of vehicle alone (*p < 0.005, paired t-test) and 2 μg/mL L1-Fc (**p < 0.002, paired t-test). Ethanol pretreatment of 25 mm for 1 h prior to addition of L1-Fc significantly reduced phosphoERK1/2 both in the vehicle alone (†p < 0.001, paired t-test) and 20 μg/mL L1-Fc-added cells (††p < 0.0004, paired t-test). PhosphoERK1/2 was significantly greater in the 20 μg/mL L1-Fc + ethanol cells compared with the vehicle alone + ethanol cells (‡p < 0.024, paired t-test).
It has been proposed that L1 promotes neurite outgrowth by signaling via the FGFR1 (Doherty and Walsh 1996). Binding of bFGF to FGFR1 also activates ERK1/2 (Jung et al. 2003). To determine the site of ethanol inhibition, we sought to determine if L1 activation of ERK1/2 in CGN was dependent on the activation of FGFR1. The FGFR1 was identified in anti-phosphotyrosine immunoprecipitates of CGN stimulated with 50 ng/mL bFGF by showing that the immunoreactivity of a band with the expected molecular weight of 123 kDa was absent when a specific peptide inhibitor of the primary antibody was added to the immunoblotting solution (Fig. 4a). Addition of bFGF to culture media resulted in a rapid tyrosine phosphorylation of the FGFR1 in CGN. Tyrosine phosphorylation of FGFR1 occurred 2 min following the addition of the growth factor, and was reduced or absent at 10 min post-addition (Figs 4b and c). Tyrosine phosphorylation of FGFR1 was examined at 2 and 10 min following L1 clustering to coincide with both the phosphorylation of FGFR1 and ERK1/2 activation. No increase in tyrosine phosphorylation of the FGFR1 was seen at 2 or 10 min following the addition of clASCS4 (Figs 4b and c). An increase in the activation of ERK1/2 at 10 min after addition of either bFGF or clASCS4 occurred (Figs 4d and e). The increase of phosphoERK1/2 and no tyrosine phosphorylation of FGFR1 after addition of clASCS4 indicates that signaling via L1 was occurring in the absence of FGFR1 activation.
Fig. 4.
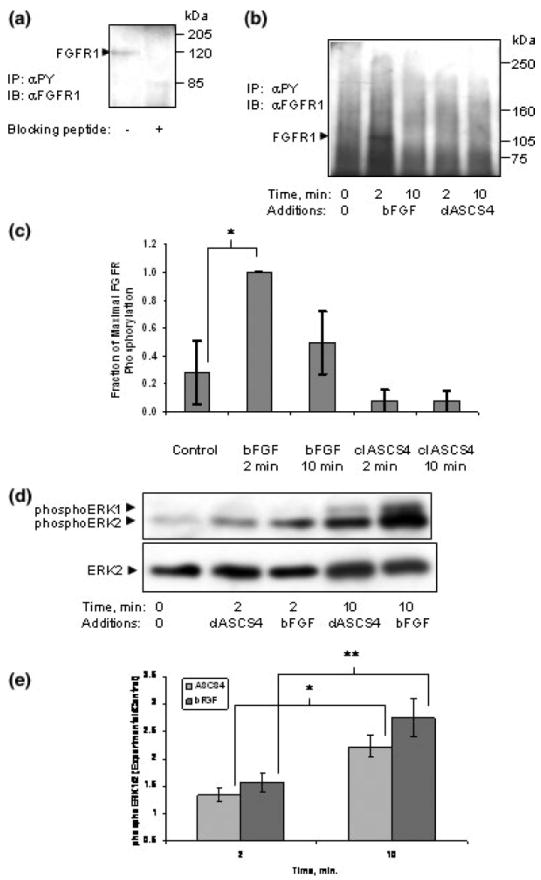
(a) FGFR1 is present in cerebellar granule cells. FGFR1 was detected by immunoblot in immunoprecipitates of cerebellar granule cells using anti-phosphotryosine as the immunoprecipitating antibody (left panel). To confirm the identity of the band at 123 kDa, the blocking peptide for the anti-FGFR1 antibody was added to the primary antibody solution (1 μL blocking peptide: 4 μL antibody, both 200 μg/mL) before incubation with the blot. The blocking peptide eliminated FGFR1 immunoreactivity (right panel). (b) Activation of the L1 signaling cascade does not increase phosphorylation of the FGFR1. Cerebellar granule cells grown in serum-free defined media were treated with either bFGF or cross-linked ASCS4 (ASCS4). Cells were lysed at indicated times. Lysates containing equal amounts of protein were immunoprecipitated for phosphotyrosine-containing proteins. A representative immunoblot of four separate experiments is shown. Tyrosine phosphorylated FGFR1 was found maximally at 2 min following addition of bFGF. (c) Densitometric quantification of phosphorylated FGFR1 shown in (b) is plotted as relative densitometric units relative to the maximal amount found at 2 min of added bFGF. The values of four separate experiments are shown. The bar indicates the mean of the values ± SE. Tyrosine phosphorylated bFGFR1 was significantly increased above control only after 2 min of added bFGF (*p < 0.026, paired t-test). (d) Activation of L1 or FGFR1 increases phosphoERK1/2 at 10 min. The supernatant from immunoprecipitates obtained from experiments in (b) above were analyzed for ERK1/2 activation with anti-phosphoERK1/2 antibodies. To assess for the amount of protein in cell lysates, the phosphoERK blots were stripped and probed for ERK2. The blot shown is representative of four experiments. (e) Both activation of L1 or FGFR1 significantly increases phosphoERK1/2 at 10 min. Immunoblots obtained for phosphoERK1/2 analysis were scanned and the ratio of phosphoERK1/2 to total ERK2 was calculated for each lane. Each ratio was normalized to the ‘nothing-added’ lane to give the relative densitometric units. The mean ± SE are represented for four experiments. The asterisks indicate a significant increase from 2 to 10 min following addition of crosslinked antibodies or bFGF (*p < 0.025, **p < 0.01, paired t-test). Both the 2- and 10-min time points were also significantly increased over ‘nothing added’ (ASCS4, p < 0.04 and 0.005; bFGF, p < 0.02 and 0.007, respectively, paired t-test).
To further determine if FGFR1 activity is required for L1 activation of ERK1/2, FGFR1 tyrosine kinase activity was inhibited and the effect on L1 activation of ERK1/2 was investigated. FGFR1 tyrosine kinase is inhibited by DMBI, a novel inhibitor of FGFR1 as well as platelet-derived growth factor receptor (Zaman et al. 1999). First, the concentration of DMBI that completely inhibited bFGF activation of ERK1/2 without reducing total amounts of ERK2 was determined to be 60 μm, similar to that previously reported (Fig. 5a) (Zaman et al. 1999). Dimethylsulfoxide (DMSO), the vehicle for DMBI, had no effect on baseline phospho-ERK1/2 (Figs 5a–c). Addition of 60 μm DMBI had no effect on clASCS4 activation of ERK1/2, while in the same experiment significantly inhibited ERK1/2 activation by bFGF (Figs 5b and c). These results suggest that, in cultured CGN, L1 signaling to ERK1/2 occurs independently of FGFR1. Therefore, ethanol must inhibit L1-mediated neurite outgrowth by an effect on the events downstream of L1 clustering, such as signal transduction or protein trafficking.
Fig. 5.
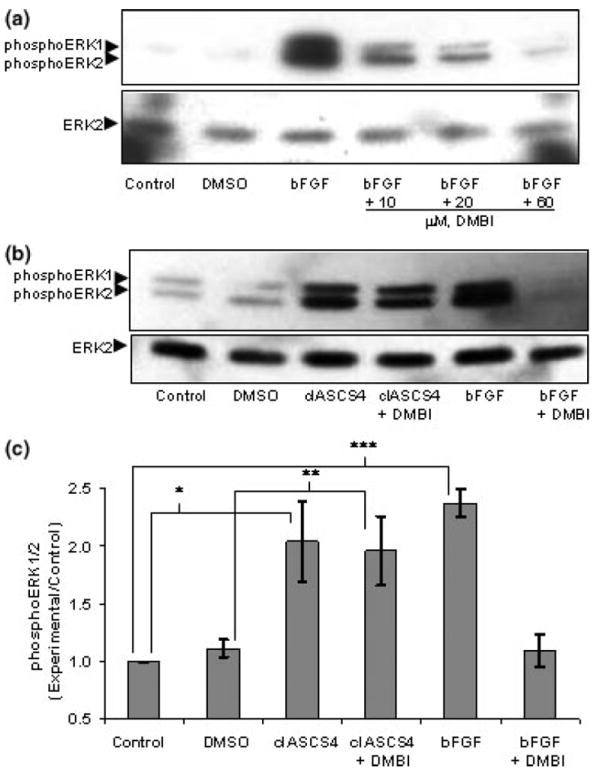
(a) DMBI inhibits bFGF activation of ERK1/2. Serum-starved cerebellar granule cells were treated with or without increasing concentrations of DMBI for 20 min prior to addition of 50 ng/mL bFGF. ERK1/2 activation (phosphoERK1/2) was assayed by western blot analysis after addition of nothing (control), DMSO, and DMBI in DMSO. DMBI (60 μm) almost completely inhibited bFGF activation of ERK1/2 without changing the amount of ERK2. (b) DMBI does not inhibit L1 activation of ERK1/2. Serum-starved cerebellar granule cells were treated with nothing (control), DMSO or 60 μm of DMBI in DMSO for 20 min, then either clASCS4 or bFGF was added for 10 min. ERK1/2 activation was assayed by western blot analysis. Western blots showed that addition of clASCS4 increased activation of ERK1/2 (phosphoERK1/2) in the presence or absence of DMBI. In contrast, DMBI prevented bFGF activation of ERK1/2. (one of three representative blots shown) (c) Densitometric quantification of ERK1/2 phosphorylation corrected for total ERK2 shown in (b) is plotted as relative densitometric units relative to the ‘nothing-added’ control. The values of three separate experiments are shown. The bar indicates the mean of the values ± SE. DMSO did not change baseline phosphoERK1/2. Addition of 60 μm of DMBI in DMSO did not inhibit clASCS4 activation of ERK1/2 (*p < 0.05, **p < 0.03, paired t-test). bFGF significantly increased phosphoERK1/2 (***p < 0.004, paired t-test). However, no significant increase in phosphoERK1/2 was seen with bFGF and 60 μm DMBI.
Discussion
The present study sheds new light on the mechanism of ethanol inhibition of L1-mediated neurite outgrowth by showing (i) that the signal transduction pathway activated by clustering of L1 is inhibited by ethanol, and (ii) that, in this system, L1 does not cause a measurable activation of FGFR1, nor does inhibition of FGFR1 tyrosine kinase inhibit L1 activation of ERK1/2. Therefore, L1 probably does not activate ERK1/2 via FGFR1 in this cell type. Thus, in this simple, well-characterized system, ethanol inhibits L1-mediated neurite outgrowth by disrupting the downstream events following L1 activation that lead to activation of ERK1/2.
The effect of ethanol on L1-mediated ERK1/2 activation was robust. The effect was not dependent on the method of L1 activation, either using cross-linked monoclonal antibodies or L1-Fc chimeras. In addition, the activation was reduced by 60–70%, similar to the 75% inhibition with the MEK inhibitor, PD98059 reported by Schmid et al. (2000). Thus, the ethanol effect was equivalent to that seen with inhibition of MEK which resulted in a 50% reduction in neurite length (Schmid et al. 2000), similar to our previous findings (Bearer et al. 1999b).
One limitation of this study is relating these observations to our previous work showing the ethanol inhibition of neurite outgrowth. To promote neurite outgrowth, L1 is postulated to undergo a continuous recycling pathway in the growth cone with subsequent activation/deactivation of ERK (Kamiguchi and Lemmon 2000a). Ethanol effects on L1 activation of ERK1/2 would be difficult to interpret in extending neurites because of the ongoing activation/deactivation of ERK. In order to measure the effect of ethanol on L1 activation of ERK, we used an assay system where the recycling pathway of L1 does not occur. Ethanol pre-incubation would simulate the conditions within the neuron during neurite outgrowth in the presence of ethanol. L1 is ‘triggered’ by clustering to generate a burst of ERK activation which can then be detected by western blot. We anticipate that ethanol continues to inhibit L1 activation of ERK1/2 during the course of the neurite outgrowth assay. However, inhibition of ERK1/2 activation may alter gene expression in the neuron (Schmid et al. 2000), which may then influence overall neurite outgrowth, independent of any further effect on ERK1/2.
Our failure to find an activation of FGFR1 during L1-mediated activation of ERK1/2 was not surprising. The molecular structure of FGFR1 has the putative binding site for L1 buried within the molecule and not accessible for binding (Plotnikov et al. 1999), as is the putative binding site for FGFR1 in the L1 molecule (Bateman et al. 1996). In addition, the strongest evidence implicating FGFR1 in the downstream signaling cascade of L1 is the inhibition of L1-mediated neurite outgrowth when cells over express a dominant negative form of FGFR1 (Saffell et al. 1997). However, an alternative explanation for this result is the sequestration of a common docking/signaling component by the dominant negative construct required for ERK1/2 activation (Kamiguchi and Lemmon 2000b).
There are several possible sites of action for the inhibition of L1 activation of ERK1/2 by ethanol, including (i) inhibition of endocytosis, (ii) inhibition of L1 clustering and subsequent protein–protein interactions, (iii) inhibition of L1 phosphorylation/dephosphorylation, and (iv) inhibition of the signal cascade between L1 and ERK1/2.
Endocytosis of L1 following homophilic binding has been shown to be a prerequisite of ERK1/2 activation (Schaefer et al. 1999; Schmid et al. 2000). Transfection of cells with a dominant–negative form of dynamin (K44A) inhibits clathrin-mediated endocytosis (van der Bliek et al. 1993; Damke et al. 1994). Transfection of K44A dynamin into L1-transfected 3T3 cells inhibits L1 endocytosis by over 80% (Kamiguchi et al. 1998). Crosslinking L1 with anti-L1 polyclonal antibodies activates ERK1/2 in L1-transfected 3T3 cells, but is almost completely inhibited in L1-transfected cells co-transfected with dynamin K44A (Schaefer et al. 1999). Thus, ethanol could potentially inhibit endocytosis, resulting in inhibition of L1-mediated ERK1/2 activation. The effects of ethanol on endocytosis have been studied in different systems with mixed results. Ethanol impairs endocytosis of both the asialoglycoprotein receptor in hepatocytes (Dalton et al. 2003) and formaldehyde-modified albumin in liver endothelial cells (Duryee et al. 2003). In contrast, chronic ethanol exposure was associated with an enhanced endocytosis of the α1 subunit of GABAA receptors in the cerebral cortex (Kumar et al. 2003). If ethanol inhibits L1 endocytosis in this model, then the amount of L1 on the cell surface would increase, and, one would expect, cell adhesion would increase (Long et al. 2001). However, while ethanol has been shown to either inhibit (Wilkemeyer and Charness 1998; Wilkemeyer et al. 2000; Wilkemeyer et al. 2003), or have no effect on (Vallejo et al. 1997; Bearer et al. 1999b), L1-mediated cell adhesion/aggregation, it has never been reported to increase cell adhesion. Hence, ethanol inhibition of L1 endocytosis is unlikely to be the underlying mechanism.
A second possibility is that ethanol disrupts the multimerization in cis of L1 which is required for activation of the signaling cascade. Activation of ERK1/2 requires multivalent binding of L1 with either cross-linking antibodies or divalent/multivalent L1-Fc as shown here and by Schmid et al. (2000) and Schaefer et al. (1999). Artificially trimerized full-length L1 is significantly better at promoting cell adhesion and neurite outgrowth (Hall et al. 2000). In addition, clustering of L1 appears to promote cis interaction with other proteins, such as integrins (Silletti et al. 2000). Clustering of the palmitoylated Ig superfamily member, NCAM, has been shown to activate the tyrosine kinase fyn via its incorporation into lipid rafts (Niethammer et al. 2002). While palmitoylation of L1 has not yet been described, a close family member, neurofascin, is palmitoylated on a highly conserved cysteine residue in the transmembrane domain (Ren and Bennett 1998). Neurofascin and neurofascin lacking this cysteine residue localize in distinct fractions within low-density detergent-resistant membrane fractions (DRMs) enriched in signaling molecules, suggesting that native neurofascin is targeted to DRMs. It is expected that L1 would be similarly palmitoylated and targeted to DRMs. Indeed the presence of L1 in lipid rafts has been recently described (Nakai and Kamiguchi 2002), and it is known that L1 is phosphorylated by src family kinases which associate with DRMs (Schaefer et al. 2003). Disruption of lipid rafts by depletion of cellular cholesterol or sphingolipids reduces neurite outgrowth of cerebellar granule cells on L1 and N-cadherin, but not laminin (Nakai and Kamiguchi 2002). L1 appears to co-localize in cross-linked lipid rafts. Using a novel technique where localized DRMs can be disrupted, DRMs in the P region of the growth cone influenced the rate of neurite outgrowth, whether in the filopodia or the lamellipodia (Nakai and Kamiguchi 2002). Ethanol may inhibit L1 signal transduction by inhibiting the multimerization of L1 by a conformational change at the FN3 domain (Silletti et al. 2000) or by perturbing its association with lipid rafts. CD14 partitioning into lipid rafts has been shown to be altered by ethanol (Dai et al. 2005). Ethanol inhibition of L1 cis multimerization would be consistent with its observed effects on L1-mediated homophilic binding, neurite outgrowth and signal transduction.
A third possibility for the mechanism of ethanol inhibition of L1 ERK1/2 activation is an effect on the dephosphorylation–phosphorylation of tyrosine 1176 in the cytoplasmic domain of L1. Clustering of L1 induces a rapid dephosphorylation of this tyrosine, enabling AP2 to bind to L1 and for endocytosis to occur (Schaefer et al. 2003). The phosphatase involved is unknown. Following endocytosis, the non-receptor tyrosine kinase p60src re-phosphorylates Y1176 (Schaefer et al. 2003). This dephosphorylation–phosphorylation cycle appears necessary for L1 activation of ERK1/2. L1 clustering does not activate ERK1/2 in cerebellar neurons from src−/− mice (Schmid et al. 2000). While the dephosphorylation–phosphorylation of Y1176 is clearly a potential target for ethanol, other kinases/phosphatases may also be critical targets. L1 clustering on the cell surface is known to activate both kinases and phosphatases (Klinz et al. 1995; Schaefer et al. 1999) and to change the phosphorylation state of L1 (Buchstaller et al. 1996; Zisch et al. 1997).
Finally, the inhibitory effect of ethanol on L1-mediated ERK1/2 activation may be an effect on any of the members of the signal cascade leading from endocytosed L1 to ERK1/2. This cascade involves P13-knase, Rac1, MEK and ERK1/2 as elegantly described by Schmid et al. (2000). Not only may the effect of ethanol be on each enzyme, but it could also potentially be an effect on scaffolding proteins as well. In a recent study, phospholipase D2 (PLD2) was activated by anti-L1 in cerebellar granule neurons (Watanabe et al. 2004). Inhibition of ERK1/2 activation blocked anti-L1 mediated activation of PLD2, implying that PLD2 activation is downstream of ERK1/2. Ethanol at 104 mm specifically inhibited PLD2 and not PLD1. While PLD2 may be a site for the inhibitory action of ethanol, our results suggest that it may be a secondary site. The IC50 for ethanol to inhibit L1-mediated neurite outgrowth is 3–5 mm (Bearer et al. 1999b). The results presented here show complete inhibition of L1 activation of ERK1/2 at an ethanol concentration of 25 mm.
Our results implicate ethanol in disrupting events which occur following L1 homophilic binding that lead to ERK1/2 activation, and that this pathway is independent of the FGFR1. Additional experiments will be required to explore these possibilities.
Acknowledgments
We thank Bob Miller and Laura Nagy for their review of this manuscript. This work was supported by National Institutes of Health Grant AA-11839 to CFB and EY-05285 to VL. VL holds the Walter G. Ross Chair in Developmental Neuroscience at the University of Miami.
Abbreviations used
- bFGF
basic fibroblast growth factor
- BSA
bovine serum albumin
- CGN
cerebellar granule neurons
- clASCS4
cross-linked ASCS4 monoclonal antibody
- DMBI
3-(4-dimethylamino-benzylide-nyl)-2-indolinone
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethylsulfoxide
- DRM
detergent-resistant membrane
- ERK1/2
extracellular signal-regulated kinase 1 and 2
- FAS
fetal alcohol syndrome
- FBS
fetal bovine serum
- FGFR1
fibroblast growth factor receptor 1
- HBSS
Hank’s balanced salt solution
- HRP
horseradish peroxidase
- Ig
immunoglobulin
- L1
L1 cell adhesion molecule
- MEK
mitogen-activated protein kinase kinase
- NCAM
neural cell adhesion molecule
- PLD1
phospholipase D1
- PLD2
phospholipase D2
- SDS–PAGE
sodium dodecyl sulfate – polyacrylamide gel electrophoresis
References
- Abel E, Sokol R. A revised conservative estimate of the incidence of FAS and its economic impact. Alcoholism: Clin Exp Res. 1991;15:514–524. doi: 10.1111/j.1530-0277.1991.tb00553.x. [DOI] [PubMed] [Google Scholar]
- Bateman A, Jouet M, MacFarlane J, Du J-S, Kenwrick S, Chothia C. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 1996;15:6050–6059. [PMC free article] [PubMed] [Google Scholar]
- Bearer CF. Mechanisms of brain injury: L1 cell adhesion molecule as a target for ethanol-induced pre-natal brain injury. Seminars Pediatric Neurol. 2001;8:100–107. doi: 10.1053/spen.2001.25227. [DOI] [PubMed] [Google Scholar]
- Bearer C, Buck K, Swick A, O’Riordan M. Ethanol inhibits both L1-mediated neuritogenesis and neurite outgrowth. Alcoholism: Clin Exp Res. 1999a;23:29A. [Google Scholar]
- Bearer CF, Swick AR, O’Riordan MA, Cheng G. Ethanol inhibits L1-mediated neurite outgrowth in post-natal rat cerebellar granule cells. J Biol Chem. 1999b;274:13 264–13 270. doi: 10.1074/jbc.274.19.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie C, Siegel R. Developmental cues modulate GABAA receptor subunit mRNA expression in cultured cerebellar granule neurons. J Neuroscience. 1993;13:1784–1792. doi: 10.1523/JNEUROSCI.13-04-01784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Redeimeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummendorf T, Hubert M, Treubert U, Leuschner R, Tarnok A, Rathjen F. The axonal recognition molecule F11 is a multifunctional protein: specific domains mediate interactions with Ng-CAM and restrictin. Neuron. 1993;10:711–727. doi: 10.1016/0896-6273(93)90172-n. [DOI] [PubMed] [Google Scholar]
- Buchstaller A, Kunz S, Berger P, Kunz B, Ziegler U, Rader C, Sonderegger P. Cell adhesion molecules NgCAM and Axonin-1 form heterodimers in the neuronal membrane and cooperate in neurite outgrowth promotion. J Cell Biol. 1996;135:1593–1607. doi: 10.1083/jcb.135.6.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani V, Chedotal A, Schachner M, Faivre-Sarraith C, Rougon G. Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signalling pathways in axonal guidance. Neuron. 2000;27:237–249. doi: 10.1016/s0896-6273(00)00033-7. [DOI] [PubMed] [Google Scholar]
- Dai Q, Zhang J, Pruett SB. Ethanol alters cellular activation and CD14 partitioning in lipid rafts. Biochem Biophys Res Comm. 2005;332:37–42. doi: 10.1016/j.bbrc.2005.04.088. [DOI] [PubMed] [Google Scholar]
- Dalton SR, Wiegert RL, Baldwin CR, Kassel KM, Casey CA. Impaired receptor-mediated endocytosis by the asialoglycoprotein receptor in ethanol-fed mice: implications for studying the role of this receptor in alcoholic apoptosis. Biochem Pharmacol. 2003;15:535–543. doi: 10.1016/s0006-2952(02)01555-1. [DOI] [PubMed] [Google Scholar]
- Damke H, Baba T, Warnock DE, Schmid SL. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty P, Walsh F. CAM–FGF receptor interactions: a model for axonal growth. Mol Cell Neurosci. 1996;8:99–111. doi: 10.1006/mcne.1996.0049. [DOI] [PubMed] [Google Scholar]
- Duryee MJ, Klassen LW, Freeman TL, Willis MS, Tuma DJ, Thiele GM. Chronic ethanol consumption impairs receptor-mediated endocytosis of MAA-modified albumin by liver endothelial cells. Biochem Pharmacol. 2003;15:1045–1054. doi: 10.1016/s0006-2952(03)00416-7. [DOI] [PubMed] [Google Scholar]
- Felsenfeld D, Hynes M, Skoler K, Furley A, Jessell T. TAG-1 can mediate homophilic binding, but neurite outgrowth on TAG-1 requires an L1-like molecule and β1 integrins. Neuron. 1994;12:675–690. doi: 10.1016/0896-6273(94)90222-4. [DOI] [PubMed] [Google Scholar]
- Hall H, Bozic D, Fauser C, Engel J. Trimerization of cell adhesion molecule L1 mimics clustered L1 expression on the cell surface: influence on L1–ligand interactions and on promotion of neurite outgrowth. J Neurochem. 2000;75:336–346. doi: 10.1046/j.1471-4159.2000.0750336.x. [DOI] [PubMed] [Google Scholar]
- Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Hockberger PE, Tseng H-T, Connor JA. Immunocytochemical and electrophysiological differentiation of rat cerebellar granule cells in explant cultures. J Neurosci. 1987;7:1370–1383. doi: 10.1523/JNEUROSCI.07-05-01370.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Cotman SL, Halfter W, Cole GJ. The heparan sulfate proteoglycan agrin modulates neurite outgrowth mediated by FGF-2. J Neurobiol. 2003;55:261–277. doi: 10.1002/neu.10213. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H. The mechanism of axon growth. Mol Neurobiol. 2003;28:219–227. doi: 10.1385/MN:28:3:219. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci. 2000a;20:3676–3686. doi: 10.1523/JNEUROSCI.20-10-03676.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V. IgCAMs: bidirectional signals underlying neurite growth. Curr Opin Cell Biol. 2000b;12:598–605. doi: 10.1016/s0955-0674(00)00138-1. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Yoshihara F. The role of endocytic L1 trafficking in polarized adhesion and migration of nerve growth cones. J Neurosci. 2001;21:9194–9203. doi: 10.1523/JNEUROSCI.21-23-09194.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinz SG, Schachner M, Maness PF. L1 and N-CAM antibodies trigger protein phosphatase activity in growth cone-enriched membranes. J Neurochem. 1995;65:84–95. doi: 10.1046/j.1471-4159.1995.65010084.x. [DOI] [PubMed] [Google Scholar]
- Kuhn T, Stoeckli E, Condrau M, Rathjen F, Sonderegger P. Neurite outgrowth on immobilized axonin-1 is mediated by a heterophilic interaction with L1(G4) J Cell Biol. 1991;115:1113–1126. doi: 10.1083/jcb.115.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kralie JE, O’Buckley TK, Grobin AC, Morrow AL. Chronic ethanol consumption enhances internalization of α1 subunit-containing GABAA receptors in cerebral cortex. J Neurochem. 2003;86:700–708. doi: 10.1046/j.1471-4159.2003.01894.x. [DOI] [PubMed] [Google Scholar]
- Lemmon V, Farr K, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–1603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- Lemmon V, Burden S, Payne H, Elmslie G, Hlavin M. Neurite growth on different substrates: permissive versus instructive influences and the role of adhesive strength. J Neurosci. 1992;12:818–826. doi: 10.1523/JNEUROSCI.12-03-00818.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner J, Rathjen F, Schachner M. L1 mono- and polyclonal antibodies modify cell migration in early post-natal mouse cerebellum. Nature. 1983;305:427–430. doi: 10.1038/305427a0. [DOI] [PubMed] [Google Scholar]
- Long KE, Asou H, Snider MD, Lemmon V. The role of endocytosis in regulating L1-mediated adhesion. J Biol Chem. 2001;276:1285–1290. doi: 10.1074/jbc.M006658200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Tsiotra P, Karagogeos D, Hortsch M. Cis-activation of L1-mediated ankyrin recruitment by TAG-1 homophilic cell adhesion. J Biol Chem. 1998;273:33 354–33 359. doi: 10.1074/jbc.273.50.33354. [DOI] [PubMed] [Google Scholar]
- Milev P, Maurel P, Haring M, Mrgolis RK, Margolis RU. TAG-1/axonin-1 is a high-affinity ligand of neurocan, phosphacan/protein-tryosine phosphatase ζ/β, and N-CAM. J Biol Chem. 1996;271:15 716–15 723. doi: 10.1074/jbc.271.26.15716. [DOI] [PubMed] [Google Scholar]
- Nakai Y, Kamiguchi H. Migration of nerve growth cones requires detergent-resistant membranes in a spatially defined and substrate-dependent manner. J Cell Biol. 2002;159:1097–1108. doi: 10.1083/jcb.200209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M. Co-signaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol. 2002;157:521–532. doi: 10.1083/jcb.200109059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov AN, Schlessinger J, Hubbard SR, Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Ramanathan R, Wilkemeyer M, Mittal B, Perides G, Charness M. Alcohol inhibits cell–cell adhesion mediated by human L1. J Cell Biol. 1996;133:381–390. doi: 10.1083/jcb.133.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Bennett V. Palmitoylation of neurofascin at a site in the membrane-spanning domain highly conserved among the L1 family of cell adhesion molecules. J Neurochem. 1998;70:1839–1849. doi: 10.1046/j.1471-4159.1998.70051839.x. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Sadoul R, Faissner A, Schachner M. Biochemical characterization of different molecular forms of the neural cell adhesion molecule L1. J Neurochem. 1988;50:510–521. doi: 10.1111/j.1471-4159.1988.tb02941.x. [DOI] [PubMed] [Google Scholar]
- Saffell J, Williams E, Mason I, Walsh F, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. 1997;18:231–242. doi: 10.1016/s0896-6273(00)80264-0. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Roonprapunt C, Grumet M. Purification of Ig-fusion proteins from medium containing Ig. Biotechniques. 1998;25:382–385. doi: 10.2144/98253bm09. [DOI] [PubMed] [Google Scholar]
- Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JMJ. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37 965–37 967. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- Schaefer AW, Kamei Y, Kamiguchi H, Wong EV, Rapoport I, Kirchhausen T, Beach CM, Landreth G, Lemmon SK, Lemmon V. L1 endocytosis is controlled by a phosphorylation–dephosphorylation cycle stimulated by outside-in signaling by L1. J Cell Biol. 2003;157:1223–1232. doi: 10.1083/jcb.200203024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch U, Lohse M, Schachner M. Neural cell adhesion molecules influence second messenger systems. Neuron. 1989;3:13–20. doi: 10.1016/0896-6273(89)90111-6. [DOI] [PubMed] [Google Scholar]
- Silletti S, Mei F, Sheppard D, Montgomery AMP. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J Cell Biol. 2000;149:1485–1501. doi: 10.1083/jcb.149.7.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton K, Howe C, Battaglia F, editors. Summary Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. National Academy Press; Washington DC: 1996. p. 57. [Google Scholar]
- Vallejo Y, Hortsch M, Dubreuil R. Ethanol does not inhibit the adhesive activity of Drosophila neuroglian or human L1 in Drosophila S2 tissue culture cells. J Biol Chem. 1997;272:12 244–12 247. doi: 10.1074/jbc.272.18.12244. [DOI] [PubMed] [Google Scholar]
- Von Bohlen Und Halbach F, Taylor J, Schachner M. Cell type-specific effects of the neural adhesion molecules L1 and N-CAM on diverse second messenger systems. Eur J Neurosci. 1992;4:896–909. doi: 10.1111/j.1460-9568.1992.tb00116.x. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Ymazaki M, Miyazaki H, Arikawa C, Itoh K, Sasaki T, Maehama T, Frohman MA, Kanaho Y. Phospholipase D2 functions as a downstream signaling molecule of MAP kinase pathway in L1-stimulated neurite outgrowth of cerebellar granule neurons. J Neurochem. 2004;89:142–151. doi: 10.1111/j.1471-4159.2004.02308.x. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer MF, Charness ME. Characterization of ethanol-sensitive and -insensitive fibroblast cell lines expressing human L1. J Neurochem. 1998;71:2382–2391. doi: 10.1046/j.1471-4159.1998.71062382.x. [DOI] [PubMed] [Google Scholar]
- Wilkemeyer MF, Sebastian AB, Smith SA, Charness ME. Antagonists of alcohol inhibition of cell adhesion. Proc Natl Acad Sci USA. 2000;97:3690–3695. doi: 10.1073/pnas.050545697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkemeyer MF, Chen S-Y, Menkari CE, Brenneman DE, Sulik KK, Charness ME. Differential effects of ethanol antagonism and neuroprotection in peptide fragment NAPVSIPQ prevention of ethanol-induced developmental toxicity. Proc Natl Acad Sci USA. 2003;100:8843–8548. doi: 10.1073/pnas.1331636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E, Doherty P, Turner G, Reid R, Hemperly J, Walsh F. Calcium influx into neurons can solely account for cell contact-dependent neurite outgrowth stimulated by transfected L1. J Cell Biol. 1992;119:883–892. doi: 10.1083/jcb.119.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Kenwrick S, Willems P, Lemmon V. Mutations in the cell adhesion molecule L1 cause mental retardation. Trends Neurosci. 1995;18:168–172. doi: 10.1016/0166-2236(95)93896-6. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Koo EH, Selkoe DJ. Cell surface amyloid β-protein precursor colocalizes with β1 integrins at substrate contact sites in neural cells. J Neurosci. 1997;17:1004–1010. doi: 10.1523/JNEUROSCI.17-03-01004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman GJ, Vink PM, van den Doelen AA, Veeneman GH, Theunissen HJ. Tyrosine kinase activity of purified recombinant cytoplasmic domain of platelet-derived growth factor β-receptor (β-PDGFR) and discovery of a novel inhibitor of receptor tyrosine kinases. Biochem Pharmacol. 1999;57:57–64. doi: 10.1016/s0006-2952(98)00271-8. [DOI] [PubMed] [Google Scholar]
- Zheng J, Buxbaum R, Heidemann S. Measurements of growth cone adhesion to culture surfaces by micromanipulation. J Cell Biol. 1994;127:2049–2060. doi: 10.1083/jcb.127.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisch AH, Stallcup WB, Chong LD, Dahlin-Huppe K, Voshol J, Schachner M, Pasquale EB. Tyrosine phosphorylation of L1 family adhesion molecules: implication of the Eph kinase Cek5. J Neurosci Res. 1997;47:655–665. doi: 10.1002/(sici)1097-4547(19970315)47:6<655::aid-jnr12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]