

Abstract
Alopecia areata (AA) is a common autoimmune disease resulting from damage of the hair follicle by T cells. The immune pathways required for autoreactive T cell activation in AA are not defined limiting clinical development of rational targeted therapies1. Genome-wide association studies (GWAS)2 implicated ligands for the NKG2D receptor (product of the KLRK1 gene) in disease pathogenesis. Here, we show that cytotoxic CD8+NKG2D+ T cells are both necessary and sufficient for the induction of AA in mouse models of disease. Global transcriptional profiling of mouse and human AA skin revealed gene expression signatures indicative of cytotoxic T cell infiltration, an interferon-γ (IFN-γ) response and upregulation of several γ-chain (γc) cytokines known to promote the activation and survival of IFN-γ–producing CD8+NKG2D+ effector T cells. Therapeutically, antibody-mediated blockade of IFN-γ, interleukin-2 (IL-2) or interleukin-15 receptor β (IL-15Rβ) prevented disease development, reducing the accumulation of CD8+NKG2D+ T cells in the skin and the dermal IFN response in a mouse model of AA. Systemically administered pharmacological inhibitors of Janus kinase (JAK) family protein tyrosine kinases, downstream effectors of the IFN-γ and γc cytokine receptors, eliminated the IFN signature and prevented the development of AA, while topical administration promoted hair regrowth and reversed established disease. Notably, three patients treated with oral ruxolitinib, an inhibitor of JAK1 and JAK2, achieved near-complete hair regrowth within 5 months of treatment, suggesting the potential clinical utility of JAK inhibition in human AA.
Alopecia areata is a T cell–mediated autoimmune disease characterized phenotypically by hair loss and, histologically, by infiltrating T cells surrounding the hair follicle bulb (reviewed in ref. 1). Previous studies have shown that transfer of total T cells (but not B cells or sera) can cause the disease in human xenograft models3, as well as in C3H/HeJ mice4, a mouse strain that develops spontaneous AA with considerable similarity to human AA. Broad-acting intralesional steroids are the most commonly used therapy for AA, with varying success. Progress in developing effective, rationally targeted therapies has been limited by our lack of mechanistic understanding of the underlying key T cell inflammatory pathways in AA.
We2 and others5 have previously identified a cytotoxic subset of CD8+NKG2D+ T cells within the infiltrate surrounding human AA hair follicles, as well as concomitant upregulation in the follicle itself of the ‘danger signals’ ULBP3 (ref 2) and MICA5, two NKG2D ligands (NKG2DLs) whose importance in disease pathogenesis has also been suggested by genome-wide association studies2.
To determine the contribution of CD8+NKG2D+ T cells to AA pathogenesis, we used the C3H/HeJ mouse model6, which spontaneously develops alopecia and recapitulates many pathologic features of human AA7. In lesional skin biopsies from alopecic mice, CD8+NKG2D+ T cells infiltrate the epithelial layers of the hair follicle, which overexpress the NKG2DLs, H60 and Rae-1, analogous to what has been observed in skin biopsies of human AA2 (Fig. 1a,b and Supplementary Fig. 1a,b). Flow cytometric analysis of the CD45+ leukocyte population in the skin revealed a marked increased number of CD8+NKG2D+ T cells in the skin of diseased C3H/HeJ mice, in conjunction with cutaneous lymphadenopathy and increased total cellularity, as compared with disease-free C3H/HeJ mice (Fig. 1c,d). Other cell types, including CD4+ T cells4 and mast cells8, were present in much smaller numbers (Supplementary Fig. 1c and data not shown).
Figure 1.
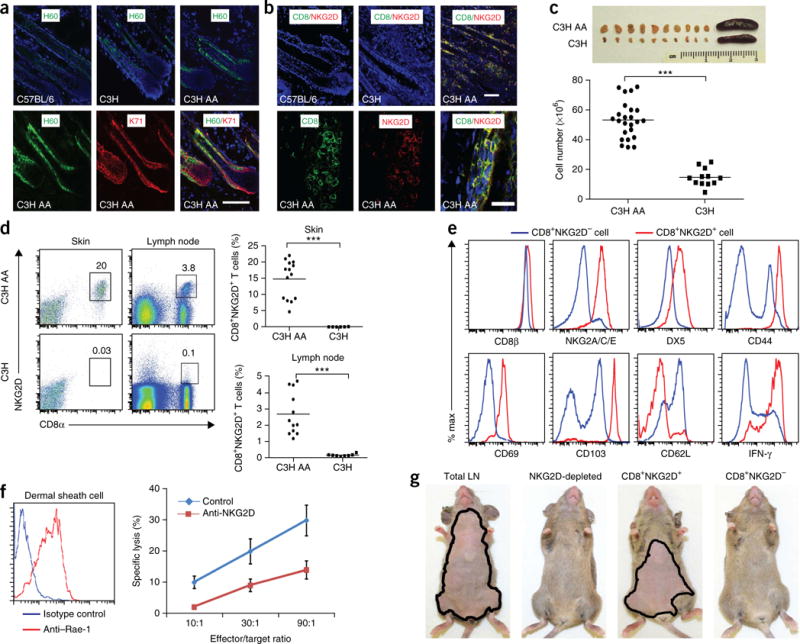
CD8+NKG2D+ cytotoxic T lymphocytes accumulate in the skin and are necessary and sufficient to induce disease in AA mice. (a) Immunofluorescence staining of NKG2D ligand (H60) in the hair follicle inner root sheath (marked by K71). Scale bar, 100 μm. (b) CD8+NKG2D+ cells in hair follicles of C57BL/6, healthy C3H/HeJ and C3H/HeJ AA mice. Top scale bar, 100 μm; bottom scale bar, 50 μm. (c) Cutaneous lymphadenopathy and hypercellularity in C3H/HeJ AA mice. (d) Frequency (number shown above boxed area) of CD8+NKG2D+ T cells in the skin and skin-draining lymph nodes in alopecic mice versus ungrafted mice. (e) Immunophenotype of CD8+NKG2D+ T cells in cutaneous lymph nodes of C3H/HeJ alopecic mice. (f) Left, Rae-1t–expressing dermal sheath cells grown from C3H/HeJ hair follicles. Right, dose-dependent specific cell lysis induced by CD8+NKG2D+ T cells isolated from AA mice cutaneous lymph nodes in the presence of blocking anti-NKG2D antibody or isotype control. Effector to target ratio given as indicated. Data are expressed as means ± s.d. (g) Hair loss in C3H/HeJ mice injected subcutaneously with total lymph node (LN) cells, CD8+NKG2D+ T cells alone, CD8+NKG2D− T cells or lymph node cells depleted of NKG2D+ (5 mice per group). Mice are representative of two experiments. ***P < 0.001 (Fisher’s exact test). For c,d,f, n and number of repeats are detailed in the Supplementary Methods.
The immunophenotype of the skin-infiltrating CD8+ T cells in mice with AA was similar to that of the CD8+NKG2D+ population found in the cutaneous lymph nodes: CD8αβ+ effector memory T cells (TEM, CD8hiCD44hiCD62LlowCD103+) bearing several natural killer (NK) immunoreceptors, including CD49b and NKG2A, NKG2C and NKG2E (Fig. 1e and Supplementary Fig. 1d). These CD8+ TEM cells expressed high levels of IFN-γ and exhibited NKG2D-dependent cytotoxicity against ex vivo-expanded syngeneic dermal sheath target cells (Fig. 1f). Gene expression analysis of the CD8+NKG2D+ T cells isolated from alopecic C3H/HeJ lymph node cells using RNA-seq demonstrated a transcriptional profile characteristic of effector cytotoxic T lymphocytes (CTLs)9,10 and identified several additional NK-specific transcripts (Supplementary Table 1 and Supplementary Fig. 2).
We next evaluated the requirement of these CD8+ TEM cells in disease pathogenesis. Transfer of cytotoxic CD8+NKG2D+ cells or total lymph node cells from diseased mice induced AA in all five healthy C3H/HeJ recipients by 14 weeks after transfer, whereas lymph node cell populations depleted of NKG2D+ cells were unable to transfer disease (Fig. 1g). Thus, CD8+NKG2D+ T cells are the dominant cell type in the dermal infiltrate and are necessary and sufficient for T cell–mediated transfer of AA.
To characterize the transcriptional profile of AA lesional skin from C3H/HeJ mice as well as human AA, we performed Affymetrix microarray analyses to identify differentially expressed genes in skin between individuals with AA and skin from control individuals without disease (Supplementary Fig. 3 and Supplementary Tables 2 and 3) Three gene expression signatures were identified in lesional skin: IFN response genes, such as those encoding the IFN-inducible chemokines CXCL-9, CXCL-10 and CXCL-11 (Supplementary Figs. 3 and 4), several key CTL-specific transcripts, such as those encoding CD8A and granzymes A and B, and γc cytokines and their receptors, such as the transcripts for interleukin-2 (IL-2) and IL-15, in both human and mouse AA skin. As IL-2Rα was previously shown to be expressed on infiltrating lymphocytes surrounding human AA hair follicles11, we performed immunofluorescence analysis for both IL-15 and its chaperone receptor IL-15Rα to identify the source of IL-15 in the skin. We detected a marked upregulation of both components in AA hair follicles in both human and mouse AA and found IL-15Rβ expressed on infiltrating CD8+ T cells in humans (Supplementary Figs. 5 and 6).
IL-2 and IL-15 are well-known drivers of cytotoxic activity by IFN-γ–producing CD8+ effector T cells and NK cells12,13 and have been implicated in the induction and/or maintenance of autoreactive CD8+ T cells14–16. To test the efficacy of IFN-γ– and γc-targeted therapies in vivo, we used the well-established graft model of AA, in which skin grafts from mice with spontaneous AA are transferred onto the backs of unaffected 10-week-old recipient C3H/HeJ mice. In this model, AA develops reliably in 95–100% of grafted recipients within 6–10 weeks7, allowing us to test interventions aimed at either preventing or reversing disease.
The role of IFN-γ in AA was previously investigated using both knockout studies and administration of IFN-γ, where IFN-γ–deficient mice were resistant and exogenous IFN-γ precipitated disease17,18. Administration of neutralizing antibodies to IFN-γ at the time of grafting prevented AA development in grafted recipients and abrogated major histocompatibility complex (MHC) upregulation and CD8+NKG2D+ infiltration in the skin (Fig. 2a–c). Likewise, a role for IL-2 in AA pathogenesis was previously established using genetic experiments in which IL-2 haploinsufficiency on the C3H/HeJ background conferred resistance to disease by about 50% using the graft model19, and this role is supported by our genome-wide association studies in humans2. Systemically administered blocking antibodies to either IL-2 (Fig. 2d–f) or IL-15Rβ (Fig. 2g–i) prevented AA in grafted mice, blocked the accumulation of CD8+NKG2D+ T cells in the skin and abrogated MHC upregulation. However, IL-21 blockade failed to prevent the development of AA in grafted C3H/HeJ mice (Supplementary Fig. 7). Notably, none of these blocking antibodies given alone was able to reverse established AA (data not shown).
Figure 2.
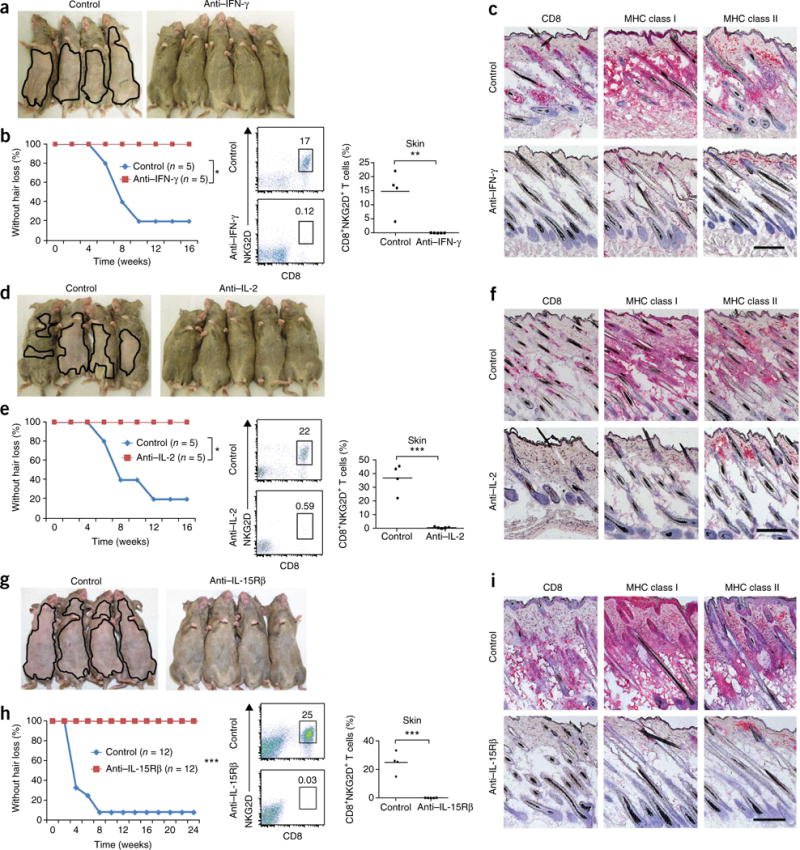
Prevention of AA by blocking antibodies to IFN-γ, IL-2 or IL-15Rβ. C3H/HeJ grafted mice were treated systemically from the time of grafting. (a–h) AA development in C3H/HeJ grafted mice treated systemically from the time of grafting with antibodies to IFN-γ (a,b), IL-2 (d,e) and IL-15Rβ (g,h). Frequency (number shown above boxed area) of CD8+NKG2D+ T cells in the skin of mice treated with antibodies to IFN-γ (b), IL-2 (e) and IL-15Rβ (h) compared to PBS-treated mice. (*P < 0.05, **P < 0.01, ***P < 0.001, statistical methods described in the Supplementary Methods. Immunohistochemica staining of skin biopsies showing CD8 and MHC class I and II expression in skin of mice treated with isotype control antibody or with antibodies to IFN-γ (c), IL-2 (f) or IL-15Rβ (i). Scale bars, 100 μm. For each experiment, n and number of repeats are detailed in the Supplementary Methods.
We next asked whether we could recapitulate the effects of type I cytokine blockade by intervening downstream using small-molecule inhibitors of JAK kinases, which signal downstream of a wide range of cell surface receptors. In particular, IFN-γ receptors and γc family receptors signal through JAK1/2 and JAK1/3, respectively20. JAK activation was shown by the presence of phosphorylated signal transducer and activator of transcription (STAT) proteins (pSTAT1, pSTAT3 and to a lesser extent pSTAT5) in human and mouse alopecic hair follicles, but not in normal hair follicles (Supplementary Fig. 8). In in vitro–cultured dermal sheath cells from C3H/HeJ mice, exogenous IFN-γ increased STAT1 activation, whereas IFN-γ plus TNF-α increased surface IL-15 expression (Supplementary Fig. 9a,b). Ruxolitinib, a US Food and Drug Administration (FDA)–approved small-molecule inhibitor of the JAK1/2 kinases (JAK selectivity is JAK1 = JAK2>Tyk2>>>JAK3)21 critical for IFN-γR signaling inhibited these responses (Supplementary Fig. 9a,b). In cultured CTL effectors from C3H/HeJ mice, the FDA-approved small-molecule JAK3 inhibitor tofacitinib (JAK3>JAK1>>JAK2 selectivity)22 blocked IL-15–triggered pSTAT5 activation (Supplementary Fig. 9c). Tofacitinib also blocked killing of dermal sheath cells (Supplementary Fig. 9d) and IL-15-induced upregulation of granzyme B and IFN-γ expression (Supplementary Fig. 9e).
To test whether inhibition of these signaling pathways would be therapeutically effective in vivo, we systemically administered ruxolitinib (Fig. 3a–c) and tofacitinib (Fig. 3f–h) at the time of grafting and found that they prevented the development of AA and the expansion of CD8+NKG2D+ T cells in all grafted recipients. The skin of mice treated with either drug showed no histological signs of inflammation (Fig. 3d,i). Global transcriptional analysis of whole-skin biopsies showed that both drugs also blocked the dermal inflammatory signature, as measured by Alopecia Areata Disease Activity Index (ALADIN, Fig. 3e,j), and Gene Expression Dynamic Index (GEDI) analysis (Supplementary Fig. 10)23.
Figure 3.
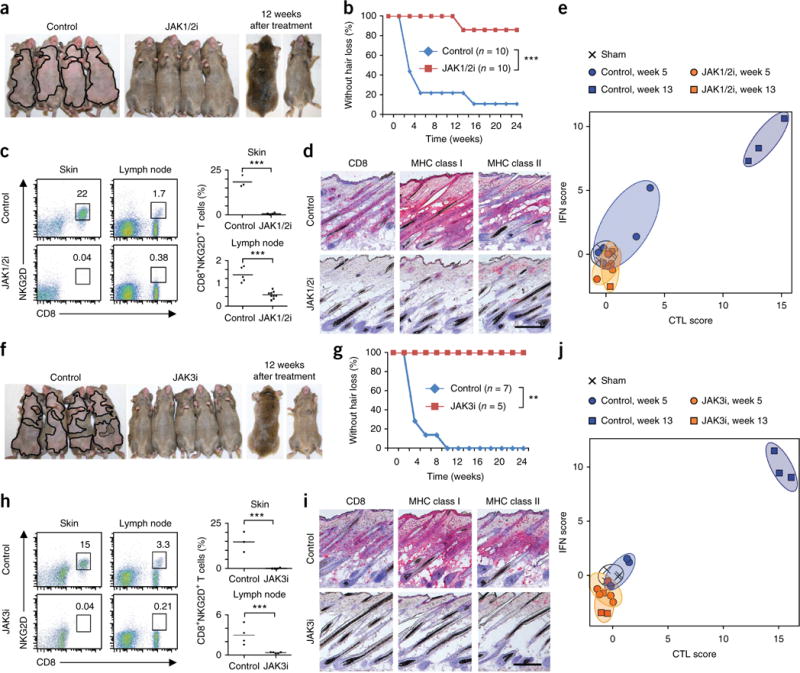
Systemic JAK1/2 or JAK3 inhibition prevents the onset of AA in grafted C3H/HeJ mice. (a–j) AA development in C3H/HeJ grafted mice treated systemically from the time of grafting with ruxolitinib (JAK1/2i) (a,b) or tofacitinib (JAK3i) (f,g) (**P < 0.01). Frequency (number shown above boxed area) of CD8+NKG2D+ T cells in skin and cutaneous lymph nodes of mice treated with PBS or with JAK1/2i (c) or JAK3i (h) (***P < 0.001, statistical methods described in Supplementary Methods). Immunohistochemical staining of skin biopsies showing CD8 and MHC class I and II expression in skin of mice treated with PBS or with JAK1/2i (d) or JAK3i (i). ALADIN score of transcriptional analysis from mice treated with PBS or with JAK1/2i (e) or JAK3i (j), given as log2 mean expression Z-scores as indicated in the Supplementary Methods. Hair regrowth after an additional 12 weeks after treatment withdrawal is also shown. (a,f). Scale bars, 100 μm. For each experiment, n and number of repeats are detailed in the Supplementary Methods.
We next asked whether systemic tofacitinib treatment could reverse established disease by initiating therapy 7 weeks after grafting, a time point at which all mice had developed extensive AA. Systemic therapy resulted in substantial hair regrowth all over the body, reduced the frequency of CD8+NKG2D+ T cells and reversed histological markers of disease (Supplementary Fig. 11), all of which persisted 2–3 months after the cessation of treatment.
Next, to test a more clinically relevant route of delivery, we asked whether topical administration of protein tyrosine kinase inhibitors could reverse established AA in mice with kinetics similar to those of systemic delivery. In established disease, we found that topical ruxolitinib and topical tofacitinib were both highly effective in reversing disease in treated lesions (applied to back skin). A full coat of hair emerged in the ruxolitinib- or tofacitinib-treated mice by 7 weeks of treatment (data not shown), and we observed complete hair regrowth within 12 weeks following topical therapy (Fig. 4a,b). Topical therapy was associated with a markedly reduced proportion of CD8+NKG2D+ T cells in the treated skin and lymph node (Fig. 4c), normalization of the ALADIN transcriptional signature (Fig. 4d), reversal of histological markers of disease (Fig. 4e) and correction of the GEDI in all treated mice (Supplementary Fig. 12). Notably, untreated areas on the abdomen remained alopecic (Fig. 4a and Supplementary Fig. 13), demonstrating that topical therapy acted locally and that the observed therapeutic effects were not the result of systemic absorption. These effects were visible as early as 2–4 weeks after the onset of treatment (Supplementary Fig. 14) and persisted 2–3 months after the cessation of treatment (Fig. 4a).
Figure 4.
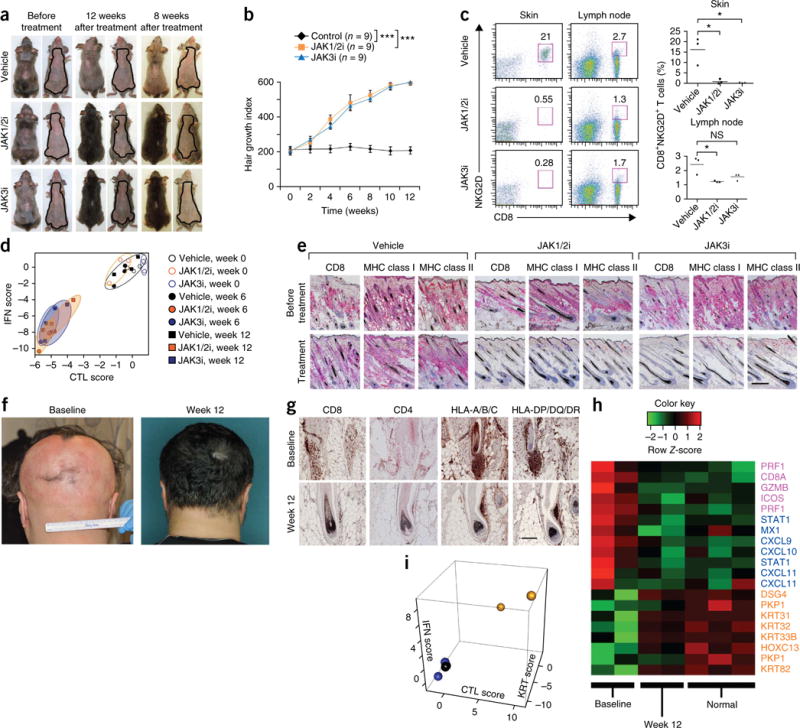
Reversal of established AA with topical small-molecule inhibitors of the downstream effector kinases JAK1/2 or JAK3, and clinical results of patients with AA. (a) Three mice per group with long-standing AA (at least 12 weeks after grafting) treated topically on the dorsal back with 0.5% JAK1/2i (center), 0.5% JAK3i (bottom) or vehicle alone (Aquaphor, top) by daily application for 12 weeks. This experiment was repeated three times. Hair regrowth at an additional 8 weeks after treatment withdrawal is also shown. (b) Time course of hair regrowth index shown as weeks after treatment. (c) The frequency (number shown above boxed area) of CD8+NKG2D+ T cells in the skin of mice treated with JAK1/2i or JAK3i compared to vehicle control mice (mean ± s.e.m., n = 3 per group, *P < 0.05, **P < 0.01, statistical methods described in the Supplementary Methods). NS, not significant. (d) The ALADIN score shows treatment-related loss of CTL and IFN signatures, given as log2 mean expression Z-scores as indicated in the Supplementary Methods. (e) Immunohistochemical staining of mouse skin biopsies shows treatment-related loss of expression of CD8 and MHC class I and II markers. Scale bar, 100 μm. (f) Treatment of patient 3 with AA, who had hair loss involving >80% of his scalp at baseline, with ruxolitinib and hair regrowth after 12 weeks of oral treatment. (g) Clinical correlative studies of biopsies obtained before treatment (baseline) and after 12 weeks of treatment of patient 2, including immunostains for CD4, CD8 and human leukocyte antigen (HLA) class I (A, B, C) and class II (DP, DQ, DR). Scale bar, 200 μm. (h,i) RNA microarray analysis from treated patients 1 and 2 with AA (before treatment versus after treatment versus 3 normal subjects) presented as a heatmap (h) and as a cumulative ALADIN index (i). KRT, hair follicle keratins.
To test the efficacy of JAK inhibitors in human subjects with AA, we treated three patients with moderate to severe disease orally with ruxolitinib, 20 mg twice daily. Ruxolitinib is currently FDA-approved for the treatment of myelofibrosis24,25, a disease driven by wild-type and mutant JAK2 signaling downstream of hematopoietic growth factor receptors. In addition, small clinical studies using topical ruxolitinib in psoriasis26 have demonstrated anti-inflammatory activity that may be due to interruption of the IL-17 signaling axis. All three ruxolitinib-treated patients exhibited near-complete hair regrowth within 3 to 5 months of oral treatment (Fig. 4f and Supplementary Figs. 15 and 16). Comparison of biopsies obtained at baseline and after 12 weeks of treatment demonstrated reduced perifollicular T cell infiltration, reduced follicular expression of human leukocyte antigen class I and class II expression (Fig. 4g) and normalization of the ALADIN inflammatory and hair keratin signatures following treatment (Fig. 4h,i).
Taken together, our data suggest CD8+NKG2D+ T cells promote AA pathogenesis, acting as cytolytic effectors responsible for autoimmune attack of the hair follicle (Supplementary Fig. 17). We postulate that IFN-γ produced by CD8 T cells leads to the collapse of immune privilege in the hair follicle27, inducing further production of IL-15 (ref. 28) (Supplementary Figs. 5 and 6) and a feed-forward loop that promotes type I cellular autoimmunity. The clinical response of a small number of patients with AA to treatment with the JAK1/2 inhibitor ruxolitinib suggests future clinical evaluation of this compound or other JAK protein tyrosine kinase inhibitors currently in clinical development is warranted in AA29.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Mice
7- to 10-week-old female C57BL/6 and C3H/HeJ mice (Jackson Laboratories, Bar Harbor, ME) were used and maintained under specific pathogen-free conditions. Experiments were performed in compliance with institutional guidelines as approved by the Institutional Animal Care and Use Committee of Columbia University Medical Center.
Transfer of alopecia areata using grafted C3H/HeJ skin
Normal-haired C3H/HeJ mice were grafted at 8 weeks of age (during the second telogen) with skin from a C3H/HeJ mouse that developed AA spontaneously, as described previously7. In brief, mice spontaneously affected with AA were euthanized, and full thickness skin grafts of approximately 2 cm in diameter were removed and grafted to normal-haired C3H/HeJ mice. All experiments included 10 grafted mice (5 treated and 5 untreated) and 3 sham grafted mice (mice grafted with autologous grafts). Hair loss typically began at around 4–6 weeks after grafting. No sham grafted mouse developed hair loss.
Flow cytometric analysis of skin and cutaneous lymph nodes
To make a single-cell suspension of mouse skin, fat was removed from the overlying skin in cold PBS and then incubated in collagenase type I (2 mg/ml in PBS) at 32 °C for 75 min. After digestion, the skin was minced in RPMI/10% FBS, filtered through a 70-μM cell strainer and centrifuged at 1100g for 5 min. The pellet was resuspended in RPMI/10% FBS, filtered through a 40-μM cell strainer and spun at 400g for 5 min. The pellet was resuspended in FACS buffer (PBS/5% BSA), DAPI to gate on live cells and staining antibodies (listed in Supplementary Methods). Cutaneous lymph nodes were pooled, minced in RPMI, filtered through a 40-μM cell strainer, centrifuged at 400g for 5 min, stained and analyzed on a BD LSR II flow cytometer.
Transfer of T cell populations into recipient C3H/HeJ mice
For positive selection of T cell populations, lymph node cells were obtained from 5 C3H/HeJ alopecic mice, stained with anti-CD4, anti-CD8 and anti-NKG2D antibodies) and then sorted (BD Influx) to obtain two fractions: CD8+NKG2D+ T cells and CD8+NKG2D− T cells. Antibody dilutions are given in the Supplementary Methods. Three to five 7-week-old C3H/HeJ mice per group were injected subcutaneously with two million sorted cells of each population. For negatively selected populations, NKG2D+ cells were depleted by incubating total lymph node cells from 3 alopecic C3H/HeJ mice with biotinylated anti-NKG2D (CX5) and then with streptavidin-conjugated beads (Miltenyi) before removal on a Miltenyi magnetic column. Five million cells (either CD8/NKG2D-depleted or total lymph node cells) were suspended in 100 ul PBS and transferred into each of 5 mice by subcutaneous injection.
Prevention and treatment studies in mice
For prevention studies, mice were treated beginning on the day of grafting (n = 5–10 mice per group). For anti-IFN-γ experiments, i.p. injections of hamster isotype control IgG or hamster polyclonal anti-IFN-γ IgG (BioXCell) 300 μg in 100 μl PBS were administered ten times weekly for 12 weeks. For anti-IL-2 experiments, i.p. injections of control rat isotype control IgG or simultaneous administration of two anti-IL-2 rat monoclonal antibodies (BioXCell) (250 μg clone S4B6 and 250 μg JES6-1 together in 100 μl PBS) were administered three times weekly for 12 weeks. For anti-IL-15-Rβ experiments, i.p. injections of rat isotype control IgG or anti-IL-15Rβ antibody (Biolegend, clone TM-β1) (200 μg in 100 μl PBS) were administered two times weekly for 12 weeks. For JAK1/2i experiments, mice were administered vehicle (0.5% methylcellulose; Sigma-Aldrich) or vehicle-containing 50 mg/kg of JAK1/2i ruxolitinib (ChemieTek) daily by oral gavage for 12 weeks. For JAK3i experiments, mice were implanted subcutaneously with Alzet osmotic mini-pumps (pumps, model 2004, Durect Corporation) on the back of each mouse to deliver vehicle (poly(ethylene glycol) (PEG)300) or vehicle containing the JAK3i tofacitinib (Abmole) at 15 mg/kg/day for 12 weeks.
For topical treatment studies, grafted mice with long-standing AA (more than 8 weeks) were treated once daily for 12 weeks to affected skin on the dorsal back with vehicle (10% DMSO in Aquaphor) or vehicle containing JAK inhibitors, initially dissolved in DMSO and then diluted 1:10 in Aquaphor, to achieve 0.5% JAKi ointment). Full-thickness skin biopsies were excised from the dorsal surface of each mouse at interim time points, and skin samples were either snap frozen in liquid nitrogen for RNA extraction or snap frozen in OCT for immunostaining. Hair status was examined twice weekly.
Human clinical studies
We initiated a single center, proof-of-concept clinical trial in the Clinical Trials Unit in the Department of Dermatology at the Columbia University Medical Center entitled “An Open-Label Pilot Study to Evaluate the Efficacy of ruxolitinib in Moderate to Severe Alopecia Areata,” ClinicalTrials.gov identifier NCT01950780. The protocol for this intervention trial was reviewed and approved by the Institutional Review Board at Columbia University and conducted under the Declaration of Helsinki principles. Informed consent was received before inclusion in the study. Eligibility criteria included >30% hair loss for at least 3 months in duration with no evidence for hair regrowth at the time of enrollment. The first three treated patients are described here. The ruxolitinib dose was 20 mg orally twice daily for 3–6 months. Skin punch biopsies were obtained at baseline and after 12 weeks of treatment. Consent for photography was obtained for the patients shown in Figure 4 and in the Supplementary Figures 15 and 16.
Immunohistochemistry and immunofluorescence
8 μM acetone-fixed frozen mouse skin or human skin sections were air-dried and stained overnight with anti-mouse or anti-human antibodies (see Supplementary Methods) at 4 °C in a moist chamber. Human hair follicles were microdissected and embedded in OCT compound before sectioning and staining (see Supplementary Methods).
Primary dermal sheath and lymphokine-activated killer (LAK) cell culture
Dermal sheath (DS) cells were isolated from microdissected mouse vibrissa follicles and cultured in 20% FBS DMEM with 5 ng/ml murine FGF (Pepro Tech). LAK cells were generated from bulk splenocytes plated at 4 × 106 in 6-well plates with 50 ng/ml murine IL-15 (Pepro Tech), 50 nM JAK3i (tofacitinib) or 50 ng/ml murine IL-15 plus 50 nM JAK3i and incubated at 37 °C in a 5% CO2 incubator for 96 h.
In vitro cytotoxicity assays
Determination of specific killing of target cells was performed using CFSE-labeled DS cells as targets mixed with different ratios of effector cells incubated for 5 h at 37 °C 5% CO2 with or without neutralizing rat anti-mouse NKG2D antibody (20 ug/ml) (Biolegend, CX5). Specific lysis of DS cells was determined flow cytometrically by measuring cell death of CFSE + DS cells using Annexin V/7-AAD.
Gene expression sample preparation in human and mouse skin and T cells
Total RNA was isolated using the miRNeasy Mini Kit (Qiagen Inc., Valencia, CA, USA) with on-column DNA digestion using the RNase-free DNase set (Qiagen, Inc.). For RNA-seq analysis CD3+CD8+CD44+NKG2D+ and CD3+CD8+CD44+NKG2D− cells were flow-sorted from lymph nodes of alopecic C3H/HeJ mice. RNA was extracted as above and prepared for RNA-seq using the TruSeq RNA Sample Prep Kit v2. Samples were sequenced on the HiSeq 2000 sequencer (Illumina, San Diego, CA) for 50 cycles. RNA-seq files were demultiplexed by the Rockefeller University Genomics Core Facility.
For global transcriptional profiling in mouse skin, total extracted RNA was processed using the 3′ IVT Express Kit from Affymetrix. Resulting biotinylated cDNA samples were hybridized to the Mouse Genome 430 2.0 gene chips and subsequently washed, stained with streptavidin-phycoerythrin and scanned on an HP GeneArray Scanner (Hewlett-Packard Company, Palo Alto, CA). For gene expression studies, mice grafted with autologous healthy skin were included as sham-operated controls.
For human AA samples, perilesional punch biopsies from 5 patients with patchy alopecia areata who were not undergoing local or systemic treatments were collected and compared to scalp biopsies from 5 unrelated unaffected individuals. All procedures were performed under Institutional Review Board–approved protocols at Columbia University and conducted under the Declaration of Helsinki principles. Informed consent was received before inclusion in the study. Extracted total RNA was reverse transcribed and amplified using the Ovation RNA Amplification V2 kit (NuGEN Technologies, Inc., San Carlos, CA). Amplified cDNA was biotinylated with the Encore Biotin Module (NuGEN Technologies) and then hybridized to the U133 Plus 2.0 gene chips. RT-PCR confirmations done as described in the Supplementary Methods. Primer sequences are given in Supplementary Tables 4 and 5.
Statistical analyses
Statistical methods for each figure are given in the Supplementary Methods. No statistical method was used to predetermine sample size. The investigators were not blinded to allocation during experiments and outcome assessment. The experiments were not randomized.
Supplementary Material
Acknowledgments
We thank the National Alopecia Areata Registry, as well as M. Duvic, V. Price, M. Hordinsky and D. Norris, and the National Alopecia Areata Foundation. We thank J. Sundberg, T. Behrens, D. Bickers, J. O’Shea, T. Waldmann, B. Jabri, D. Raulet, L. Lanier, T. Spies, M. Hayden, R. Paus, P. Green, B. Lebwohl, D. Accili and C. Jahoda for stimulating discussions. We are grateful for clinical support from M. Furniss, C. Clark and G. Ulerio and expert technical assistance from M. Zhang, E. Chang, H. Lam and J. Huang. This work was supported in part by US Public Health Service National Institutes of Health NIAMS grants R01AR056016 (to A.M.C.) and R21AR061881 (to A.M.C and R.C.), a Shared Instrumentation Grant for the LSR II Flow Cytometer (S10RR027050) to R.C. and the Columbia University Skin Disease Research Center (P30AR044535), as well as the Locks of Love Foundation and the Alopecia Areata Initiative. J.E.C. is supported by the T32GM082271 Medical Genetics Training Grant (issued to A.M.C.). A.J., C.A.H., S.H. and A.d.J. are recipients of Career Development Awards from the Dermatology Foundation, and A.J. is also supported by the Louis V. Gerstner Jr Scholars Program.
Footnotes
Accession codes. Microarray and RNA-seq data were deposited in Gene Expression Omnibus with accession numbers GSE45657, GSE45512, GSE45513, GSE45514, GSE45551 and GSE58573.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
L.X., Z.D. and A.J. were responsible in large part for performing the studies reported herein and participated in the design, execution and interpretation of the data. C.A.H. was responsible for establishing the C3H/HeJ graft model. A.d.J., S.H., G.M.D., L.R. and P.S. were involved in additional molecular and cell biological experiments. W.G. performed immunofluorescence and morphometric studies. L.P. and J.E.C. performed biostatistical analysis of all data sets. J.M.-W. was instrumental in human sample acquisition and analysis. A.M.C. and R.C. were responsible for conception, design, oversight, execution and interpretation of data for this study. All authors contributed to drafts, writing, figure preparation and editing of the final manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Gilhar A, Etzioni A, Paus R. Alopecia areata. N Engl J Med. 2012;366:1515–1525. doi: 10.1056/NEJMra1103442. [DOI] [PubMed] [Google Scholar]
- 2.Petukhova L, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113–117. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilhar A, Ullmann Y, Berkutzki T, Assy B, Kalish RS. Autoimmune hair loss (alopecia areata) transferred by T lymphocytes to human scalp explants on SCID mice. J Clin Invest. 1998;101:62–67. doi: 10.1172/JCI551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McElwee KJ, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25− cells promote systemic alopecia areata and CD4+/CD25+cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947–957. doi: 10.1111/j.0022-202X.2005.23692.x. [DOI] [PubMed] [Google Scholar]
- 5.Ito T, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008;128:1196–1206. doi: 10.1038/sj.jid.5701183. [DOI] [PubMed] [Google Scholar]
- 6.Sundberg JP, Cordy WR, King LE., Jr Alopecia areata in aging C3H/HeJ mice. J Invest Dermatol. 1994;102:847–856. doi: 10.1111/1523-1747.ep12382416. [DOI] [PubMed] [Google Scholar]
- 7.McElwee KJ, Boggess D, King LE, Jr, Sundberg JP. Experimental induction of alopecia areata-like hair loss in C3H/HeJ mice using full-thickness skin grafts. J Invest Dermatol. 1998;111:797–803. doi: 10.1046/j.1523-1747.1998.00380.x. [DOI] [PubMed] [Google Scholar]
- 8.Bertolini M, et al. Abnormal interactions between perifollicular mast cells and CD8+ T cells may contribute to the pathogenesis of alopecia areata. PLoS ONE. 2014;9:e94260. doi: 10.1371/journal.pone.0094260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Best JA, et al. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bezman NA, et al. Molecular definition of the identity and activation of natural killer cells. Nat Immunol. 2012;13:1000–1009. doi: 10.1038/ni.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brajac I, Gruber F, Petrovecki M, Malnar-Dragojevic D. Interleukin-2 receptor α-chain expression in patients with alopecia areata. Acta Dermatovenerol Croat ADC. 2004;12:154–156. [PubMed] [Google Scholar]
- 12.Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood. 2001;97:14–32. doi: 10.1182/blood.v97.1.14. [DOI] [PubMed] [Google Scholar]
- 13.Ye W, Young JD, Liu CC. Interleukin-15 induces the expression of mRNAs of cytolytic mediators and augments cytotoxic activities in primary murine lymphocytes. Cell Immunol. 1996;174:54–62. doi: 10.1006/cimm.1996.0293. [DOI] [PubMed] [Google Scholar]
- 14.Meresse B, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med. 2006;203:1343–1355. doi: 10.1084/jem.20060028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol. 2010;185:5693–5703. doi: 10.4049/jimmunol.1002188. [DOI] [PubMed] [Google Scholar]
- 16.Meresse B, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 17.Freyschmidt-Paul P, et al. Interferon-γ-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515–521. doi: 10.1111/j.1365-2133.2006.07377.x. [DOI] [PubMed] [Google Scholar]
- 18.Gilhar A, Kam Y, Assy B, Kalish RS. Alopecia areata induced in C3H/HeJ mice by interferon-γ: evidence for loss of immune privilege. J Invest Dermatol. 2005;124:288–289. doi: 10.1111/j.0022-202X.2004.23580.x. [DOI] [PubMed] [Google Scholar]
- 19.Freyschmidt-Paul P, et al. Reduced expression of interleukin-2 decreases the frequency of alopecia areata onset in C3H/HeJ mice. J Invest Dermatol. 2005;125:945–951. doi: 10.1111/j.0022-202X.2005.23888.x. [DOI] [PubMed] [Google Scholar]
- 20.O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis. 2013;72(suppl. 2):ii111–ii115. doi: 10.1136/annrheumdis-2012-202576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintás-Cardama A, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghoreschi K, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eichler GS, Huang S, Ingber DE. Gene Expression Dynamics Inspector (GEDI): for integrative analysis of expression profiles. Bioinformatics. 2003;19:2321–2322. doi: 10.1093/bioinformatics/btg307. [DOI] [PubMed] [Google Scholar]
- 24.Verstovsek S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison C, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 26.Punwani N, et al. Preliminary clinical activity of a topical JAK1/2 inhibitor in the treatment of psoriasis. J Am Acad Dermatol. 2012;67:658–664. doi: 10.1016/j.jaad.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 27.Paus R, Nickoloff BJ, Ito TA. A ‘hairy’ privilege. Trends Immunol. 2005;26:32–40. doi: 10.1016/j.it.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 28.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6:595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 29.Dolgin E. Companies hope for kinase inhibitor JAKpot. Nat Rev Drug Discov. 2011;10:717–718. doi: 10.1038/nrd3571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.