

Abstract
Globally, the cancer associated deaths are generally attributed to the spread of cancerous cells or their features to the nearby or distant secondary organs by a process known as metastasis. Among other factors, the metastatic dissemination of cancer cells is attributed to the reactivation of an evolutionary conserved developmental program known as epithelial to mesenchymal transition (EMT). During EMT, fully differentiated epithelial cells undergo a series of dramatic changes in their morphology, along with loss of cell to cell contact and matrix remodeling into less differentiated and invasive mesenchymal cells. Many studies provide evidence for the existence of EMT like states in prostate cancer (PCa) and suggest its possible involvement in PCa progression and metastasis. At the same time, the lack of conclusive evidence regarding the presence of full EMT in human PCa samples has somewhat dampened the interest in the field. However, ongoing EMT research provides new perspectives and unveils the enormous potential of this field in tailoring new therapeutic regimens for PCa management. This review summarizes the role of many transcription factors and other molecules that drive EMT during prostate tumorigenesis.
Keywords: Epithelial mesenchymal transition, prostate cancer, transcription factors, endoplasmic reticulum, stress, signaling, Y Box Protein-1
INTRODUCTION
Epithelial cancers including prostate cancer (PCa) exhibit an innate property to spread to a distant organ. In PCa, metastatic potential of the primary tumor to spread the distant sites of the body is responsible for the majority of cancer related deaths [1]. The metastatic phenotype is not expressed by all the epithelial cells instead there are some specific populations that express it and they must escape the restraints of the primary tumor site. This process requires specialized degrading enzymes that can break the basement membranes for invasion which is prerequisite step for the metastasis [2, 3]. During invasion, the metastasis capable epithelial cells undergo a series of actin cytoskeleton reorganization that supports and facilitates both invasion and migration of cells. Cancer cells leave the primary tumor mass mainly by losing cell-cell contact that results in altered cell shape by a phenomenon of epithelial-to-mesenchymal transition (EMT) [4, 5].
1. EMT: AN ESSENTIAL DEVELOPMENTAL PROCESS
At the time of embryonic development, cells travel very long distances to reach their final destinations. To achieve this, epithelial cells rely heavily on a very fine tuned and highly regulated EMT program that converts them into a mesenchymal state [6]. Tissues arise mainly from developmental transition of epithelial cells to mesenchymal or stromal cells; a process known as EMT which most of the times is subsequently followed by a reverse process known as mesenchymal to epithelial (MET) transition [7]. In general, epithelial cells are in very close contact with their immediate environment and also with their axis of polarity via sequential arrangements of adherent junctions, desmosomes and tight junctions [8]. In contrast, mesenchymal cells are loosely structured within a three-dimensional extracellular matrix that also comprises connective tissues [9]. For communicational purposes, the gap junctions are used by epithelial cells. In general, the developmental EMT is divided into three different types [5]:
1.1. Primary EMT
The well-recognized examples of primary EMT are gastrulation and neural crest development [9, 10, 11]. Gastrulation is an evolutionary conserved process for body plan development. During this epithelial shaped epiblast gives rise to mesenchymal shaped mesoderm. The cell undergoes sequential shifts in shape, in which there is first internalization of mesendoderm, followed by convergence to midline and eventually extension along the anteroposterior axis [12–14]. It is also noteworthy that some of these elements have been found to be evolutionary conserved. There are many evolutionary conserved transcription factors that induce EMT during gastrulation. In invertebrates both Snail and Twist play key roles [15]; Snail suppresses E-cadherin transcription, helps in both ventral furrow formation and delamination of primary mesenchyme cells [16]. Snail mediated mitotic block is necessary for gastrulation to occur.
Similarly, in vertebrates the snail is important for gastrulation to proceed. Deficiency of snail in embryos leads to failure of complete gastrulation and cells fail to migrate due to unsuccessful events of EMT [17, 18]. Additionally, EMT associated factors also help in cellular invasion mainly via contributing in the process of basal membrane degradation executed by metalloproteases [19, 20]. Post gastrulation the next step is the formation of neural crest; here precursor cells delaminate from border regions of both neural and non-neural ectodermal zones thereby give rise to different structural derivatives [21]. Apart from these main developmental processes, EMT is also involved in somitogenesis [22, 23], endocardium and endocardial cushion formation [24, 25].
1.2. Secondary EMT
During the course of development, primary EMT is followed by the differentiation events in order to generate cell types of different origins [26]. For example, post primary EMT, neural crest cells differentiate and establish into neurons, bone cells and mesodermal cells of different types [9]. Reversibly, all these cellular populations are converted back to epithelial type mainly by using the MET phenomenon. However, this is further followed by a secondary EMT event to produce mesenchymal type cells with a highly constrained differentiation potential. Similarly a secondary EMT process of both ventral and dorsal epithelia gives rise to endocardial progenitors, hematopoietic stem cells and connective tissue of body wall muscle. In addition, the endoderm, ectoderm and mesoderm give rise to other important structures that includes mammary gland [27, 28], mesothelium [29], kidney [30–32], liver [33], pancreas [34–36], etc.
1.3. Tertiary EMT
A classic example of the tertiary type of EMT is development of the heart, which requires successive cycles of EMT followed by MEt also [37]. Initially, mesodermal cells at the time of gastrulation get specified for this process along with other cardiac progenitors to become a two layered epithelium. This is followed by another round of EMT, which also includes formation of endothelial cell linings of the heart. Subsequently, this process further leads to the development into a four compartment structure of the heart. Interestingly, a group of specialized endothelial cells may undergo a tertiary EMT to form endocardium that later assembles into the atrioventricular valvuloseptal complex [38–40].
2. DEVELOPMENTAL EMT HIJACKED BY CANCER CELLS
In the neoplastic environment distinct cell populations are found which encompass highly populated differentiated epithelial cells to less numbered dedifferentiated mesenchymal cells. However, in order to disseminate from the local tumor environment epithelial cells must shift transiently into a mesenchymal state for which these neoplastic epithelial cells hijack the evolutionary conserved EMT process. This transient mesenchymal cell possesses multiple features that are important for metastasis. These involve the ability to invade primary tumors in vicinity, extravasate, survival during movement and formation of micrometastasis in distant organs. The role of EMT in cancer cell invasion, migration and metastasis is now well established in multiple cancer types like breast, ovary, colon, and lung and also in prostate [41]. Mostly in all these type of cancers the expression of EMT related molecules is found to be well correlated with high grade tumors especially those that have poor prognosis [42]. Looking at the scientific data it becomes apparent that cancer cells hijack and use the EMT program for metastatic colonization.
2.1. EMT in Prostate Cancer
There is considerable data to suggest that EMT contributes to PCa progression and metastasis. Nauseef and Henry [43] reviewed this evidence and explored studies that have aimed to better define the role and mechanisms of EMT in PCa. Conclusive evidence to suggest a role for physiologic EMT in human PCa is still lacking, although the expression of EMT related markers in human PCa samples is now well established its association with PCa on the basis of pathological findings is quite difficult due to the possibility of misinterpretation related to compartmental staining between the stromal and the basal layers. Also, no lineage tracking has been done so far to understand how this dynamic process moves on with the progression of age. Another aspect of this ambiguity is that majority of EMT related data is based on PCa cell lines of human origin.
3. MOLECULAR MECHANISMS OF EMT INDUCTION IN PCA
Many molecular events directly or indirectly govern the program of EMT in PCa (Fig. 1). Below we discuss some of these important and established molecular mechanisms that play an important role in EMT induction in PCa.
Fig. 1. Important role of EMT in PCa progression.

The role of EMT in migration, invasion and cell survival is well established. However the impact of EMT on anoikis resistance and immune suppression is still quite not established and debatable.
Growth Factor Signalings
There are different types of growth signals that include transforming growth factor beta (TGF-β), endothelial growth factor (EGF), hepatocyte growth factor (HGF), insulin-like growth factor 1 (IGF-1), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) that are known to contribute in EMT in various cancers including PCa.
3.1. TGF-β Signaling
The TGF-β family includes three TGF-βs, two activins, several bone morphogenetic proteins (BMPs), homodimers and heterodimers of ligands that act in a sequential manner through binary combinations of transmembrane dual specificity kinase receptors. Generally, during the period of development, the expression of all isoforms TGFβ1, TGFβ2 and TGFβ3 is associated with EMT-like events however, only TGFβ1 induces EMT in wound healing, fibrosis and cancer [44].
A role for EMT is also suggested in the development of benign prostatic hyperplasia (BPH). Hu et al [45] showed that treatment of BPH-1 cells with normal prostate stromal WPMY-1 cells results in accumulation of mesenchymal-like cells. To assess the role of TGF-β they treated the cells in the presence of anti-TGF-β antibody and observed upregulation of E-cadherin and CK5/8 levels and down-regulation of p-SMAD3. These results showed that stromal cell supernatant was able to induce EMT in BPH-1 cells, possibly through secreting TGF-β1 to activate Smad signaling. In addition, Slabakova et al. [46] also showed that TGF-β induces expression of Snai2/Slug, a well-known transcription factor involved in EMT in BPH-1 cells. In PCa TGF-β can induce the nuclear accumulation of nuclear factor-kappa B (NF-κB) along with morphological change towards a mesenchymal type. However, EMT like features have been shown to be blocked by the inhibitor of NF-κB demonstrating that NF-κB is an important mediator of TGF-β mediated EMT in PCa [47]. There is evidence for the involvement of other important molecules like STAT3, PAR-4 and NEDD9 in TGF-β induced EMT in PCa. STAT3 expression was induced by TGF-β treatment which further mediated the induction of TWIST1 and HIF1α expression in different PCa cell lines [48, 49]. Similarly Chaudhry et al [50] showed upregulation of prostate apoptosis response-4 (PAR-4) along with EMT related markers in PCa cell lines after TGF-β treatment. PAR-4 upregulation was also observed along with Smad2 and IκB-α in the presence of each TGF-β isoforms, suggesting PAR-4 an important target of TGF-β signaling. Disruption of TGF-β signaling reduces the PAR-4 expression. It has been reported that the overexpression of PAR-4 results in the upregulation of vimentin and Snail expression together in simultaneous with cell migration. However, Par-4 silencing by Si-RNA resulted in decrease of these proteins and prevented also the TGF-β-induced EMT. Recently, Morimoto et al [51] showed critical role of NEDD9, a Crk-associated substrate (Cas) family protein in TGF-β-induced EMT in PCa. Importantly, the Knockdown of endogenous NEDD9 expression completely diminished the TGF-β-triggered tumor invasion in several PCa cell lines. In addition to the EMT inducing potential of TGF-β, other components of this signaling like TGF-β receptor type 2 (TGF-βR2) have been shown to influence EMT in TRAMP mouse model of PCa. The In vivo disruption of TGF-β signaling was shown to accelerate the pathologic malignant phenotype of prostate of the TRAMP mouse model by altered prostate growth and by inducing EMT [52]. Overall, current evidences related to the TGF-β induced EMT in PCa is well recognized but there are important issues that remain unanswered such as a conclusive evidence of EMT from human tissues and role of TGF-β (stromal and/or epithelial) in EMT induction in PCa.
3.2. FGF Signaling
It has been reported that both FGF and its receptor FGFR1 are upregulated in PCa [53]. However, to assess the role of FGFR1 activation on PCa progression in an inducible FGFR1 model that express a prostate-specific, inducible chimeric version of FGFR lead towards the phenotypic switching of epithelial cells towards a mesenchymal phenotype along with development of 100% adenocarcinoma. Also in these mice, lymph node and liver metastasis was evident and the metastatic foci retain mesenchymal features. To identify the possible molecules that contribute to iFGFR1 induced EMT a gene expression study in tumors derived from these mice was done that showed a transcription factor SOX-9 as a possible mechanism mediating this effect [54]. This study clearly confirms EMT inducing potential of this pathway. In addition to FGFR1 other receptors of this pathway like FGFR4 were also found to be co-expressed with matrix metalloproteinase (MMP-14) in certain PCa cell lines. Overexpression of MMP-14 was also shown to induce an EMT-like state in PCa [55], however the precise mechanism for the EMT induction by MMP-14 is not well defined.
Recent literature suggests that the switching between alternatively spliced isoforms may lead towards the imbalanced FGFR signaling. In addition, the alternative splicing of the third Ig-like domain determines the ligand-binding specificity of the receptor and generates the IIIb or the IIIc isoform of the FGFRs [56]. Mostly IIIb isoforms are expressed in epithelial cells, whereas IIIc isoforms are expressed in mesenchymal cells. Exon switching in epithelial cells from the epithelial FGFR2 IIIb isoform to the mesenchymal FGFR2 IIIc isoform by the alternative splicing has been also reported in rat models of prostate [57], suggesting an additional mechanism for FGF induced EMT in Pca.
3.3. IGF1 Signaling
The IGF1 induced EMT in PCa is not extensively studied however, Graham et al. showed that treatment of PCa cell lines with soluble IGF1 results in the upregulation of Zinc finger E-box-binding homeobox 1 (ZEB1), a transcription factor mostly involved in EMT induction [58]. ZEB1 has been mainly known for E-cadherin repression, thereby in enhancing the metastasis. IGF1 treatment also resulted in the upregulation of the MAPK pathway. Based on these observations, it was proposed that IGF1 activation of ZEB1 in PCa cells was MEK/ERK dependent. The evidence of involvement of IGF signalling in EMT in PCa has been recently supported by studies of Chen et al, who showed that EMT-related gene signature of circulating tumor cells (CTCs) included both IGF1 and IGF2 along with some other signaling molecules [59].
3.4. EGF Signaling
The aberrant expression of epidermal growth factor receptor (EGFR) in both androgen independent and metastatic PCa, that are also known to possess heightened EMT related features, is well established [60]. Many studies have shown the EGF mediated phenotypic switching in PCa. For example, the treatment of EGF alone or in combination with TGF-β results in epithelial to mesenchymal phenotype in ARCaP cells. Similarly, Gan et al showed that EGF treatment to PCa cell lines promotes loss in cell-cell besides the down-regulation of E-cadherin and the upregulation of the transcriptional repressor i.e Snail which forms the typical characteristics of EMT [61]. EGF induced EMT was found to be mainly dependent on Akt activation, as inhibition of Akt signaling abolished EGF driven EMT in PCa cell lines [62, 63]. In addition, EGF was shown to selectively induce the protein degradation of epithelial origins in neoplasm; or LIM domain and actin binding 1, LIMA-1 (EPLIN), a putative suppressor of EMT in PCa [64]. Further mechanistic analysis revealed that EGF activated the phosphorylation, ubiquitination, and degradation of EPLIN through an extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent signaling cascade. This study highlighted a novel molecular mechanism for EGF mediated regulation of EMT in PCa. Recently Cho et al showed that EGF induced upregulation of both TWIST1 and STAT3 transcription factors, known to be critical regulators of EMT in PCa [48].
3.5. PI3K-AKT/RAS/MAPK Signaling
Unlike the TGFβ-induced EMT, receptor tyrosine kinases (RTKs) mediated EMT requires other important signalling nodes like AKT, mechanistic target of rapamycin (mTOR) and Mitogen-activated Protein (MAP) Kinases (MAPKs). Lim et al showed that BMP7 induced EMT in PCa cell lines requires both AKT and Extracellular Signal-Regulated Kinase (ERK) signaling [65]. Similarly, Mulholland et al showed that Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis in PCa. Mice with prostate conditional Pten deletion with conditional activatable K-ras (G12D/WT) model showed accelerated PCa progression, accompanied by EMT and macrometastasis with complete penetrance. Also, a progenitor subpopulation with mesenchymal properties from the Ras/AKT activated mice prostate was found to be highly metastatic upon orthotopic transplantation [66]. This study not only underlined the significance of Ras signaling in metastatic PCa but also uncovered the importance of EMT in these events during PTEN loss. In addition PI3K-AKT/mTOR/MAPK signaling was also involved in induction of EMT during PCa radiotherapy. EMT in association with cancer stem cells (CSCs) was found to be an important mechanism for radioresistance during PCa treatment [67]. Ma et al recently showed that both p38 MAPK and ERK are involved in hepatocyte-mediated phenotypic switching in PCa [68].
4. EXTRACELLULAR SIGNALS TO REGULATE EMT
Many of the extracellular signals like Wnt, Hedgehog and Notch are well known to support EMT in a variety of cancers including PCa. However, we briefly discuss here Wnt signaling as there are strong evidences to support Wnt role in the induction of EMT in PCa.
4.1. Wnt Signaling
It is well established that the wingless-type (Wnt) pathway has developmental role in tissues and organisms. Both canonical and noncanonical Wnt signaling is involved in the development of prostate gland [69]. However, the aberrant activation of the Wnt pathway has been found to be involved in the progression of PCa. Jiang et al first reported that activation of the Wnt/beta-catenin signaling pathway correlates with the characteristic of EMT and also positively influences invasiveness and proliferation in PCa [70]. Similarly knockdown of an essential Wnt signaling component, β-catenin in PCa cells results in dramatic reversal of EMT induced by hypoxia inducible factor-1α [71]. Additionally Wnt signaling was shown to be involved in EMT induced by SOX2. SOX2 promotes metastasis of breast and PCa cells by promoting EMT mainly through WNT/β-catenin, but not TGF-β or Snail1 signaling [72]. On the contrary, forced expression of naturally-occurring Wnt inhibitor, WIF1 in PCa cells is associated with reduced expression of EMT transcription factors e.g. Slug and Twist and morphological transitions from mesenchymal to epithelial features [73]. All these studies strongly establish a role for Wnt signaling in either inducing or supporting EMT in PCa.
4.2. Androgen Related Signaling
Prostate is an androgen-dependent tissue and requires androgenic and androgen receptor (AR) signaling axis for normal functioning. However, deregulated androgen signaling is one of the most important factors in PCa progression. Initial evidences suggested an inverse relationship between AR levels and androgen-mediated EMT induction [74]. In another study, it was reported that androgen deprivation induces N-cadherin expression and N-cadherin increased in castration-resistant human tumors with established metastases [75]. This was further demonstrated by Sun et al who showed that androgen deprivation induces EMT in both normal prostate and PCa [76]. Additionally, targeting AR in PCa with siRNA promoted PCa cell migration and invasion mediated by CCL2-dependent STAT3 activation and EMT pathways [77]. Though it is now well established that AR signaling axis is a potent modulator of EMT but the interplay between AR signaling and expression of EMT related transcription factors needs to be further explored. Very recently a study suggested AR as a critical regulator of ZEB2 expression during EMT in PCa [78]. Apart from AR, the AR splice variants are found to be expressed in higher amounts in castration-resistant PCa which also showed increased EMT related events [79]. It is therefore speculative to suggest any correlation between AR splice variants and EMT in these settings. In line with this a recent study showed that overexpression of AR splice variants 3 (AR3) modulates the expression of TGF-β and IGF1 pathways, both well-known EMT related signaling pathways. Some EMT-associated genes were also up-regulated in AR splice variants 3 transgenic (AR3Tg) mice prostates [80]. Owing to the role of AR signaling axis in PCa it is not surprising that EMT is heavily modulated AR signaling axis, however, this field still lacks the mechanistic explanations how AR modulate EMT in PCa.
4.3. Heat Shock Proteins
Molecular chaperones including heat shock proteins (HSPs) are important components of core stress response machinery and involved in protein homeostasis, prosurvival signaling and transcriptional networks. HSP27 and 90 are the two most well established proteins that are known to be involved in metastatic PCa. Hance et al provided preliminary evidence that secretory form HSP90 (referred here as extracellular HSP90) induces EMT like events in PCa cells. In support of their concept, they showed that metastatic PCa cells exhibited increased eHsp90 expression relative to their lineage-related nonmetastatic counterparts. Treatment of PCa cells with extracellular Hsp90 promoted cell proliferation and shifted cellular morphology towards a mesenchymal phenotype [81]. Conversely, inhibition of eHsp90 attenuated motility, blocked migration, and reversed cell morphology towards an epithelial phenotype. Shiota et al showed a role for HSP27 mediated regulation of EMT in PCa. To confirm the EMT inducing potential of Hsp27, two epithelial cell lines and ARCaPE were tailored to stably overexpress Hsp27. Hsp27 overexpression in both these cell lines downregulated E-cadherin and increase in vimentin and fibronectin expression. On the contrary, HSP27 knockdown using siRNA in the PCa cell line DU145 resulted in increased E-cadherin and decreased vimentin and fibronectin expression. Hsp27 overexpressing cells also showed upregulated mesenchymal markers such as p-GSK-3β, vimentin, fibronectin, Twist, and N-cadherin at both protein and mRNA levels [82]. Though the evidence correlating HSPs with EMT in PCa is few, it is clear that HSPs are important players during EMT. However, it would be of great interest to examine how HSPs can influence the EMT related events in other conditions like androgen deprivation and drug resistance since HSPs are known to play an important role in therapy resistance.
4.4. Epigenetic Factors
Genetic mutations are a known and one of the major causes in oncogenesis; however, rapidly increasing and overwhelming evidence and experimentation shows the alterations also occur at the epigenetic level. The main epigenetic events related to genetic processes (transcription, replication, recombination, and segregation) are DNA methylation, histone modifications, chromatin remodeling and microRNAs [83]. Recent literature supports the involvement of epigenetics in EMT in a variety of cancers including PCa [41]. The histone methyltransferase MMSET/WHSC1 (Multiple Myeloma SET domain), was recently identified as one of the important epigenetic factors, capable of inducing an EMT like state in PCa cells. Forced expression of MMSET in normal prostate RWPE-1 cells induced TWIST1 expression. Chromatin immunoprecipitation (CHIP) analysis showed that MMSET binds directly to TWIST1 locus, suggesting a direct role of MMSET in the regulation of TWIST1 expression [84]. The classical epithelial marker E-cadherin is often found to be silenced in PCa due to promoter methylation [85–86]. It is intriguing to note that methylation of E-cadherin is a priming event that helps in creating a permissive environment for outgrowth and continued morphogenesis of prostatic ducts at different stages of the development [87]. This again tells us how PCa cells hijack this evolutionary conserved process to decrease the expression of E-cadherin. Another well-known histone methyltransferase i, e EZH2, a component of polycomb repressive complex 2 (PRC2), has been implicated in EMT and is known to repress the E-cadherin expression. EZH2 is also known to suppress disabled homolog 2-interacting protein (DAB2IP) [88]. Forced suppression of DAB2IP was found to activate EMT [89] and its expression was also found to be lost in PCa samples generally through an epigenetic mechanism [90]. Furthermore, BMI, a component of PRC1 is known to modulate EMT and found to be upregulated in PCa [91, 92]. However, it remains to be known whether BMI is able to induce EMT like state in PCa.
4.5. microRNAs
MicroRNAs (miRNAs) are non-coding RNA molecules that consist of 21–23 nucleotides and can bind selectively to mRNAs. Generally, miRNAs are involved in regulation of gene expression mainly through participating in post-transcriptional silencing of target genes. They express differentially in site, tissue and developmental context. miRNAs are known to regulate various important physiological and pathological processes such as differentiation, proliferation, migration, invasion, survival and EMT. Like most cellular genes; the expression of miRNAs is influenced by transcription factor binding, chromosomal rearrangements, genetic and epigenetic alterations [93].
Several miRNAs have been shown to directly target families of EMT transcription factors and are known to facilitate EMT like state in PCa. Based on their possible targets, miRNAs can act both as EMT inducers or inhibitors. For example, the most studied miRNA-200 family which consists of miR-200a, miR-200b, miR-200c, miR-141 and miR-449, are significantly down-regulated during PCa progression and act as tumor suppressors in PCa mainly by inhibiting EMT [94, 95]. Kong et al showed that miR-200 can inhibit the platelet-derived growth factor-D (PDGF-D)-induced EMT in PC3 cells via targeting both ZEB1 and ZEB2 [96]. In benign prostate hyperplasia (BPH) miR-200 can reverse the TGFb-induced EMT phenotype [44]. Similarly, Liu et al evaluated miRNA expression associated with tumorigenesis and EMT in a model of Pten- and TP53-null prostate adenocarcinoma and found that both miRNA-200 in association with miRNA-1 was reduced with progression of prostate adenocarcinoma, and identified Slug as one of the phylogenetically conserved targets of these miRNAs [97]. Forced expression of miRNA-200 inhibited both EMT and tumorigenesis in human and mouse model systems [98]. Other well-known tumor suppressor miRNAs like miRNA-203 and 205 were also found to inhibit EMT in PCa. Both miRNA-203 and 205 inhibit PCa progression via their ability to restore epithelial phenotype of PCa cell lines [99].
In addition, miR-143/-145 cluster that consists of miR-143 and miR-145 are known to inhibit EMT and down-regulated in metastatic PCa. Forced expression of both miRNAs in PC3 cells represses fibronectin and enhances E-cadherin expression and both can reverse EMT [100]. Similarly, miRNA-29b expression was also found to be lower in PCa cells (PC3 and LNCaP) with regard to immortalized prostate epithelial cells [101]. Here, N-cadherin, Twist and Snail expression was down-regulated in PC3 cells expressing miR-29b [102]. Additionally, loss of well-known miR-100 enhances migration, invasion, EMT and stemness properties in PCa cells through targeting Argonaute 2 [103]. Very recently, Gandelini et al demonstrated that miRNA-205 is down-regulated in PCa cells upon cancer associated fibroblasts (CAF) stimulation, mainly via direct transcriptional repression by HIF-1. In this case, forced expression of miR-205 in PCa cells reversed CAF-induced EMT [104]. These studies clearly highlight the enigmatic roles of miR-NAs in EMT. miRNAs can regulate different components of EMT like transcription, post transcription and signaling that are important for EMT induction in epithelial subtypes. These qualities make miRNAs an important prognostic marker and therapeutic tool in EMT-associated PCa progression.
5. OTHER FACTORS
Apart from the some conventional well known factors recent studies had highlighted role of other factors that are involved in EMT-associated with PCa. Wu et al showed monoamine oxidase –A (MAOA) induces EMT in PCa epithelial cells [105]. Knockdown and overexpression studies for MAOA in PCa cell lines showed that MAOA induces EMT mainly via activation of VEGF and its co-receptor neuropilin-1. Authors found MAOA-dependent stimulation of neuropilin-1 expression promoted AKT/FOXO1/TWIST1 signaling, permitting FOXO1 binding at the TWIST1 promoter. Authors also found MAOA/VEGF-A/TWIST1 pathway was activated in high-grade PCa specimens. Other studies had also established the role of VEGF in EMT associated with PCa [106, 107].
6. TRANSCRIPTION FACTORS DRIVING EMT IN PCA
EMT inducers discussed above relay the necessary signals for epithelial switching to the downstream molecules, known as executioners (or transcription factors) that finally accomplish EMT (Fig. 2). Some of the well-established transcription factors (TFs) include Twist1, Snail, Slug, Zeb1, Zeb2 etc. The role of a specific transcription factor in EMT depends on the site specific microenvironment, cell type and functionality of other signaling pathways within the cell [108]. The main function of these EMT related transcription factors is to repress the expression of epithelial associated genes and induction of mesenchymal genes [109]. The information related to the functions and molecular targets of the established TFs in PCa is well known and has been reviewed extensively [43, 110, 111]. Here, we focus more on some of the newly identified TFs like YB-1, p63, OVOL family and their role during EMT in PCa.
Fig. 2. Schematic representation of the induction of EMT by different transcription factors.
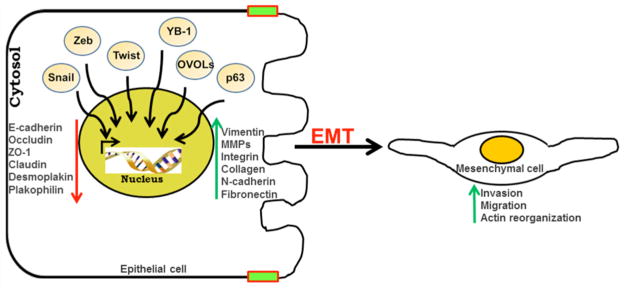
In the presence of a stimulus the transcription factor moves into the nucleus, interacts at different gene loci and further modulates the expression of genes related to migration, invasion and EMT.
6.1. Y Box Protein-1
YB-1 is encoded by the YBX1 gene; a member of DNA- and RNA-binding proteins which contains a highly conserved nucleic-acid-binding motif known as the cold shock domain (CSD) that binds to both types of nucleic acids. Initially, YB-1 was recognized as a repressor factor that binds to an inverted CCAAT box of MHC class II promoters, however using DNA probes it was later found that YB-1 also binds to enhancers of some genes. The YB-1 protein is remarkably multifunctional, and is associated with a variety of oncogenic features like cell proliferation, migration, invasion, EMT, cell cycle progression, DNA damage repair, angiogenesis, and genomic instability in different type of cancers. These features strongly suggest that YB-1 should be considered an oncogene [112, 113]. YB-1 is linked with EMT related factors like Twist1 and clusterin clearly suggesting some involvement of YB-1 in EMT during PCa progression [114, 115]. However, no direct evidence is available for this presumption.
Recently, we observed that the forced expression of YB-1 in prostate epithelial cells induced a mesenchymal morphology that was associated with down-regulation of epithelial markers. Moreover, silencing of YB-1 in different PCa cells was shown to reverse the mesenchymal phenotype to an epithelial morphology, reducing cell migration, invasion and proliferation [1]. Also, a significant inverse correlation between phospho-YB-1Ser102 and E-cadherin in these samples was observed [1]. Overall, it may be concluded that YB-1 has the potential to induce EMT in prostate epithelial cells and has an inverse association with E-cadherin expression.
6.2. p63
The p63 is a part of a well-known family of transcription factors that also include p53 and p73. Due to the presence of two promoters, p63 encodes two proteins; one that contains TAp63 and another ΔNp63 which lacks an N-terminal transactivating (TA) domain homologous to that present in p53. However, the C terminus generates three isoforms (α, β, and γ) of both TAp63 and ΔNp63 due to alternative splicing. Interestingly, ΔNp63α via a second C-terminal TA domain can transactivate a spectrum of genes distinct from that recognized by the N-terminal TA domain [116, 117]. Mice lacking p63 fail to develop prostate. Moreover, p63 is normally expressed in basal cells of the adult prostate but it is nearly lost in differentiated luminal cells. Similarly, PCa cells also barely express p63. Both type of p63 isoforms i.e. TAp63 and ΔNp63 have been shown to regulate the expression of EMT related genes such as ZEB1 and vimentin in PCa. It was found that the effects of p63 on EMT markers are mediated mainly through miR-205 and p53 mutations [118], because mutant p53 can suppress the actions of p63. It has been reported that p63 is highly down-regulated when epithelial EP156T cells underwent EMT to become EPT1 cells [119] and over-expression of ΔNp63 in mesenchymal type cells led to gain of several epithelial characteristics [120, 121].
6.3. OVOLs
Ovo gene family is evolutionarily conserved zinc-finger’s that encodes transcription factors which regulates gene expression in various developmental processes [122]. In Drosophila, the ovo functions for epidermal denticle formation and oogenesis. However, in mammals, three ovo homologues are found, known as OVOL 1, OVOL 2, and OVOL 3. In mice, OVOL 1 is tightly regulated by wnt signaling and is required for hair follicle differentiation, kidney and male germ cell development [123, 124].
In humans, OVOL1 is highly responsive to TGF-β1/BMP7 stimulation through Smad4-dependent pathway. All the members of this gene family act downstream of signaling pathways required for diverse processes during both early and late stages of embryonic development [125]. Recently OVOLs were found to be positively correlated with the E-cadherin expression in primary tumors of PCa. Further analysis by using Oncomine database demonstrated negative correlation of OVOLs with the protein expression of ZEB1 and Vimentin. This transcriptional repression of ZEB1 was mainly regulated by OVOL2 via direct interaction of OVOL2 with in the promoter region of ZEB1. However, OVOL1 fails to show the same effect on ZEB1 expression [126]. Overall these results clearly suggest that the mesenchymal or epithelial state of prostate cells is controlled, in part, by a regulatory feedback loop between the OVOLs and ZEB1 [127].
7. SOME IMPORTANT QUESTIONS
Even though a role for EMT has been observed in PCa development and progression the evidence is not compelling enough for specific targeting of the components involved in EMT. Based on this fact we discuss some issues that need to be addressed. Can EMT be used as a biomarker for staging PCa? Much of the current literature suggests that EMT related events are correlated with high grade and metastatic PCa. The same was also found to be true during the progression of hormone independent PCa. However, the clinical relevance of EMT is frequently questioned due to the lack of confirmatory evidences of full EMT in different grades of human PCa samples. Also right now there is no direct scientific evidence or report that confirms the role of EMT specifically in bone metastasis cases in PCa. All these observations create a limitation in using EMT as a biomarker for high grade and metastatic PCa. Can EMT be developed as a therapeutic target for PCa management? Classical cytotoxic chemotherapy or androgen ablation has proved less beneficial in PCa because of the reappearance of the tumor acquired through drug resistance along with some signs of EMT and stemness. Newer strategies are being used to not only inhibit proliferation but also EMT and stem cell-like properties to prevent the metastatic spread. A similar strategy could be employed against PCa to target cancer stem cells and EMT. In this direction, large numbers of studies have tried design inhibitors of EMT in PCa but with limited success. The reasons for this could be: (a) EMT may not be appropriate therapeutic target, since metastasis also contains evidences of epithelial subtype, (b) The role of EMT in metastatic dissemination is still debatable (c) Failure of desirable effects of EMT inhibition in reversing current aspects of therapy resistance. Though EMT can be induced by various signaling pathways and regulatory networks; however, it is mainly executed by TFs and their targeting remains a challenge due to their intracellular localization. Does PCa microenvironment interact with EMT? Since, tumor microenvironment plays an influential role in determining EMT. In this context, many of the tumor microenvironment components like tumor associated macrophages (TAMs), myeloid-derived suppressor cells, regulatory T cells (MDSCs) and cancer-associated fibroblasts (CAFs) have been observed to be associated with EMT in a variety of epithelial cancers. However, the role of PCa microenvironment components in EMT related events or vice versa remains unclear. It would be interesting to understand if such unique paracrine crosstalk occurs in Pca or other such cancers.
SUMMARY
The ongoing research suggests several important functions of EMT that have been correlated with therapy resistance, stemness, tumor recurrence in PCa. Therefore, it is not surprising that the research on EMT has got attention and is on forefront of PCa research. Many established (Snail, Twist and Zeb families) along with some newly identified TFs (YB-1, p63 and OVOLs) are being linked with EMT. However, a difficulty in therapeutically targeting these TFs remains a challenge for the development of an effective EMT inhibitor. Also the dependency of EMT on different cell type and initiating signalling pathways poses another major challenge in finding new druggable EMT targets. Furthermore, the biggest challenge still is the EMT characterization in PCa samples, as it is heavily affected by the transient and reversible nature of the process. Beside this, current focus of PCa research on tumor microenvironment had another dimension on EMT perspective, as it is clear now, signals arising from microenvironment play a key role in governing EMT and thereby may have a direct impact on either tumor progression or possible clinical outcomes in PCa patients. Future efforts shall focus to delineate the mysteries of EMT will form a basis for developing novel anti-metastatic therapeutic approaches, as well as prognostic or diagnostic markers for PCa management.
Acknowledgments
We apologize to those researchers whose work was not included in this review due to space limitations. The studies included in this review from the author’s laboratory were supported by USPHS Grant RO1 CA160867 to HM. AH was recipient of visiting faculty fellowship (IUSSTF Fellowship/2012/19-2012) from Indo-US S&T Forum.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
Send Orders for Reprints to reprints@benthamscience.ae
References
- 1.Khan MI, Adhami VM, Lall RK, et al. YB-1 expression promotes epithelial-to-mesenchymal transition in prostate cancer that is inhibited by a small molecule fisetin. Oncotarget. 2014;5:2462–74. doi: 10.18632/oncotarget.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 3.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 5.Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22 :361–68. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts BJ, Pashaj A, Johnson KR, et al. Desmosome dynamics in migrating epithelial cells requires the actin cytoskeleton. Exp Cell Res. 201;317:2814–22. doi: 10.1016/j.yexcr.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Nakaya Y, Sheng G. EMT in developmental morphogenesis. Cancer Lett. 2013;341:9–15. doi: 10.1016/j.canlet.2013.02.037. [DOI] [PubMed] [Google Scholar]
- 11.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–49. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakaya Y, Sukowati EW, Wu Y, Sheng G. RhoA and microtubule dynamics control cell-basement membrane interaction in EMT during gastrulation. Nat Cell Biol. 2008;10:765–75. doi: 10.1038/ncb1739. [DOI] [PubMed] [Google Scholar]
- 13.Nakaya Y, Sheng G. Epithelial to mesenchymal transition during gastrulation: an embryological view. Dev Growth Differ. 2008;50 :755–66. doi: 10.1111/j.1440-169X.2008.01070.x. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer-Vaquer M, Viotti AK, Hadjantonakis Transitions betweenepithelial and mesenchymal states and the morphogenesis of the earlymouse embryo. Cell Adhes Migr. 2010;4:447–57. doi: 10.4161/cam.4.3.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliveri P, Tu Q, Davidson EH. Global regulatory logic for specification of an embryonic cell lineage. Proc Natl Acad Sci USA. 2008;105:5955–962. doi: 10.1073/pnas.0711220105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu SY, Ferkowicz M, McClay DR. Ingression of primary mesenchyme cells of the sea urchin embryo: a precisely timed epithelial mesenchymal transition. Birth Defects Res C Embryo Today. 2007;81:241–52. doi: 10.1002/bdrc.20113. [DOI] [PubMed] [Google Scholar]
- 17.Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001;21:8184–188. doi: 10.1128/MCB.21.23.8184-8188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 19.Jorda M, Olmeda D, Vinyals A, et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci. 2005;118:3371–385. doi: 10.1242/jcs.02465. [DOI] [PubMed] [Google Scholar]
- 20.Haraguchi M, Okubo T, Miyashita Y, et al. Snail regulates cell-matrix adhesion by regulation of the expression of integrins and basement membrane paroteins. J Biol Chem. 2008;283:23514–523. doi: 10.1074/jbc.M801125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Kalcheim C, Ben-Yair R. Cell rearrangements during development of the somite and its derivatives. Curr Opin Genet Dev. 2005;15:371–80. doi: 10.1016/j.gde.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Christ B, Huang R, Scaal M. Amniote somite derivatives. Dev Dyn. 2007;236:2382–96. doi: 10.1002/dvdy.21189. [DOI] [PubMed] [Google Scholar]
- 24.Ishii Y, Langberg J, Rosborough K, Mikawa T. Endothelial cell lineages of the heart. Cell Tissue Res. 2009;335:67–73. doi: 10.1007/s00441-008-0663-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–35. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 26.Kovacic JC, Mercader N, Torres M, et al. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gjorevski N, Nelson CM. Integrated morphodynamic signalling of the mammary gland. Nat Rev Mol Cell Biol. 2011;12:581–93. doi: 10.1038/nrm3168. [DOI] [PubMed] [Google Scholar]
- 28.Cowin P, Wysolmerski J. Molecular mechanisms guiding embryonic mammary gland development. Cold Spring Harb Perspect Biol. 2010;2:a003251. doi: 10.1101/cshperspect.a003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mutsaers SE. Mesothelial cells: their structure, function and role in serosal repair. Respirology. 2002;7:171–91. doi: 10.1046/j.1440-1843.2002.00404.x. [DOI] [PubMed] [Google Scholar]
- 30.Dressler GR. Advances in early kidney specification, development and patterning. Development. 2009;136:3863–874. doi: 10.1242/dev.034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4:a008300. doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schluter MA, Margolis B. Apical lumen formation in renal epithelia. J Am Soc Nephrol. 2009;20:1444–452. doi: 10.1681/ASN.2008090949. [DOI] [PubMed] [Google Scholar]
- 33.Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell. 2010;18:175–89. doi: 10.1016/j.devcel.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–65. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 35.Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009;326:4–35. doi: 10.1016/j.ydbio.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 36.Villasenor A, Chong DC, Henkemeyer M, Cleaver O. Epithelial dynamics of pancreatic branching morphogenesis. Development. 2010;137:4295–305. doi: 10.1242/dev.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012;139:3471–486. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 38.Bertrand S, Escriva H. Evolutionary crossroads in developmental biology: amphioxus. Development. 2011;138:4819–830. doi: 10.1242/dev.066720. [DOI] [PubMed] [Google Scholar]
- 39.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–48. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 40.Eichmann A, Yuan L, Moyon D, et al. Vascular development: from precursor cells to branched arterial and venous networks. Int J Dev Biol. 2005;49:259–67. doi: 10.1387/ijdb.041941ae. [DOI] [PubMed] [Google Scholar]
- 41.Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22:361–68. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;15:2192–206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol. 201;8:428–39. doi: 10.1038/nrurol.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Principe DR, Doll JA, Bauer J, et al. TGF-β: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. 2014;10(2):djt369. doi: 10.1093/jnci/djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu S, Yu W, Lv TJ, et al. Evidence of TGF-β1 mediated epithelial-mesenchymal transition in immortalized benign prostatic hyperplasia cells. Mol Membr Biol. 2014;31:103–10. doi: 10.3109/09687688.2014.894211. [DOI] [PubMed] [Google Scholar]
- 46.Slabáková E, Pernicová Z, Slavíčková E, et al. TGF-β1-induced EMT of non-transformed prostate hyperplasia cells is characterized by early induction of SNAI2/Slug. Prostate. 2011;71:1332–343. doi: 10.1002/pros.21350. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Q, Helfand BT, Jang TL, et al. Nuclear factor-kappaB-mediated transforming growth factor-beta-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res. 2009;15:3557–567. doi: 10.1158/1078-0432.CCR-08-1656. [DOI] [PubMed] [Google Scholar]
- 48.Cho KH, Jeong KJ, Shin SC, et al. STAT3 mediates TGF-β1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett. 2013;336:167–73. doi: 10.1016/j.canlet.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 49.Cho KH, Choi MJ, Jeong KJ, et al. A ROS/STAT3/HIF-1α signaling cascade mediates EGF-induced TWIST1 expression and prostate cancer cell invasion. Prostate. 2014;74:528–36. doi: 10.1002/pros.22776. [DOI] [PubMed] [Google Scholar]
- 50.Chaudhry P, Fabi F, Singh M, et al. Prostate apoptosis response-4 mediates TGF-β-induced epithelial-to-mesenchymal transition. Cell Death Dis. 2014;5:e1044. doi: 10.1038/cddis.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morimoto K, Tanaka T, Nitta Y, et al. NEDD9 crucially regulates TGF-β-triggered epithelial-mesenchymal transition and cell invasion in prostate cancer cells: involvement in cancer progressiveness. Prostate. 2014;74:901–10. doi: 10.1002/pros.22809. [DOI] [PubMed] [Google Scholar]
- 52.Pu H, Collazo J, Jones E, et al. Dysfunctional transforming growth factor-beta receptor II accelerates prostate tumorigenesis in the TRAMP mouse model. Cancer Res. 2009;69:7366–374. doi: 10.1158/0008-5472.CAN-09-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5:1063–071. [PubMed] [Google Scholar]
- 54.Acevedo VD, Gangula RD, Freeman KW, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12:559–71. doi: 10.1016/j.ccr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Sugiyama N, Varjosalo M, Meller P, et al. Fibroblast growth factor receptor 4 regulates tumor invasion by coupling fibroblast growth factor signaling to extracellular matrix degradation. Cancer Res. 2010;70:7851–861. doi: 10.1158/0008-5472.CAN-10-1223. [DOI] [PubMed] [Google Scholar]
- 56.Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–13. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 57.Holzmann K, Grunt T, Heinzle C, et al. Alternative Splicing of Fibroblast Growth Factor Receptor IgIII Loops in Cancer. J Nucliec Acids. 2012;2012:950508. doi: 10.1155/2012/950508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graham TR, Zhau HE, Odero-Marah VA, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 59.Chen CL, Mahalingam D, Osmulski P, et al. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013;73:813–26. doi: 10.1002/pros.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giannoni E, Bianchini F, Masieri L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 61.Zhau HE, Odero-Marah V, Lue HW, et al. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis. 2008;25:601–10. doi: 10.1007/s10585-008-9183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gan Y, Shi C, Inge L, et al. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene. 2010;29:4947–958. doi: 10.1038/onc.2010.240. [DOI] [PubMed] [Google Scholar]
- 63.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501–11. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang S, Wang X, Iqbal S, et al. Epidermal growth factor promotes protein degradation of epithelial protein lost in neoplasm (EPLIN), a putative metastasis suppressor, during epithelial-mesenchymal transition. J Biol Chem. 2013;288:1469–479. doi: 10.1074/jbc.M112.438341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lim M, Chuong CM, Roy-Burman P. PI3K, Erk signaling in BMP7-induced epithelial-mesenchymal transition (EMT) of PC-3 prostate cancer cells in 2- and 3-dimensional cultures. Horm Cancer. 2011;2:298–09. doi: 10.1007/s12672-011-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mulholland DJ, Kobayashi N, Ruscetti M, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cnacer Res. 2012;72:1878–889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang L, Graham PH, Hao J, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma B, Wells A. The Mitogen-activated Protein (MAP) Kinases p38 and Extracellular Signal-regulated Kinase (ERK) Are Involved in Hepatocyte-mediated Phenotypic Switching in Prostate Cancer Cells. J Bio Chem. 2014;289:11153–161. doi: 10.1074/jbc.M113.540237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang L, Pu Y, Hu WY, et al. The role of Wnt5a in prostate gland development. Dev Bio. 2009;328:188–99. doi: 10.1016/j.ydbio.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang YG, Luo Y, He DL, et al. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol. 2007;14:1034–039. doi: 10.1111/j.1442-2042.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- 71.Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of β-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1α. Cancer Invest. 2011;29 :377–82. doi: 10.3109/07357907.2010.512595. [DOI] [PubMed] [Google Scholar]
- 72.Li X, Xu Y, Chen Y, Chen S, et al. SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/β-catenin signal network. Cancer Lett. 2013;336:379–89. doi: 10.1016/j.canlet.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 73.Yee DS, Tang Y, Li X, et al. The Wnt inhibitory factor 1 restoration in prostate cancer cells was associated with reduced tumor growth, decreased capacity of cell migration and invasion and a reversal of epithelial to mesenchymal transition. Mol Cancer. 2010;9:162. doi: 10.1186/1476-4598-9-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matuszak EA, Kyprianou N. Androgen regulation of epithelial–mesenchymal transition in prostate tumorigenesis. Expert Rev Endocrinol Metab. 2011;6:469–82. doi: 10.1586/eem.11.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jennbacken K, Tesan T, Wang W, et al. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr Relat Cancer. 2010;17:469–79. doi: 10.1677/ERC-10-0015. [DOI] [PubMed] [Google Scholar]
- 76.Sun Y, Wang BE, Leong KG, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72:527–36. doi: 10.1158/0008-5472.CAN-11-3004. [DOI] [PubMed] [Google Scholar]
- 77.Izumi K, Fang LY, Mizokami A, et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med. 2013;5:1383–401. doi: 10.1002/emmm.201202367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacob S, Nayak S, Fernandes G, et al. Androgen receptor as a regulator of ZEB2 expression and its implications in epithelial-to-mesenchymal transition in prostate cancer. Endocr Relat Cancer. 2014;21:473–86. doi: 10.1530/ERC-13-0514. [DOI] [PubMed] [Google Scholar]
- 79.Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun F, Chen HG, Li W, et al. Androgen receptor splice variant AR3 promotes prostate cancer via modulating expression of autocrine/paracrine factors. J Biol Chem. 2014;289:1529–39. doi: 10.1074/jbc.M113.492140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hance MW, Dole K, Gopal U, et al. Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J Biol Chem. 2012;287:37732–744. doi: 10.1074/jbc.M112.389015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shiota M, Bishop JL, Nip KM, et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013;73:3109–119. doi: 10.1158/0008-5472.CAN-12-3979. [DOI] [PubMed] [Google Scholar]
- 83.Stadler SC, Allis CD. Linking epithelial-to-mesenchymal-transition and epigenetic modifications. Semin Cancer Biol. 2012;22:404–10. doi: 10.1016/j.semcancer.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ezponda T, Popovic R, Shah MY, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32:2882–890. doi: 10.1038/onc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li LC, Zhao H, Nakajima K, et al. Methylation of the E-cadherin gene promoter correlates with progression of prostate cancer. J Urol. 2001;166:705–09. [PubMed] [Google Scholar]
- 86.Saha B, Kaur P, Tsao-Wei D, et al. Unmethylated E-cadherin gene expression is significantly associated with metastatic human prostate cancer cells in bone. Prostate. 2008;68:1681–688. doi: 10.1002/pros.20836. [DOI] [PubMed] [Google Scholar]
- 87.Keil KP, Abler LL, Mehta V, et al. DNA methylation of E-cadherin is a priming mechanism for prostate development. Dev Biol. 2014;387:142–53. doi: 10.1016/j.ydbio.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cao Q, Yu J, Dhanasekaran SM, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27:7274–284. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xie D, Gore C, Liu J, et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci USA. 2010;107:2485–90. doi: 10.1073/pnas.0908133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen H, Toyooka S, Gazdar AF, Hsieh JT. Epigenetic regulation of a novel tumor suppressor gene (hDAB2IP) in prostate cancer cell lines. J Bio Chem. 2003;278:3121–130. doi: 10.1074/jbc.M208230200. [DOI] [PubMed] [Google Scholar]
- 91.Ammirante M, Kuraishy AI, Shalapour S, et al. An IKKα-E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013;27:1435–440. doi: 10.1101/gad.220202.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Siddique HR, Parray A, Zhong W, et al. BMI1, stem cell factor acting as novel serum-biomarker for Caucasian and African-American prostate cancer. PLoS One. 2013;8:e52993. doi: 10.1371/journal.pone.0052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25 :200–07. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watahiki A, Wang Y, Morris J, et al. MicroRNAs associated with metastatic prostate cancer. PLoS One. 2011;6:e24950. doi: 10.1371/journal.pone.0024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fang YX, Gao WQ. Roles of microRNAs during prostatic tumorigenesis and tumor progression. Oncogene. 2014;33:135–47. doi: 10.1038/onc.2013.54. [DOI] [PubMed] [Google Scholar]
- 96.Kong D, Li Y, Wang Z, et al. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27:1712–721. doi: 10.1002/stem.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elsa JS, Graça I, Baptista T, et al. Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics. 2013;8:548–558. doi: 10.4161/epi.24519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu YN, Yin JJ, Abou-Kheir W, et al. miR -1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32:296–306. doi: 10.1038/onc.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Viticchiè G, Lena AM, Latina A, et al. miR -203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle. 2011;10:1121–131. doi: 10.4161/cc.10.7.15180. [DOI] [PubMed] [Google Scholar]
- 100.Bhatnagar N, Li X, Padi SK, et al. Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis. 2010;1:e105. doi: 10.1038/cddis.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peng X, Guo W, Liu T, et al. Identification of miRs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT. Plos One. 2011;6:e20341. doi: 10.1371/journal.pone.0020341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ru P, Steele R, Newhall P, et al. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012;11:1166–173. doi: 10.1158/1535-7163.MCT-12-0100. [DOI] [PubMed] [Google Scholar]
- 103.Wang M, Ren D, Guo W, et al. Loss of miR-100 enhances migration, invasion, epithelial-mesenchymal transition and stemness properties in prostate cancer cells through targeting Argonaute 2. Int J Oncol. 2014;45:362–72. doi: 10.3892/ijo.2014.2413. [DOI] [PubMed] [Google Scholar]
- 104.Gandellini P, Giannoni E, Casamichele A, et al. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antiox Redox Sig. 2014;20:1045–059. doi: 10.1089/ars.2013.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu JB, Shao C, Li X, et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J Clin Invest. 2014;124:2891–908. doi: 10.1172/JCI70982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mak P, Leav I, Pursell B, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell. 2010;17:319–32. doi: 10.1016/j.ccr.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gonzalez-Moreno O, Lecanda J, et al. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Exp Cell Res. 2010;316:554–67. doi: 10.1016/j.yexcr.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 108.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 109.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–94. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 110.Smith BN, Odero-Marah VA. The role of Snail in prostate cancer. Cell Adeh Migr. 2012;6:433–41. doi: 10.4161/cam.21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions ofTwist1 and underlying molecular mechanisms. Cell Res. 2012;22:90–106. doi: 10.1038/cr.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Davies AH, Dunn SE. YB-1 drives preneoplastic progression: Insight into opportunities for cancer prevention. Oncotarget. 2011;2:401–406. doi: 10.18632/oncotarget.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lasham A, Print CG, Woolley AG, Dunn SE, Braithwaite AW. YB-1: oncoprotein, prognostic marker and therapeutic target? Biochem J. 2013;449:11–23. doi: 10.1042/BJ20121323. [DOI] [PubMed] [Google Scholar]
- 114.Shiota M, Izumi H, Onitsuka T, et al. Twist promotes tumor cell growth through YB-1 expression. Cancer Res. 2008;68:98–105. doi: 10.1158/0008-5472.CAN-07-2981. [DOI] [PubMed] [Google Scholar]
- 115.Pauliina M, Munne, Gu Y, et al. TP53 supports basal-like differentiation of mammary epithelial cells by preventing translocation of deltaNp63 into nucleoli. Sci Rep. 2014;4:4663. doi: 10.1038/srep04663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, Yan W, Chen X. P63 regulates tubular formation via epithelial-to-mesenchymal transition. Oncogene. 2014;33:1548–57. doi: 10.1038/onc.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shiota M, Zoubeidi A, Kumano M, et al. Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol Cancer Res. 2011;9:1755–66. doi: 10.1158/1541-7786.MCR-11-0379. [DOI] [PubMed] [Google Scholar]
- 118.Straub WE, Weber TE, Schäfer B, et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis. 2010;1:e5. doi: 10.1038/cddis.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Candi E, Agostini M, Melino G, Bernassola F. How the TP53 Family Proteins TP63 and TP73 Contribute to Tumorigenesis: Regulators and Effectors. Hum Mutat. 2014;35:702–14. doi: 10.1002/humu.22523. [DOI] [PubMed] [Google Scholar]
- 120.Tucci P, Agostini M, Grespi F, et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc Natl Acad Sci USA. 2012;109:15312–317. doi: 10.1073/pnas.1110977109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Olsen JR, Oyan AM, Rostad K, et al. p63 attenuates epithelial to mesenchymal potential in an experimental prostate cell model. PLoS One. 2013;8:e62547. doi: 10.1371/journal.pone.0062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li B, Dai Q, Li L, et al. Ovol2, a mammalian homolog of Drosophila ovo: gene structure, chromosomal mapping, and aberrant expression in blind-sterile mice. Genomics. 2002;80:319–25. doi: 10.1006/geno.2002.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mackay DR, Hu M, Li B, Rhéaume C, Dai X. The mouse Ovol2 gene is required for cranial neural tube development. Dev Biol. 2006;291:38–52. doi: 10.1016/j.ydbio.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Unezaki S, Horai R, Sudo K, Iwakura Y, Ito S. Ovol2/Movo, a homologue of Drosophila ovo, is required for angiogenesis, heart formation and placental development in mice. Genes Cells. 2007;12:773–85. doi: 10.1111/j.1365-2443.2007.01084.x. [DOI] [PubMed] [Google Scholar]
- 125.Zhang T, Zhu Q, Xie Z, et al. The zinc finger transcription factor Ovol2 acts downstream of the bone morphogenetic protein pathway to regulate the cell fate decision between neuroectoderm and mesendoderm. J Biol Chem. 2013;288:6166–77. doi: 10.1074/jbc.M112.418376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roca H, Hernandez J, Weidner S, et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS One. 2013;8:e76773. doi: 10.1371/journal.pone.0076773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee B, Villarreal-Ponce A, Fallahi M, et al. Transcriptional mechanisms link epithelial plasticity to adhesion and differentiation of epidermal progenitor cells. Dev Cell. 2014;29:47–58. doi: 10.1016/j.devcel.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]