

Synopsis
Alopecia Areata (AA) is a recurrent autoimmune type of hair loss that affects about 5.3 million people in the United States alone. Despite being the most prevalent autoimmune disease, affecting more individuals than most other autoimmune diseases combined, the molecular and cellular mechanisms underlying this complex disease are still poorly understood, and rational treatments are lacking. It is currently accepted that AA is an autoimmune disease that occurs in genetically susceptible individuals and that environmental factors play a role in the development and progression of the disease. However, further efforts are necessary to clearly pinpoint the causes and molecular pathways leading to this disease and, most importantly, to find evidence-based treatments to treat AA. Here, we will focus on the central role of genetics for gaining insight into disease pathogenesis and setting the stage for the rational development of novel effective therapeutic approaches. This is an exciting new era marking the beginning of translational research in AA based on genetic findings.
1. Alopecia Areata: a common autoimmune disease
1.1 Epidemiology of AA
Alopecia Areata (AA) is a frequent autoimmune disease with a lifetime risk of about 1.7% in the general population, including males and females across all ethnic groups (1). The prevalence of AA is 0.1–0.2%, worldwide as well as in the US and is responsible for 0.7–3% of patients seen by dermatologists (2, 3). The peak incidence of AA appears to occur among 15–29 year-old individuals, with as many as 44% of people having onset of disease before their 20s and less than 30% of patients having onset after their 40s. The clinical course of AA is highly unpredictable, and it can occur at any age from birth to late decades of life.
1.2 Clinical findings and etiology
AA is a nonscarring type of hair loss that usually manifests suddenly as one or more well-defined circumscribed areas that can occur anywhere in the body, although they are most frequently seen on the scalp (90% of cases) (3–5). The disease can then remit spontaneously or, alternatively, the initial patches can coalesce and progress to cover the entire scalp (alopecia totalis or AT) or even the entire body surface (alopecia universalis or AU). The clinical heterogeneity and unpredictable course of AA is a major source of distress for affected individuals.
Epidemiological studies in AA have demonstrated that a history of autoimmune disease increases the risk of AA (6). Specific reported associations include AA with thyroid disease,(4, 7–11) celiac disease (CeD) (12), rheumatoid arthritis (RA) (7, 8, 13), vitiligo (4, 7–9, 14, 15) and type 1 diabetes (T1D) (7, 10, 13, 16). Although the exact pathophysiology of AA is not clear, AA is a T-lymphocyte mediated autoimmune condition that occurs in genetically susceptible individuals (17). Histopathological findings have revealed inflammatory perifollicular infiltrates of T-cells around anagen (growth phase) hair follicles (“swarm of bees”), which consist of both CD4+ and CD8+ T-cells (18, 19). The focus of the attack by the infiltrating lymphocytes is the base of the hair follicle, such that the stem cell compartment is spared from destruction and, therefore, hair regrowth remains possible. Increased levels of autoantibodies, cytokine abnormalities and increased prevalence of autoimmune comorbidities have been described in AA patients (20, 21). An acute onset of disease has been documented in affected individuals at times of profound stress, grief or fear (22), indicating that besides the autoimmune and genetic components, environmental (non-genetic) factors may contribute to trigger the manifestation of this disease, thus supporting the multi-factorial etiology of AA.
1.3 Lack of cure or preventive treatment for AA
AA can cause tremendous emotional and psychosocial distress in affected individuals and their families. Patients can experience profound feelings and frustrations such as loneliness, isolation, anger, depression, and embarrassment, among many others. However, despite the high prevalence and extreme psychosocial burden of AA, there are no evidence-based treatments as yet, and treatment of AA is symptomatic and directed toward halting disease activity (23). A comprehensive Cochrane analysis assessment of 17 randomized control trials (RCTs) involving a total of 540 participants found no proven treatment for AA (24). These trials included 6–85 individuals each, and treatments included topical and oral corticosteroids, topical cyclosporine, photodynamic therapy, and topical minoxidil. Even though topical corticosteroids and minoxidil have been widely used to reduce inflammation and/or stimulate hair growth, respectively, and appear to be safe, there is no convincing evidence that they are beneficial in the long term. Most trials have been poorly reported and/or are small and underpowered, resulting in inconclusive results (17). No RCTs were found on the use of other drugs that are also commonly used for the treatment of AA, such as diphencyprone, dinitrochlorobenzene, dithranol or intralesional corticosteroids.
Because of this lack of evidence-based treatment for AA and due to the high rate of spontaneous remission, particularly in individuals with a recent onset and small patches of hair loss, the initial approach typically includes topical or intralesional steroids or observation. However, approximately 20–30% of cases fail to resolve using these strategies and only one-third of all cases achieve complete recovery for about 10–15 years (25).
2. The impact of genetic studies in AA research
2.1 Candidate gene and linkage studies
Genetics and disease gene discovery are promising and robust strategies to gain critical insights into pathogenic mechanisms in general. In the case of AA, initial evidence supporting a complex, polygenic basis for disease was supported by multiple studies revealing; (1) positive family history (26–28), (2) twin concordance (29, 30), (3) familial aggregation (26), (4) human leukocyte antigen (HLA) associations (31, 32) and (5) studies in animal models (33). The first genetic studies in AA were candidate gene association studies. In these studies, a single gene is chosen base upon a previous hypothesis about its function (usually in another autoimmune disease) and then tested for association in a small sample of cases and controls. These studies were fruitful as they identified associations to genes residing in the HLA region (HLA-DQB1, HLA-DRB1, HLA-A, HLA-B, HLA-C, NOTCH4, MICA) and also outside of this region (PTPN22, AIRE) (34).
More recently, it became possible to survey the entire genome in a nonbiased way for evidence of genetic contributions to disease using genome-wide genetic studies. These studies, which identify genes based on their position in the genome (as opposed to their function), are very powerful to uncover disease mechanisms when not much is known about the disease etiology. Strong evidence for linkage is one of the most reliable evidences that a disease has a genetic component, although these studies usually result in association of large regions of the genome, each containing many genes, and do not allow identification of particular causal variants. We previously performed a genome-wide linkage study and found several regions that co-segregate with AA among families, providing compelling evidence that the disease is in fact polygenic (35).
2.2 Genome-wide association studies (GWAS) and AA
In recent years, the identification of genetic markers for which an allele exists at a higher frequency among a group of unrelated cases, relative to a group of unrelated controls, has become possible with genome-wide association studies (GWAS). This technique, which most frequently uses single nucleotide polymorphism (SNP) markers, can identify much smaller regions or linkage disequilibrium (LD) blocks, compared to linkage studies. GWAS can survey between 500,000 and one million genetic markers among hundreds or thousands of people, counting alleles at each SNP, to identify regions in the genome where the distribution of alleles is skewed in the presence of disease.
We recently completed the first GWAS in AA interrogating allele frequencies across 500,000 SNPs between a group of 1054 unrelated AA patients and 3278 population-based controls (17). We identified a number of 139 SNPs that exceeded genome-wide significance (p<5×10−7), which clustered in eight regions across the genome and implicated genes of the immune system, as well as genes that are unique to the hair follicle: (1) 2q33.2 containing the CTLA4 gene; (2) 4q27 containing the IL2/IL21 locus; (3) 6p21.32 containing the HLA class II region; (4) 6q25.1 which harbors the ULBP gene cluster; (5) 9q31.1 containing syntaxin 17 (STX17); (6) 10p15.1 containing IL2RA; (7) 11q13 containing peroxiredoxin 5 (PRDX5); and (8) 12q13 containing Eos. Interestingly, a number of risk loci identified in our GWAS were shared with other forms of autoimmunity, such as RA, T1D, CeD, systemic lupus erythematosus (SLE), multiple sclerosis (MS) and psoriasis, in particular CTLA4, IL2/IL2RA, IL21, NKG2D ligands and genes critical to the function of regulatory T cells. Importantly, all of our loci have been replicated (36).
2.3 Immune genes associated with AA
2.3.1 CTLA4 on chromosome 2q33.2
Interestingly, extensive previous evidence arising from genetic studies exists for the involvement of CTLA4 in at least 30 different autoimmune diseases, including T1D, MS, Graves disease (GD), RA, CeD and SLE. Independent evidence supporting these findings is the association of CTLA4 with four different autoimmune diseases, which have been found in six independent GWAS according to the NIH database of GWAS (http://www.genome.gov/gwastudies). These studies associated CTLA4 with AA (our study) (17, 37), RA (38, 39), T1D (40, 41) and CeD (42). Importantly, our GWAS findings regarding the association of CTLA4 with AA have been replicated for the first time in an independent cohort (37). This group genotyped 22 SNPs across the CTLA4 locus in about 1200 cases and 1200 controls and demonstrated statistically significant association of six SNPs. One of these SNPs, rs231775, occurs in the coding region of CTLA4 and results in an amino acid substitution. The biological function of CTLA4 will be described below (see section 4.1).
2.3.2 IL2/IL21 on chromosome 4q27
IL2 and IL21 are two closely related cytokines and their encoded genes are adjacent, with similar exon/intron structures, which suggests that they might have arisen by gene duplication (43, 44). Although both cytokines are known to promote the function of effector CD8+ T cells, an antagonistic relationship between the actions of IL2 and IL21 on the differentiation of CD8+ T cells has been demonstrated (45).
Many of the immunosuppressive drugs commonly used to treat autoimmune diseases function either by inhibiting the production of IL-2 by antigen-activated T cells (corticosteroids, cyclosporine, tacrolimus, sirolimus) or by blocking IL-2 receptor signaling. Cyclosporine, tacrolimus and sirolimus have antiproliferative properties by inhibiting response to IL-2 and thereby preventing expansion and function of antigen-selected T cells.
2.3.3 HLA on chromosome 6p21.32
The HLA super locus, residing on chromosome 6, contains a large number of genes related to the immune system, including genes encoding cell-surface antigen-presenting proteins among many others. The HLA class II region contains genes responsible for foreign antigen presentation to T-cells, which specifically stimulate proliferation of helper T cells, which later stimulate antibody production by B cells or activation of other immune cells. There are different HLA types, which are inherited, and some of which have been overwhelmingly associated with autoimmune diseases. Several candidate gene studies have also previously demonstrated HLA associations with AA (34).
In our GWAS, we found the strongest association between the HLA class II locus and AA. One hundred and five of our 139 SNPs that exceeded statistical significance reside in the HLA and genome-wide, both the most significant SNP, rs9275572 (p = 1.38×10−35), as well as the SNP with the greatest magnitude of effect, rs11752643 (OR = 5.56; 95%CI: 3.03–10.00) were found here.
2.3.4 ULBP3/ULBP6 on chromosome 6q25.1
Our GWAS was the first to identify the cytomegalovirus UL16-binding protein (ULBP) gene cluster in association with an autoimmune disease. This gene cluster encodes activating ligands for the natural killer cell receptor NKG2D that have not been previously implicated in any autoimmune disease, and therefore had been missed from candidate gene studies.
NKG2D is a homodimeric activating receptor that is expressed on the surface of not only all natural killer (NK) cells, but also of most NKT cells, all human CD8+ cytotoxic T cells and activated mouse CD8+ T cells, subsets of gamma-delta T cells and, under certain circumstances such as in RA patients, CD4+ T cells. Although NKG2D is expressed constitutively on human and mouse NK cells, cytokines such as IL-15, IL-2 and IFN-gamma upregulate its expression on human NK cells (46). The NKG2D receptor is remarkable in that it can bind to a wide range of different ligands, which are distantly related homologs of MHC class I proteins.
Our GWAS results point to the specific LD block containing ULBP3 and ULBP6 as being strongly implicated in AA. Although our data shows for the first time the involvement of the ULBP family of NKG2D ligands (NKG2DLs) in the pathogenesis of an autoimmune disease (17), other NKG2DLs have been associated with various autoimmune disorders (47). For example, specific MICA alleles are overrepresented in RA, inflammatory bowel disease and T1D patients, implicating a role in disease pathogenesis (48, 49). Likewise, MICB polymorphisms are also associated with CeD, ulcerative colitis, and MS (50–52). By probing the role of ULBP3 in disease pathogenesis, we also showed that its expression in lesional scalp from patients with AA is markedly upregulated in the hair follicle dermal sheath during active disease.
2.3.5 IL2RA on chromosome 10p15.1
The IL-2-receptor α (IL2RA or CD25) is an important regulatory T cell marker, and genetic polymorphisms of IL2RA gene are associated with a number of autoimmune diseases, such as T1D, MS, GD, SLE, RA, systemic sclerosis and now AA (17, 53–57). Regulatory T cells function to maintain the immune tolerance and have been reported to have an important role in several autoimmune diseases.
Genetic variants in the IL2RA locus differentially confer risk to MS and T1D, demonstrating genetic heterogeneity of the associated polymorphisms and risk alleles between MS and T1D (56, 58). Additionally, several independent genetic variants of IL2RA correlate with the levels of a soluble form of the IL2 receptor (sIL2RA) in human serum. High concentrations of sIL2RA are found in sera from healthy individuals and have been reported to be increased in subjects with autoimmune disease, inflammation and infection (59–61). However, T1D susceptibility genotypes of IL2RA have been associated with reduced circulating levels of sIL2RA, suggesting that an inherited lower immune responsiveness predisposes to this autoimmune disease (55). These results suggest some discordance between sIL2RA levels and disease susceptibility and further studies addressing the causality of sIL2RA in autoimmune disease will be needed. Independent biological pathways contributing to disease susceptibility may include IL2RA transcriptional regulation, cell surface expression levels of IL2RA and serum sIL2RA levels (56).
One of the most remarkable findings in our GWAS in AA was the presence of association at both the IL2 locus and its receptor IL2RA, suggesting a prominent role for this signalling pathway in AA disease pathogenesis.
2.3.6 Eos/ERBB3 on chromosome 12q13
The Eos locus in chromosome 12q13 has been previously associated with T1D, placing this locus among the shared autoimmune loci (41). We also identified a significant GWAS association on a haplotype block on chromosome 12q13, which contains several genes, with AA. Among these, Eos emerged as a plausible candidate gene, given its know function in CD4+ regulatory T cells. These cells are known to maintain immunological self-tolerance and immune homeostasis by suppressing aberrant immune responses, a process that is dependent on the transcription factor Foxp3-dependent gene activation (62). Recently, the zinc-finger transcription factor of the Ikaros family – Eos – was identified as a critical mediator of Foxp3-dependent gene silencing in regulatory T cells (63). Eos silencing in these cells causes them to lose their ability to suppress immune responses, demonstrating the crucial role of Eos for regulatory T cell specification and function (63).
Despite this recent evidence making Eos a compelling candidate gene at this locus, ERBB3 has also been previously implicated as a plausible candidate gene at this locus for autoimmune diseases (41). It is possible that the underlying susceptibility allele(s) may influence both Eos and ERBB3 transcripts.
2.3.7 IL13 on chromosome 5q31 and CLEC16A on 16p13.13
As mentioned above, all loci from our initial GWAS have been corroborated by independent studies (35–37, 64). The most recent of these studies further identified IL13 and CLEC16A as two additional gene associations that showed a nominal signal in our GWAS and have now surpassed the genome wide significance threshold in a meta-analysis (36).
IL-13 is a cytokine classically associated with CD4 T helper 2 responses, although other cell types, including innate-like T cells, can also express this cytokine. As such, it is thought to be important in the pathogenesis allergic responses and associated diseases. Indeed, IL13-proximal SNPs have been associated with asthma(65, 66) as well as with other autoimmune diseases, including psoriasis (67, 68).
CLEC16A encodes a protein with unknown function. CLEC16A contains a putative C-type lectin domain and a likely immunoreceptor tyrosine-based activation motif, or ITAM. C-type lectins are receptors that bind carbohydrates and function as adhesion receptors and pathogen recognition receptors (69); CLEC16A may therefore be important in engendering or modulating immune responses. Interestingly, CLEC16A has been associated with MS (70, 71) and T1D (72, 73), further supporting its role as a modulator of the immune response.
2.4 Hair follicle-specific genes
2.4.1 PRDX5 on chromosome 11q13
Reactive oxygen species are a potentially deleterious byproduct of mitochondrial respiration that can damage DNA, proteins, and organelle membranes. Peroxiredoxins (PRDXs) are a family of enzymes responsible for quenching reactive oxygen species. PRDX5 is expressed in many tissues, including the hair follicle, and is induced under conditions of cellular stress. Upregulation of PRDX5 may confer an anti-apoptotic phenotype and may ultimately enable survival of aberrant cells which harbor danger signals, leading to presentation of damaged self-antigens to the immune system and subsequently autoimmune disease (74, 75). In a mouse model of diabetes, overexpression of a PRDX in islet cells protected against hyperglycemia and hypoinsulinemia, illustrating the protective function of PRDXs in an end-organ immune target (76). A GWAS meta-analysis for Crohn’s disease demonstrated a statistically significant association for the same allele that we identified in AA (77).
2.4.2 STX17 on chromosome 9q31.1
STX17 belongs to a family of molecules called syntaxins, which belong to the SNARE superfamily. SNARE complexes are involved in vesicular trafficking and membrane fusion. No other human disease associations with STX17 to date have been identified. However, an insertion with enhancer-like activity in an intronic segment of STX17 was found to be responsible for the gray hair phenotype in horses (78, 79). This finding is intriguing since AA is known to preferentially affect pigmented hair follicles and to spare gray hairs.
3. Autoimmune diseases: common cause, common treatment
It has been suspected that autoimmune diseases affecting different end organs in fact share common disease mechanisms. First, through epidemiological studies and later using genetic studies, researchers have demonstrated certain similarities at the molecular level, i.e., susceptibility regions on the chromosomes or the involvement of common genes (80). Although the etiology of autoimmune diseases is still unclear, these advances have contributed to the development of a “common cause” theory of autoimmunity.
In accordance with the common cause hypothesis of autoimmune diseases, we have uncovered a number of risk loci in common with other forms of autoimmunity, such as RA, T1D, CeD and Crohn’s disease/colitis, SLE, MS and psoriasis, in particular CTLA4, IL2/IL2RA, IL21 and genes that are crucial for the homeostasis of regulatory T cells. The genetic commonality with RA, T1D, and CeD is especially noteworthy in light of the pathogenic significance of the expression of an NK ligand in the end organ (synovial fluid, islets, gut, respectively, and hair follicle for AA), and the involvement of the NKG2DL/NKG2D pathway in the pathogenesis of each of these three diseases.
The recent successes of GWAS studies have identified susceptibility alleles in specific genes that underlie sets of autoimmune diseases and importantly, the majority of these shared genes can be mapped onto a discrete set of immunological molecular pathways. These discoveries have made possible the identification of important pathological processes and general mechanisms that dysregulate immune tolerance and might be common to several autoimmune diseases. Furthermore, these advances have important implications for translational research in autoimmune diseases. Firstly, the identification of particular drug targets that are common to multiple diseases will allow us to establish a rationale for using a drug that gained approval for treatment of one autoimmune disorder, to treat others. Secondly, the goal of personalized medicine might allow us in the future to tailor therapeutic approaches based on their susceptibility genes, as opposed to the inefficient process of trial and error.
Pursuing CTLA4 as a druggable target serves as a potential example of realizing this approach. Prior to GWAS studies, several genetic studies had implicated the costimulatory locus, which includes CTLA4, in a number of autoimmune diseases. CTLA4 effectively “turns off” immune responses (Figure 1) and is thought to prevent excessive, unnecessary bystander damage and autoimmune pathology by an untempered immune system. A soluble CTLA4-Ig recombinant fusion molecule can serve the same purpose, and this molecule demonstrated inhibition of naive CD4+ T cell activation and proliferation (81, 82), and prevention of autoimmunity and inhibition of graft rejection in animal models (83–87). Abatacept, a CTLA4-Ig approved for use in humans, has been shown to be efficacious in the treatment of RA and psoriasis (88, 89). Moreover, approximately 54 clinical trials are currently underway to test this drug (or a similar drug, belatacept) in several other autoimmune diseases as well as in organ transplantations (Table 1). Interestingly, in AA, preclinical studies using CTLA4-Ig have shown efficacy in preventing alopecia in the C3H/HeJ mouse model of disease (90). Collectively, our genetic evidence, taken together with the efficacy data in the AA mouse model, as well as the safety and efficacy of abatecept in RA and other autoimmune diseases, make a strong case for the testing of this drug in AA clinical trials (91).
Figure 1. Proposed mode of action of CTLA4.
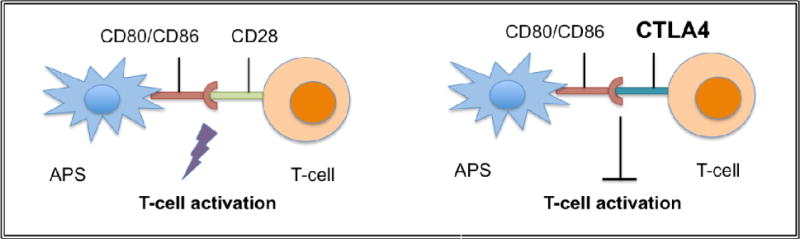
The costimulatory receptor CD28 present on the surface of T cells interacts with its ligands CD80/CD86 present on the surface of antigen presenting cells (including macrophages, dendritic cells, and B cells) driving initial T cell activation. Endogenous CTLA-4 and the therapeutic CTLA4-Ig binds CD80/86 with higher affinity than CD28 effectively competing away costimulatory drive.
Table 1.
Abatacept or belatacept clinical trials for autoimmune diseases (http://www.clinicaltrials.gov)
| Disease | No. of studies |
|---|---|
| Rheumatoid Arthritis | 23 |
| Renal Transplant | 10 |
| Type I Diabetes Mellitus | 5 |
| Lupus Nephritis | 3 |
| Solid Organ Transplant | 1 |
| Acute Graft Versus Host Disease | 1 |
| Takayasu’s Arteritis; Giant Cell Arteritis | 1 |
| Urticaria | 1 |
| Wegener’s Granulomatosis | 1 |
| Polymyositis; Dermatomyositis | 1 |
| Alopecia Totalis/Universalis | 1 |
| Uveitis | 1 |
| Multiple Sclerosis, Relapsing-Remitting | 1 |
| Ankylosing Spondylitis | 1 |
| Relapsing Polychondritis | 1 |
| Allergic Asthma | 1 |
| Juvenile Idiopathic Arthritis | 1 |
|
| |
| Total=54 | |
5. Conclusion
The great advances that genetics has brought into the study of autoimmune diseases in general, and AA in particular, have paved the way for an exciting new era of translational research that we are about to enter. The continuous discovery of molecular and signaling pathways that overlap between different autoimmune diseases greatly helps to illuminate pathologically important disease processes, arguing for unifying/general mechanisms that dysregulate immune tolerance at one of several multiple end organ sites.
Our GWAS findings in AA, together with other studies, have already opened new avenues for exploration of therapeutic strategies based on the actual underlying mechanisms of disease, with a focus on T cells as well as cells that express the NKG2D receptor. Taking advantage of the common molecular pathways as well as of the fact that a number of drugs and biologics are currently being used or tested for the treatment of other autoimmune diseases, we anticipate that new therapies for AA will be available in the near future.
Key Points.
AA can cause tremendous emotional and psychosocial distress in affected individuals and their families
In recent years, the identification of genetic markers for which an allele exists at a higher frequency among a group of unrelated cases, relative to a group of unrelated controls, has become possible with genome-wide association studies (GWAS)
It has been suspected that autoimmune diseases affecting different end organs in fact share common disease mechanisms.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Paper for Dermatologic Clinics, Issue; Hair: what is new in diagnosis and management?
References
- 1.Hon KL, Leung AK. Alopecia areata. Recent Pat Inflamm Allergy Drug Discov. 2011 May;5:98. doi: 10.2174/187221311795399291. [DOI] [PubMed] [Google Scholar]
- 2.Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992 May;128:702. doi: 10.1001/archderm.1992.01680150136027. [DOI] [PubMed] [Google Scholar]
- 3.Safavi KH, Muller SA, Suman VJ, Moshell AN, Melton LJ. 3rd, Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995 Jul;70:628. doi: 10.4065/70.7.628. [DOI] [PubMed] [Google Scholar]
- 4.Tan E, Tay YK, Goh CL, Chin Giam Y. The pattern and profile of alopecia areata in Singapore–a study of 219 Asians. Int J Dermatol. 2002 Nov;41:748. doi: 10.1046/j.1365-4362.2002.01357.x. [DOI] [PubMed] [Google Scholar]
- 5.Tan E, Tay YK, Giam YC. A clinical study of childhood alopecia areata in Singapore. Pediatr Dermatol. 2002 Jul-Aug;19:298. doi: 10.1046/j.1525-1470.2002.00088.x. [DOI] [PubMed] [Google Scholar]
- 6.Barahmani N, Schabath MB, Duvic M. History of atopy or autoimmunity increases risk of alopecia areata. J Am Acad Dermatol. 2009 Oct;61:581. doi: 10.1016/j.jaad.2009.04.031. [DOI] [PubMed] [Google Scholar]
- 7.Muller SA, Winkelmann RK. Alopecia Areata. An Evaluation of 736 Patients. Arch Dermatol. 1963 Sep;88:290. doi: 10.1001/archderm.1963.01590210048007. [DOI] [PubMed] [Google Scholar]
- 8.Shellow WV, Edwards JE, Koo JY. Profile of alopecia areata: a questionnaire analysis of patient and family. Int J Dermatol. 1992 Mar;31:186. doi: 10.1111/j.1365-4362.1992.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 9.Wang SJ, et al. Increased risk for type I (insulin-dependent) diabetes in relatives of patients with alopecia areata (AA) Am J Med Genet. 1994 Jul 1;51:234. doi: 10.1002/ajmg.1320510313. [DOI] [PubMed] [Google Scholar]
- 10.Friedmann PS. Alopecia areata and auto-immunity. Br J Dermatol. 1981 Aug;105:153. doi: 10.1111/j.1365-2133.1981.tb01200.x. [DOI] [PubMed] [Google Scholar]
- 11.Cunliffe WJ, Hall R, Stevenson CJ, Weightman D. Alopecia areata, thyroid disease and autoimmunity. Br J Dermatol. 1969 Dec;81:877. doi: 10.1111/j.1365-2133.1969.tb15967.x. [DOI] [PubMed] [Google Scholar]
- 12.Corazza GR, et al. Celiac disease and alopecia areata: report of a new association. Gastroenterology. 1995 Oct;109:1333. doi: 10.1016/0016-5085(95)90597-9. [DOI] [PubMed] [Google Scholar]
- 13.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci U S A. 2007 Jul 10;104:11694. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunliffe WJ, Hall R, Newell DJ, Stevenson CJ. Vitiligo, thyroid disease and autoimmunity. Br J Dermatol. 1968 Mar;80:135. doi: 10.1111/j.1365-2133.1968.tb12282.x. [DOI] [PubMed] [Google Scholar]
- 15.Narita T, et al. Generalized Vitiligo and Associated Autoimmune Diseases in Japanese Patients and Their Families. Allergol Int. 2011 Jul 25; doi: 10.2332/allergolint.11-OA-0303. [DOI] [PubMed] [Google Scholar]
- 16.Sipetic S, et al. Family history and risk of type 1 diabetes mellitus. Acta Diabetol. 2002 Sep;39:111. doi: 10.1007/s005920200028. [DOI] [PubMed] [Google Scholar]
- 17.Petukhova L, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010 Jul 1;466:113. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghersetich I, Campanile G, Lotti T. Alopecia areata: immunohistochemistry and ultrastructure of infiltrate and identification of adhesion molecule receptors. Int J Dermatol. 1996 Jan;35:28. doi: 10.1111/j.1365-4362.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 19.Todes-Taylor N, Turner R, Wood GS, Stratte PT, Morhenn VB. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984 Aug;11:216. doi: 10.1016/s0190-9622(84)70152-6. [DOI] [PubMed] [Google Scholar]
- 20.Gregoriou S, et al. Cytokines and other mediators in alopecia areata. Mediators Inflamm. 2010;2010:928030. doi: 10.1155/2010/928030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brajac I, et al. Treatment of alopecia areata: modern principles and perspectives. Lijec Vjesn. 2010 Nov-Dec;132:365. [PubMed] [Google Scholar]
- 22.Jelinek JE. Sudden whitening of the hair. Bull N Y Acad Med. 1972 Sep;48:1003. [PMC free article] [PubMed] [Google Scholar]
- 23.Wasserman D, Guzman-Sanchez DA, Scott K, McMichael A. Alopecia areata. Int J Dermatol. 2007 Feb;46:121. doi: 10.1111/j.1365-4632.2007.03193.x. [DOI] [PubMed] [Google Scholar]
- 24.Delamere FM, Sladden MM, Dobbins HM, Leonardi-Bee J. Interventions for alopecia areata. Cochrane Database Syst Rev. 2008:CD004413. doi: 10.1002/14651858.CD004413.pub2. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell AJ, Balle MR. Alopecia areata. Dermatol Clin. 1987 Jul;5:553. [PubMed] [Google Scholar]
- 26.Blaumeiser B, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006 Apr;54:627. doi: 10.1016/j.jaad.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 27.McDonagh AJ, Tazi-Ahnini R. Epidemiology and genetics of alopecia areata. Clin Exp Dermatol. 2002 Jul;27:405. doi: 10.1046/j.1365-2230.2002.01077.x. [DOI] [PubMed] [Google Scholar]
- 28.van der Steen P, et al. The genetic risk for alopecia areata in first degree relatives of severely affected patients. An estimate. Acta Derm Venereol. 1992 Sep;72:373. [PubMed] [Google Scholar]
- 29.Jackow C, et al. Alopecia areata and cytomegalovirus infection in twins: genes versus environment? J Am Acad Dermatol. 1998 Mar;38:418. doi: 10.1016/s0190-9622(98)70499-2. [DOI] [PubMed] [Google Scholar]
- 30.Scerri L, Pace JL. Identical twins with identical alopecia areata. J Am Acad Dermatol. 1992 Nov;27:766. doi: 10.1016/s0190-9622(08)80226-5. [DOI] [PubMed] [Google Scholar]
- 31.Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999 Dec;4:216. doi: 10.1038/sj.jidsp.5640214. [DOI] [PubMed] [Google Scholar]
- 32.de Andrade M, et al. Alopecia areata in families: association with the HLA locus. J Investig Dermatol Symp Proc. 1999 Dec;4:220. doi: 10.1038/sj.jidsp.5640215. [DOI] [PubMed] [Google Scholar]
- 33.Sundberg JP, Silva KA, Li R, Cox GA, King LE. Adult-onset Alopecia areata is a complex polygenic trait in the C3H/HeJ mouse model. J Invest Dermatol. 2004 Aug;123:294. doi: 10.1111/j.0022-202X.2004.23222.x. [DOI] [PubMed] [Google Scholar]
- 34.Gilhar A, Paus R, Kalish RS. Lymphocytes, neuropeptides, and genes involved in alopecia areata. J Clin Invest. 2007 Aug;117:2019. doi: 10.1172/JCI31942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Mir A, et al. Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007 Feb;80:316. doi: 10.1086/511442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jagielska D, et al. Follow-Up Study of the First Genome-Wide Association Scan in Alopecia Areata: IL13 and KIAA0350 as Susceptibility Loci Supported with Genome-Wide Significance. J Invest Dermatol. 2012 Apr 26; doi: 10.1038/jid.2012.129. [DOI] [PubMed] [Google Scholar]
- 37.John KK, et al. Genetic variants in CTLA4 are strongly associated with alopecia areata. J Invest Dermatol. 2011 May;131:1169. doi: 10.1038/jid.2010.427. [DOI] [PubMed] [Google Scholar]
- 38.Gregersen PK, et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet. 2009 Jul;41:820. doi: 10.1038/ng.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl EA, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010 Jun;42:508. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett JC, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009 Jun;41:703. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper JD, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet. 2008 Dec;40:1399. doi: 10.1038/ng.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dubois PC, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010 Apr;42:295. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parrish-Novak J, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000 Nov 2;408:57. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 44.Parrish-Novak J, Foster DC, Holly RD, Clegg CH. Interleukin-21 and the IL-21 receptor: novel effectors of NK and T cell responses. J Leukoc Biol. 2002 Nov;72:856. [PubMed] [Google Scholar]
- 45.Hinrichs CS, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008 Jun 1;111:5326. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010 May;235:267. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito T, et al. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008 May;128:1196. doi: 10.1038/sj.jid.5701183. [DOI] [PubMed] [Google Scholar]
- 48.Gambelunghe G, et al. MICA gene polymorphism in the pathogenesis of type 1 diabetes. Ann N Y Acad Sci. 2007 Sep;1110:92. doi: 10.1196/annals.1423.011. [DOI] [PubMed] [Google Scholar]
- 49.Kirsten H, et al. Association of MICA with rheumatoid arthritis independent of known HLA-DRB1 risk alleles in a family-based and a case control study. Arthritis Res Ther. 2009;11:R60. doi: 10.1186/ar2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez-Morera JL, et al. Genetic influence of the nonclassical major histocompatibility complex class I molecule MICB in multiple sclerosis susceptibility. Tissue Antigens. 2008 Jul;72:54. doi: 10.1111/j.1399-0039.2008.01066.x. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Xia B, Lu M, Ge L, Zhang X. MICB0106 gene polymorphism is associated with ulcerative colitis in central China. Int J Colorectal Dis. 2010 Feb;25:153. doi: 10.1007/s00384-009-0787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Rodero S, et al. MHC class I chain-related gene B promoter polymorphisms and celiac disease. Hum Immunol. 2006 Mar;67:208. doi: 10.1016/j.humimm.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 53.Martin JE, et al. The autoimmune disease-associated IL2RA locus is involved in the clinical manifestations of systemic sclerosis. Genes Immun. 2011 Oct 20; doi: 10.1038/gene.2011.72. [DOI] [PubMed] [Google Scholar]
- 54.Lindner E, et al. IL2RA gene polymorphism rs2104286 A>G seen in multiple sclerosis is associated with intermediate uveitis: possible parallel pathways? Invest Ophthalmol Vis Sci. 2011;52:8295. doi: 10.1167/iovs.11-8163. [DOI] [PubMed] [Google Scholar]
- 55.Lowe CE, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007 Sep;39:1074. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 56.Maier LM, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol. 2009 Feb 1;182:1541. doi: 10.4049/jimmunol.182.3.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carr EJ, et al. Contrasting genetic association of IL2RA with SLE and ANCA-associated vasculitis. BMC Med Genet. 2009;10:22. doi: 10.1186/1471-2350-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alcina A, et al. IL2RA/CD25 gene polymorphisms: uneven association with multiple sclerosis (MS) and type 1 diabetes (T1D) PLoS One. 2009;4:e4137. doi: 10.1371/journal.pone.0004137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makis AC, et al. Serum levels of soluble interleukin-2 receptor alpha (sIL-2Ralpha) as a predictor of outcome in brucellosis. J Infect. 2005 Oct;51:206. doi: 10.1016/j.jinf.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Giordano C, et al. Increased soluble interleukin-2 receptor levels in the sera of type 1 diabetic patients. Diabetes Res. 1988 Jul;8:135. [PubMed] [Google Scholar]
- 61.Greenberg P, et al. Requirements for T cell recognition and elimination of retrovirally-transformed cells. Princess Takamatsu Symp. 1988;19:287. [PubMed] [Google Scholar]
- 62.Hill JA, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007 Nov;27:786. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 63.Pan F, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science. 2009 Aug 28;325:1142. doi: 10.1126/science.1176077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Entz P, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006 Jul-Aug;16:363. [PubMed] [Google Scholar]
- 65.Elias JA, Lee CG. IL-13 in asthma. The successful integration of lessons from mice and humans. Am J Respir Crit Care Med. 2011 Apr 15;183:957. doi: 10.1164/rccm.201101-0080ED. [DOI] [PubMed] [Google Scholar]
- 66.Li X, et al. Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J Allergy Clin Immunol. 2010 Feb;125:328. doi: 10.1016/j.jaci.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang M, et al. Variants in the 5q31 cytokine gene cluster are associated with psoriasis. Genes Immun. 2008 Mar;9:176. doi: 10.1038/sj.gene.6364451. [DOI] [PubMed] [Google Scholar]
- 68.Nair RP, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009 Feb;41:199. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.van den Berg LM, Gringhuis SI, Geijtenbeek TB. An evolutionary perspective on C-type lectins in infection and immunity. Ann N Y Acad Sci. 2012 Apr;1253:149. doi: 10.1111/j.1749-6632.2011.06392.x. [DOI] [PubMed] [Google Scholar]
- 70.Barahmani N, et al. Serum T helper 1 cytokine levels are greater in patients with alopecia areata regardless of severity or atopy. Clin Exp Dermatol. 2009 Oct 23; doi: 10.1111/j.1365-2230.2009.03523.x. [DOI] [PubMed] [Google Scholar]
- 71.Hafler DA, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007 Aug 30;357:851. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 72.Hakonarson H, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007 Aug 2;448:591. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 73.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007 Jun 7;447:661. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kropotov A, et al. Constitutive expression of the human peroxiredoxin V gene contributes to protection of the genome from oxidative DNA lesions and to suppression of transcription of noncoding DNA. Febs J. 2006 Jun;273:2607. doi: 10.1111/j.1742-4658.2006.05265.x. [DOI] [PubMed] [Google Scholar]
- 75.Kubo E, Singh DP, Fatma N, Akagi Y. TAT-mediated peroxiredoxin 5 and 6 protein transduction protects against high-glucose-induced cytotoxicity in retinal pericytes. Life Sci. 2009 Jun 5;84:857. doi: 10.1016/j.lfs.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 76.Ding Y, et al. Overexpression of peroxiredoxin 4 protects against high-dose streptozotocin-induced diabetes by suppressing oxidative stress and cytokines in transgenic mice. Antioxid Redox Signal. 2010 Nov 15;13:1477. doi: 10.1089/ars.2010.3137. [DOI] [PubMed] [Google Scholar]
- 77.Franke A, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010 Dec;42:1118. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sundstrom E, et al. Identification of a melanocyte-specific, microphthalmia-associated transcription factor-dependent regulatory element in the intronic duplication causing hair greying and melanoma in horses. Pigment Cell Melanoma Res. 2012 Jan;25:28. doi: 10.1111/j.1755-148X.2011.00902.x. [DOI] [PubMed] [Google Scholar]
- 79.Rosengren Pielberg G, et al. A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat Genet. 2008 Aug;40:1004. doi: 10.1038/ng.185. [DOI] [PubMed] [Google Scholar]
- 80.Karopka T, Fluck J, Mevissen HT, Glass A. The Autoimmune Disease Database: a dynamically compiled literature-derived database. BMC Bioinformatics. 2006;7:325. doi: 10.1186/1471-2105-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Linsley PS, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992 Dec 1;176:1595. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tan R, Teh SJ, Ledbetter JA, Linsley PS, Teh HS. B7 costimulates proliferation of CD4−8+ T lymphocytes but is not required for the deletion of immature CD4+8+ thymocytes. J Immunol. 1992 Nov 15;149:3217. [PubMed] [Google Scholar]
- 83.Knoerzer DB, Karr RW, Schwartz BD, Mengle-Gaw LJ. Collagen-induced arthritis in the BB rat. Prevention of disease by treatment with CTLA-4-Ig. J Clin Invest. 1995 Aug;96:987. doi: 10.1172/JCI118146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pearson TC, et al. CTLA4-Ig plus bone marrow induces long-term allograft survival and donor specific unresponsiveness in the murine model. Evidence for hematopoietic chimerism. Transplantation. 1996 Apr 15;61:997. doi: 10.1097/00007890-199604150-00002. [DOI] [PubMed] [Google Scholar]
- 85.Ndejembi MP, et al. Control of memory CD4 T cell recall by the CD28/B7 costimulatory pathway. J Immunol. 2006 Dec 1;177:7698. doi: 10.4049/jimmunol.177.11.7698. [DOI] [PubMed] [Google Scholar]
- 86.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994 Aug 26;265:1225. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 87.Khoury SJ, et al. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J Immunol. 1995 Nov 15;155:4521. [PubMed] [Google Scholar]
- 88.Kremer JM, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005 Aug;52:2263. doi: 10.1002/art.21201. [DOI] [PubMed] [Google Scholar]
- 89.Abrams JR, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999 May;103:1243. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carroll JM, McElwee KJ, EKL, Byrne MC, Sundberg JP. Gene array profiling and immunomodulation studies define a cell-mediated immune response underlying the pathogenesis of alopecia areata in a mouse model and humans. J Invest Dermatol. 2002 Aug;119:392. doi: 10.1046/j.1523-1747.2002.01811.x. [DOI] [PubMed] [Google Scholar]
- 91.Sundberg JP, McElwee KJ, Carroll JM, King LE., Jr Hypothesis testing: CTLA4 co-stimulatory pathways critical in the pathogenesis of human and mouse alopecia areata. J Invest Dermatol. 2011 Nov;131:2323. doi: 10.1038/jid.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]