

Abstract
This report describes a 32-year-old woman presenting since childhood with progressive calcium pyrophosphate disease (CPPD), characterized by severe arthropathy and chondrocalcinosis involving multiple peripheral joints and intervertebral disks. Because ANKH mutations have been previously described in familial CPPD, the proband’s DNA was assessed at this locus by direct sequencing of promoter and coding regions and revealed 3 sequence variants in ANKH. Sequences of exon 1 revealed a novel isolated nonsynonymous mutation (c.13 C>T), altering amino acid in codon 5 from proline to serine (CCG>TCG). Sequencing of parental DNA revealed an identical mutation in the proband’s father but not the mother. Subsequent clinical evaluation demonstrated extensive chondrocalcinosis and degenerative arthropathy in the proband’s father. In summary, we report a novel mutation, not previously described, in ANKH exon 1, wherein serine replaces proline, in a case of early-onset severe CPPD associated with metabolic abnormalities, with similar findings in the proband’s father.
Keywords: chondrocalcinosis, pseudogout, inorganic pyrophosphate
Calcium pyrophosphate disease (CPPD) is characterized by deposition of calcium pyrophosphate (CPP) crystals within articular hyaline and fibrocartilages, as well as certain soft tissues.1 During the last decade, it has become clear that several kindreds with familial autosomal-dominant early-onset CPPD, and some sporadic cases, have mutations in the highly conserved 492-amino-acid multiple-pass transmembrane protein ANKH, a transporter of inorganic pyrophosphate (PPi) that functions to lower intracellular PPi and raise extracellular PPi.2
The human homolog (ANKH; MIM 605145) of the mouse progressive ankylosis gene maps to chromosome 5p15.1.1 The demonstration that ank mutations in mice result in pathologic calcification, and initial linkage mapping of autosomal-dominant CPPD in humans to chromosome 5p,1–3 elucidated that ANKH mutations result in human disease.4,5 Of 6 familial CPPD ANKH mutations reported to date, 2 were localized to exon 1.4,5 The previously reported mutations in codon 5 alter the amino acid proline to either threonine (Pro5Thr) or leucine (Pro5Leu).4,5 More recently, a common promoter region polymorphism in 4% of 128 sporadic cases of chondrocalcinosis (CC) was described, with a –4-base pair (bp) G-to-A transition enhancing ANKH transcription.6
We now describe a sporadic case of severe CPPD with a heterozygous Pro-to-Ser transition (CCG>TCG) in codon 5. To our knowledge, this is the first case reported with this specific mutation and further supports the role of ANKH mutations in severe familial CPPD.
MATERIALS AND METHODS
The materials in the study derived initially from an index case, as described in the next section, and additional family members in an effort to delineate whether a novel genetic basis existed to elucidate and understand the striking clinical findings.
Index Case
A 41-year-old woman presented as an outpatient with a history of onset of bilateral ankle swelling and pain dating back to age 20. The inflammatory attacks were self-limited, lasting days to weeks and recurring every few months. Subsequently, both knees became involved, and arthroscopy was performed at age 22, revealing extensive CC intraoperatively, with CPP crystals documented. Symptoms progressed despite treatment with colchicine and nonsteroidal anti-inflammatory drugs. Additional joint involvement included shoulders, elbows, and wrists with multiple attacks every few weeks. She also began to experience severe low back pain. Over the years, attacks became less self-limited and a chronic, polyarticular arthropathy evolved. Her family history is remarkable for 2 siblings and mother with no history of arthritis, although her father carried a presumptive diagnosis of severe (non–crystal-proven) gout, onset in middle age.
On presentation, examination revealed multiple swollen, warm, and markedly tender joints—including bilateral wrists, right shoulder with minimal range of motion, and bilateral ankle swelling. She was wheelchair bound, unable to ambulate or bear weight. An ankle aspiration revealed CPP crystals via compensated polarized microscopic analysis.
Laboratory tests revealed normal complete blood cell count and metabolic panel including serum alkaline phosphatase, calcium (8.8 mg/dL), magnesium (1.9 mg/dL), urate (1.6 mg/dL), copper (122 μg/dL), ceruloplasmin (36 mg/dL), ferritin (31 ng/mL), 25-hydroxy vitamin D (28 ng/mL), and thyroid function tests. Serum phosphorus was decreased (1.9 mg/dL) and intact parathyroid hormonewas elevated (93 pg/mL; reference value, <65 pg/mL), indicating mild secondary hyperparathyroidism. The level of fibroblast growth factor-23 was not elevated (100 RU/mL; reference value, <180 RU/mL). The 24-hour urine collection revealed elevated calcium (437 mg/24 hr; reference value, <275 mg/24 hr) and phosphorus (1730 mg/24 hr; reference value, <1300 mg/24 hr) with normal creatinine (1860 mg/24 hr) and uric acid (600 mg/24 hr) excretion, with a calculated creatinine clearance of 110 mL/min.
Radiographs of multiple regions revealed extensive CC of small and large joints within the articular cartilage and fibrocartilage, as well as along tendon insertions in the feet (Fig. 1). In addition, calcifications were observed throughout the spine within intervertebral disks (Fig. 1).
FIGURE 1.
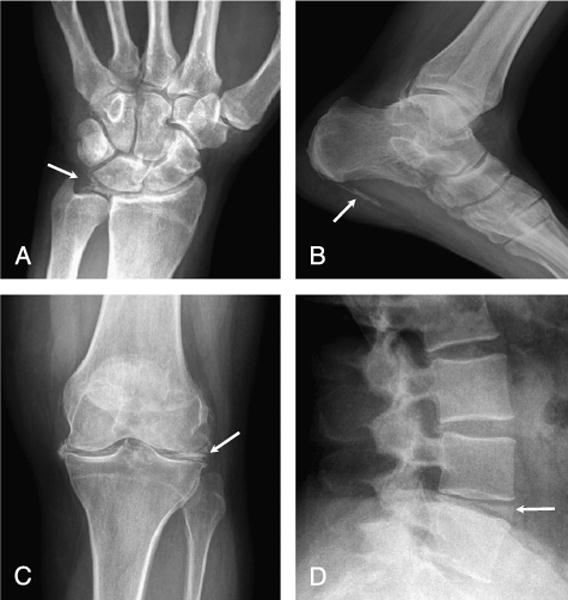
A, Left wrist: calcification of multiple intercarpal ligaments, including scapholunate, lunate-triquetral, and triangulofibrocartilage (arrow). B, Right foot: dense calcification of the plantar fascia origin (arrow) and posterior ankle joint capsule. C, Right knee: dense calcification of fibrocartilage of medial and lateral meniscus; calcification of hyaline cartilage on lateral view (not shown). D, Lumbar spine: intradiscal calcification at L4–L5.
Treatment was instituted with intra-articular steroids and oral prednisone for acute attacks and a combination of hydroxychloroquine, magnesium oxide, probenecid, and colchicine for long-term maintenance therapy. After several weeks, acute synovitis markedly improved, and for the past 12 months, only 2 minor exacerbations occurred, which responded rapidly to oral prednisolone treatment. Interestingly, we noted that abnormalities of serum phosphate, 25-OH vitamin D, and parathyroid hormone levels resolved during treatment. She has been able to continue her occupation as a teacher. Recently, radiographs (knees, hands, wrists) of the patient’s father reveal marked and extensive CC with degenerative joint disease consistent with CPPD.
Methods
A sample of DNA, extracted by a standard salting-out method from peripheral blood samples of patient and parents, was obtained after informed consent. ANKH mutations were assessed by direct sequencing of promoter and coding regions, using primers flanking intron/exon boundaries (Table 1). The whole codifying region of the gene (8203 bp) was successfully amplified and sequenced, in both directions, in the proband and parents. In addition, 550 bp of 5′-untranslated region and promoter region was also amplified and sequenced in both directions. Sequencing reactions were performed using BigDye chemistry and protocols (available from the authors), electrophoresed on ABI 3130 XL automatic sequencer (Applied Biosystems and assessed using SeqScape v2.6 (Applied Biosystems, Bedford, MA) in comparison with reference sequence NG_008273.1. Chromosome positions have been produced for build 37 version 3 of the National Center for Biotechnology Information’s genome annotation.
TABLE 1.
Primer Sequences (5′–3′) and Optimized Polymerase Chain Reaction Conditions
| Region | Forward 5′–3′ | Reverse 5′–3′ | [Mg] | °C |
|---|---|---|---|---|
| Promoter 1 | CCACGTTTCTTTCGCCCCCTC | ACTGCGGAGGAGGCAGCTTG | 3 | 60 |
| Promoter 2 | CGCCCGCCCCTGATTTCCTC | TTCTGCTGACAGCGGCTCCAT | 3 | 60 |
| Promoter 3 | GAGCAGCCGCGCTCGGAGAA | GAAGTCGATGGCTATGTTGG | 3 | 60 |
| Exon 1 | AGTCCCCTCGCGGCAGCAGA | GCTGGAAAAGTCACCCTTGA | 3 | 60 |
| Exon 2 | ACCCTATAAGCTACTTAGTG | AGAAACATTTGATAATTAGTG | 3 | 50 |
| Exon 3 | TGACCCACTCTGGGTAGAGG | CCTGCCATTAAGCTGTACACAC | 3 | 60 |
| Exon 4 | TGCAGTCCTTCACTCCACTG | CAGTTACACACGCCAGAAGG | 2 | 60 |
| Exon 5 | CCCCTGTGCTTCTGTCAGTC | CTGTCTTTCCCTGCAGACATC | 2 | 60 |
| Exon 6 | GGCTTGTGATTAACATACGAAGG | CACCGAACGAGATCTTTATGG | 3 | 60 |
| Exon 7 | CGTGCTTCGTCACCTACTGT | CCCCAACGTCACATTAACCT | 2 | 60 |
| Exon 8 | GCCCGAGAGACACTCAACAT | TGCCCCTTTACAAAAACCAA | 2 | 60 |
| Exon 9 | AGCAAGCAGGTGGCAGATCTG | AGTGAAATTTTACATTTGTG | 3 | 50 |
| Exon 10 | ACCCTGGCAGGAAGATGAG | CCCAAACTCCCTGACAACAT | 2 | 60 |
| Exon 11 | GAAGCCAGCAGATGGAGAAC | ACCATCCAACCTGGTCAGTG | 3 | 60 |
| Exon 12.1 | CAACCAAACTGCAAGGAACA | GCCGAAGTGTCATCCTGACT | 2 | 60 |
| Exon 12.2 | AGAATGAATAAGGCACGGGACG | ACAACGTCAACCGTGAGGCAG | 2 | 60 |
RESULTS
Sequencing revealed 3 sequence variants in ANKH (Fig. 2). The only nonsynonymous mutation (Pro5Ser) was located on the N-terminus of the protein, a highly conserved region across species. Bioinformatics analysis confirmed this SNP as not present in either dbSNP or the 1000 Genomes database. The effect of this nonsynonymous variant was evaluated with Polyphen and SIFT. P5S is predicted to be probably damaging by Polyphen, with a position-specific independent counts score difference of 2.100. Using sorting tolerant from intolerant, substitution at position 5 from P to S is also predicted to affect protein function. Besides this substitution, 2 other previously reported synonymous polymorphisms were also identified.
FIGURE 2.
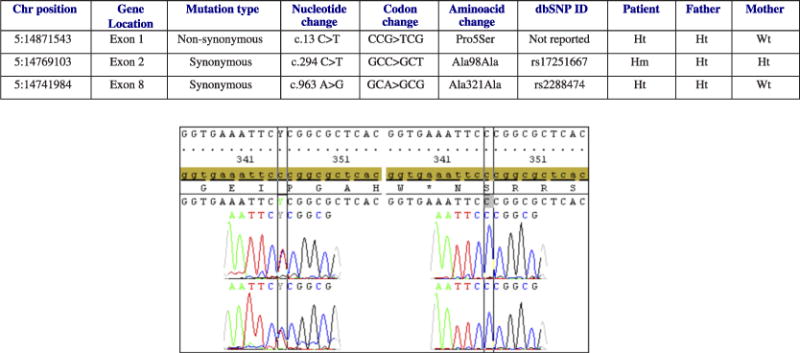
Table: ANKH genomic variants identified in the patient with CPPD deposition and her family. Hm indicates homozygous; Ht, heterozygous; Wt, wild type. Graph: left, sequences of exon 1, sense and antisense, revealed a nonsynonymous mutation (c.13 C>T), in the patient and her father, which alters the amino acid in codon 5 from proline to serine (CCG>TCG); right, control sequences of exon 1, sense and antisense, showing the wild type codon 5 (CCG) coding for proline.
DISCUSSION
Previously reported mutations in ANKH codon 5 alter amino acid proline to either threonine (Pro5Thr) or leucine (Pro5Leu).2,4,5 We now have observed a case of severe and widespread CPPD with a mutation in codon 5 leading to proline replaced by serine (Pro5Ser). To our knowledge, this is the first case described with such a mutation. The patient presented as a teenager, and radiographs of this patient, obtained approximately 20 years after disease onset, reveal striking CC involving essentially all fibrocartilage as well as extensive hyaline cartilage of diarthrodial joints, several entheses (plantar fascia), and axial regions (intervertebral disks). Involvement of the vertebral disks is rare in idiopathic CPPD but has been described in familial forms, suggesting a generalized enthesopathic pattern.
The role of ANKH mutations in the complex pathobiology of mineralization disorders remains to be fully elucidated.7 It has been proposed that certain ANKH mutations result in long-term low-grade chondrocyte “PPi leakiness,” promoting CPPD crystal deposition via extracellular matrix PPi supersaturation.1,5,8 Albeit, not all ANKH mutants1 raise extracellular PPi; nonetheless, most N-terminal mutations in association with familial CC seem to regulate PPi transport.1,5,8,9 Furthermore, some ANKH mutations modulate chondrocyte differentiation,10 whereas others (such as DeltaE490) seem to indirectly suppress PPi catabolism via impairment of TNAP expression.11
An understanding of how the ANKH mutation, as described in this patient and previous reports, results in CPPD is of paramount interest. A novel observation in our case of familial CPPD was a set of metabolic abnormalities, including fluctuating hypophosphatemia, hypercalcuria, and hyperphosphaturia along with a modest elevation of serum parathyroid hormone with normal renal function, consistent with mild secondary hyperparathyroidism of unclear etiology. Circulating vitamin D metabolites and FGF-23 levels were normal, thus unlikely to account for these laboratory abnormalities.
Although the hyperparathyroidism and secondary phosphaturia with low-serum Pi likely contributed in some fashion to CPP crystal deposition, the relationship of these metabolic disturbances and the ANKH mutation remains to be elucidated. Recently, single nucleotide polymorphisms leading to variant haplotypes in the distal region of ANKH have been described associated with elevated circulating parathyroid levels, implying that ANKH may be involved in the regulation of parathyroid hormone.12 It is known that parathyroid hormone stimulates intracellular adenylate cyclase, which leads to increased PPi production downstream. There is evidence that Pi and PPi have antagonistic roles in regulating collagen production by skeletal cells that modulate calcification13 and Pi has been postulated as an inhibitor of CPP crystal deposition, at least in vitro.14 Furthermore, autosomal recessive inheritance of a novel ANKH mutation, Leu244Ser, manifests clinically with hypophosphatemia as well as calcific joint fusion (but not CPPD), and heterozygote carriers of ANKH Leu244Ser manifest with osteoarthritis without metabolic abnormalities or pathologic calcification.15 The proposed multiple-pass transmembrane structure of ANKH, with closely aligned loops and a central ion channel,4,7 argues that altered conformation of ANKH at the N-terminal domain by nonconservative mutation, such as proline at amino acid 5 to serine could affect both PPi transport function of ANKH and ANKH linkage to Pi transport and metabolism. Pro5 in ANKH and its homolog is conserved across vertebrates including rhesus monkeys, mice, dogs, horses, elephants, chickens, lizards, stickleback, and xenopus, consistent with it being a critical component of the ANKH amino acid sequence.
In summary, we report a new mutation in ANKH exon 1, wherein serine replaces the proline, in a case of an early-onset CPPD associated with metabolic abnormalities. Further studies will be necessary to understand how different mutations of proline 5 mediate this clinical condition.
Acknowledgments
Supported by the VA Research Service and the National Institute of Health (PAG007996), Dr Terkeltaub, VA Medical Center, and University of California, San Diego.
Footnotes
The authors declare no conflict of interest.
References
- 1.Abhishek A, Doherty M. Pathophysiology of articular chondrocalcinosis-role of ANKH. Nat Rev Rheumatol. 2011;7:96–104. doi: 10.1038/nrrheum.2010.182. [DOI] [PubMed] [Google Scholar]
- 2.Couto R, Brown MA. Genetic factors in the pathogenesis of CPPD crystal deposition disease. Curr Rheumatol Rep. 2007;9:231–236. doi: 10.1007/s11926-007-0037-7. [DOI] [PubMed] [Google Scholar]
- 3.Andrew LJ, Brancolini V, de la Pena LS, et al. Refinement of the chromosome 5p locus for familial calcium pyrophosphate dihydrate deposition disease. Am J Hum Genet. 1999;64:136–145. doi: 10.1086/302186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams CJ, Zhang Y, Timms A, et al. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet. 2002;71:985–991. doi: 10.1086/343053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams CJ, Pendleton A, Bonavita G, et al. Mutations in the amino terminus of ankh in two US families with calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 2003;48:2627–2631. doi: 10.1002/art.11133. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Johnson K, Russell RG, et al. Association of sporadic chondrocalcinosis with a −4 basepair G-to-A transition in the 5′ untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 2005;52:1110–1117. doi: 10.1002/art.20978. [DOI] [PubMed] [Google Scholar]
- 7.Collins MT, Boehm M. It ANKH necessarily so. J Clin Endocrinol Metab. 2011;96:72–74. doi: 10.1210/jc.2010-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaka R, Stokes D, Dion AS, et al. P5L mutation in Ank results in an increase in extracellular inorganic pyrophosphate during proliferation and nonmineralizing hypertrophy in stably transduced ATDC5 cells. Arthritis Res Ther. 2006;8:R164. doi: 10.1186/ar2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson K, Terkeltaub R. Upregulated ank expression in osteoarthritis can promote both chondrocyte MMP-13 expression and calcification via chondrocyte extracellular PPi excess. Osteoarthritis Cartilage. 2004;12:321–335. doi: 10.1016/j.joca.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Cheng PT, Pritzker KP. Inhibition of calcium pyrophosphate dihydrate crystal formation: effects of carboxylate ions. Calcif Tissue Int. 1988;42:46–52. doi: 10.1007/BF02555838. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Tsui HW, Beier F, et al. The ANKH DeltaE490 mutation in calcium pyrophosphate dihydrate crystal deposition disease (CPPDD) affects tissue non-specific alkaline phosphatase (TNAP) activities. Open Rheumatol J. 2008;2:23–30. doi: 10.2174/1874312900802010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korostishevsky M, Vistoropsky Y, Malkin I, et al. Anthropometric and bone-related biochemical factors are associated with different haplotypes of ANKH locus. Ann Hum Biol. 2008;35:535–546. doi: 10.1080/03014460802304588. [DOI] [PubMed] [Google Scholar]
- 13.Polewski MD, Johnson KA, Foster M, et al. Inorganic pyrophosphatase induces type I collagen in osteoblasts. Bone. 2009;46:81–90. doi: 10.1016/j.bone.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thouverey C, Bechkoff G, Pikula S, et al. Inorganic pyrophosphate as a regulator of hydroxyapatite or calcium pyrophosphate dihydrate mineral deposition by matrix vesicles. Osteoarthritis Cartilage. 2009;17:64–72. doi: 10.1016/j.joca.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 15.Morava E, Kühnisch J, Drijvers JM, et al. Autosomal recessive mental retardation, deafness, ankylosis, and mild hypophosphatemia associated with a novel ANKH mutation in a consanguineous family. J Clin Endocrinol Metab. 2010;96:E189–E198. doi: 10.1210/jc.2010-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]