

Abstract
Toxoplasma gondii is an obligate, intracellular parasite with a broad host range, including humans and rodents. In both humans and rodents, Toxoplasma establishes a lifelong persistent infection in the brain. While this brain infection is asymptomatic in most immunocompetent people, in the developing fetus or immunocompromised individuals such as acquired immune deficiency syndrome (AIDS) patients, this predilection for and persistence in the brain can lead to devastating neurologic disease. Thus, it is clear that the brain-Toxoplasma interaction is critical to the symptomatic disease produced by Toxoplasma, yet we have little understanding of the cellular or molecular interaction between cells of the central nervous system (CNS) and the parasite. In the mouse model of CNS toxoplasmosis it has been known for over 30 years that neurons are the cells in which the parasite persists, but little information is available about which part of the neuron is generally infected (soma, dendrite, axon) and if this cellular relationship changes between strains. In part, this lack is secondary to the difficulty of imaging and visualizing whole infected neurons from an animal. Such images would typically require serial sectioning and stitching of tissue imaged by electron microscopy or confocal microscopy after immunostaining. By combining several techniques, the method described here enables the use of thick sections (160 µm) to identify and image whole cells that contain cysts, allowing three-dimensional visualization and analysis of individual, chronically infected neurons without the need for immunostaining, electron microscopy, or serial sectioning and stitching. Using this technique, we can begin to understand the cellular relationship between the parasite and the infected neuron.
Keywords: Neurobiology, Issue 94, Neuroscience, Confocal microscopy, Mouse, Brain, Clearing, Fluorescent proteins, Toxoplasma gondii, Apicomplexa, Infectious disease
Introduction
The overall goal of this method is to obtain high resolution, three-dimensional images of individual neurons that are infected by the obligate intracellular parasite Toxoplasma gondii.
Toxoplasma is often considered one of the most successful parasites because of its large intermediate host range, which includes humans and rodents. In both humans and rodents, after acute infection through ingestion of contaminated food or water, Toxoplasma is able to cause a persistent infection of the CNS by converting from its fast replicating form (the tachyzoite) to its slow-replicating and encysting form (the bradyzoite). In immunocompetent individuals, this latent CNS infection is thought to be relatively asymptomatic, but in immunocompromised individuals such as AIDS patients or transplant recipients, recrudescence of the parasite can lead to fatal toxoplasmic encephalitis1,2. In addition, recent studies have shown that latent infection with Toxoplasma can lead to behavioral changes in rodents3,4, though the mechanism remains unknown.
Surprisingly, despite these data highlighting the importance of the CNS-Toxoplasma interaction, relatively little is known about this relationship, especially at the cellular and molecular level. The ability to study even simple aspects of the brain-parasite interaction has been hampered in part by technologic limitations. For example, the majority of the work showing that neurons are the cells in which cysts persist has been done with electron microscopy (EM)5,6. Though EM gives high resolution, it is time consuming, labor intensive, and expensive. Immunofluorescence (IF) assays have recently been used in conjunction with confocal microscopy to confirm the work done by EM7. IF assays are technically easy to perform and relatively inexpensive, but using these techniques to understand the spatial relationship between the cyst and the infected neuron requires serial reconstruction, which is time consuming, technically difficult, and may lead to loss of valuable information. Thus, we have developed a method that can be used with the mouse model of CNS toxoplasmosis and allows us to image the entirety of infected neurons without EM or immunohistochemistry (IHC). By developing such a technique, we can begin to explore the cellular relationship between the infected cell and the cyst in a relatively quick and inexpensive manner.
The method we developed combines newer techniques for optically clearing and imaging thick brain sections by confocal microscopy8 with a system which marks in vivo cells that have been injected with parasite proteins9,10. In this system, we infect Cre-reporter mice that express a green fluorescent protein (GFP) only after Cre-mediated recombination11 with Toxoplasma strains that express a red fluorescent protein (RFP) and inject Cre recombinase into host cells9. This combination allows us to harvest the infected mouse brain after CNS infection is established, cut thick brain sections, and rapidly identify pertinent areas to image by finding the RFP+ cysts. It is important to note that as host cell expression of GFP depends solely on the injection of Cre by parasites, and not on infection, a number of the GFP+ cells do not contain parasites10. As the goal of this protocol is to be able to image whole infected neurons, the focus is only on GFP+ neurons that also contain an RFP+ cyst, but the protocol can also be used to image the GFP+/RFP- neurons.
Once the infected brain is harvested and sectioned, the sections are rendered transparent by glycerol clearing. Appropriate regions of sections are then imaged with confocal microscopy, allowing unprecedented visualization of infected host cells and the encysted parasites in their entirety. Here we provide a complete protocol for identifying, optically clearing, and imaging infected neurons.
Protocol
NOTE: Mice were bred and maintained in a temperature and humidity controlled room with 12 hr reversed light/dark cycles with food and water available ad libitum at the University of Arizona. Experiments were conducted under guidelines and approval of the Institutional Animal Care and Use Committee of the University of Arizona. All efforts were made to minimize suffering. The Cre-reporter mice are on a C57BL/6 background11 and are commercially available.
1. Mouse Infection
NOTE: The method of mouse infection with Toxoplasma gondii described below has been used in studies previously published10–12.
Grow Toxoplasma strains in human foreskin fibroblasts (HFFs) cultured in Dulbecco's High Glucose Modified Eagles Medium (DMEM) supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine (cDMEM) in a T-25 flask inside a 5% CO2 incubator until parasites have formed large parasitophorous vacuoles.
Syringe-release parasites by scraping cells from the bottom of the flask and passing the resulting solution through a 25 gauge needle connected to a 3 ml syringe 2 times inside the flask then transfer all solution to a 5ml syringe case connected to a 27 gauge needle that has been placed upside down inside a 15 ml conical tube. Connect the plunger to the syringe and pass the parasite solution into the conical tube.
Centrifuge the tube for 10 min at 300 x g. Aspirate the supernatant then resuspend the pellet in 4-6 ml sterile USP grade 1x Phosphate Buffered Saline (PBS) pH 7.4 (Stock Solution).
Load a hemocytometer with 10 µl of stock solution, wait 5-10 min for the parasites to settle then count the number of parasites in the four corner and center squares of the middle large square.
Dilute parasites to appropriate inoculum size of 100K/200 µl 1x PBS then inject mice intraperitoneally (i.p.) with a total volume of 200 µl. NOTE: For this procedure, mice were inoculated with 100K Type III (CEP)13 tachyzoites in 200 µl 1x PBS. When infecting mice with other strains of Toxoplasma, concentration of inoculum may need to be adjusted to account for level of virulence.
2. Perfusion and Brain Harvesting
NOTE: This protocol was conducted at 21 days post infection (dpi) as this represents the time point at which peak numbers of cysts are found in the mouse brain, but other time points may be used. Size, number, location, and presence or absence of cysts depends on many factors including mouse strain, parasite strain type as well as inoculum size7,14–17. Investigators will need to individually determine the appropriate inoculum for the mouse and Toxoplasma strain he/she uses.
Setup all surgical harvesting tools (Figure 1).
For each mouse, prepare and keep on ice: 20ml syringe filled with 0.9% Sodium Chloride (NaCl) + 10 U/ml Heparin in 1x sterile USP grade PBS (Heparin/Saline); 20 ml syringe filled with 4% Paraformaldehyde (PFA); Scintillation vial with 15 ml 4% PFA. For multiple mice, prepare a beaker filled with the entire amount of each solution needed for all mice then draw up a fresh syringe full of solution for each perfusion.
Anesthetize the mouse i.p with 200 µl/25 g of body weight of Ketamine/Xylazine cocktail (24 mg/ml & 4.8 mg/ml respectively) on the day of interest. Monitor the mouse for level of anesthesia by checking toe pinch reflex. Continue with protocol only once the mouse shows no response to a firm toe pinch. NOTE: Other methods of anesthesia may be used as long as the appropriate level of anesthesia is reached before proceeding with the protocol.
Place the mouse in supine position on the covered foam platform. Pin all four limbs out at the feet then spray the chest and abdomen with 70% Ethanol (EtOH) (Figure 2A). Addition of several sterile paper towels under the mouse may be used to help absorb overflow of perfusion fluids.
Lift skin just below the sternum with forceps then use scissors to make an incision. Pull skin back to expose the chest (Figure 2B).
Lift xiphoid process and make an abdominal incision just below the diaphragm, exposing the liver which is bluntly dissected away from the diaphragm. Make two incisions, one on the left and one on the right through the diaphragm and the bottom of the ribs. Extend the left and right incisions approximately 1/2-2/3 way up the ribcage, which is then reflected back to open the chest cavity (Figure 2C). NOTE: Take care not to cut any organs or major blood vessels during this procedure as doing so will compromise the quality of perfusion.
Perfuse transcardially with 10-20 ml Heparin/Saline followed by 10-20 ml 4% PFA. NOTE: If the perfusion is done properly, the liver will turn from red to tan (Figure 2D). If not done properly, blood vessels will be visible (Figure 2E).
Turn the mouse over onto its abdomen, re-pin feet then spray head and neck with 70% EtOH. Lift skin from the base of the neck and make an incision. Remove skin to expose skull (Figure 2F). Decapitate head at the level of the shoulders. Remove the skull, gently remove the brain, and place it into a scintillation vial filled with 4% PFA (Figure 2G-H). NOTE: If well perfused, the brain will appear white with no visible blood vessels (Figure 2I). If the brain is not well perfused, blood vessels will be visible (Figure 2J).
Place the scintillation vial with brain in 4% PFA at 4 °C overnight, covered from light. Rinse the brain in non-USP grade 1x PBS and transfer to a new scintillation vial filled with chilled 1x PBS. Store the brain in 1x PBS at 4 °C, covered from light. NOTE: Successful clearing and imaging has been accomplished with sections stored in 1x PBS for up to one month but it is best to section the brain within a few days as prolonged storage in PBS will reverse the PFA fixation and could lead to less than optimal images. Autofluorescence and increased blur have been noted in some sections imaged after storage in 1x PBS for one month or longer.
3. Brain Sectioning
Remove the brain from 1x PBS and place it into a brain matrix (Figure 3A) to allow for more precise and even cuts through the brain. Trim off the cerebellum and olfactory bulbs, if present, and then cut the brain coronally into 2 or 3 sections. NOTE: Trimming and cutting of the brain may be done without the aid of a brain matrix. Number of sections cut coronally will depend on the cutting distance of the Vibratome being used. The brain can be sectioned in any orientation, though sagittal and coronal sections are most commonly used for neuroanatomical identification.
Affix each brain section onto a specimen mounting block with cyanoacrylate. Affix a small block of 5% agarose gel behind the brain section for support (Figure 3B). NOTE: Other support mechanisms may be used, but without support, the Vibratome may push on the brain causing uneven sectioning.
Cut tissue into 160 µm (working distance of the objective used for imaging) thick sections with the Vibratome set to a speed of 4 and amplitude of 9. NOTE: The settings for speed and amplitude may be adjusted depending on the particular machine being used in order to obtain even, smooth sections. If the settings are not correct, the result can be uneven sections, tearing of tissue or chatter marks.
Collect tissue sections into a scintillation vial filled with fresh, chilled 1x PBS if they are going to be cleared within one week. If sections are not going to be cleared within one week, place sections into 1.5 ml microcentrifuge tubes filled with cryopreservative solution and store at -20 °C.
4. Optical Clearing of Brain Tissue Sections
NOTE: This protocol has been modified from a previously published method 8.
Prepare 25%, 50%, 75% and 90% vol/vol Glycerol in 1x PBS + 2% Tween-20 (Gl/PBST).
Remove sections from 1x PBS or, if in cryopreservative, rinse in 1x PBS and transfer to a scintillation vial containing 10 ml of 25% Gl/PBST. Place vial on a shaker at 4 °C, covered from exposure to light, for 12 hr.
Confirm that all sections have sunk to the bottom of the vial. Aspirate the 25% Gl/PBST, leaving just enough to cover the sections then fill the vial with 10 ml 50% Gl/PBST and place on a shaker at 4 °C, covered from exposure to light, for 12 hr. Repeat this process for the 75% Gl/PBST then the 90% Gl/PBST. Leave sections in the 90% Gl/PBST for 24 hr to reach maximal clearing. Over the course of the clearing process, observe gradual optical clearing (Figure 4). NOTE: This process may be expedited if solutions are changed immediately after sections have sunk to the bottom of the vial. In 75% & 90% solutions, sections will not sink all the way to the bottom of the vial but will float in the middle.
5. Mounting Brain Tissue Sections
Cut a No. 1.5 coverslip into 5 pieces to create a spacer. Use a ruler and diamond-tipped pencil to score and break the coverslip along the dashed lines shown in Figure 5A. Discard the center piece, arrange the 4 outer pieces on a plain glass slide to create a window, and then brush clear nail polish onto the seams to adhere the pieces of the spacer to the slide in the correct configuration (Figure 5B). NOTE: The spacer will be used to create the necessary space between the slide and the coverslip for mounting the cleared sections. The coverslip may be cut in different ways to create any size window necessary. Pre-cut foam spacers are also commercially available. To allow nail polish to dry completely, it is best to make slides with spacers at least one day prior to mounting sections.
Transfer sections to a slide using a plastic transfer pipet with the tip cut off. Make sure to fill in surrounding area with 90% Gl/PBST. Carefully lower a coverslip over the sections making sure not to introduce any bubbles. Excess Gl/PBST may be wicked away with a lint-free wipe.
Image GFP+ neurons containing RFP+ Toxoplasma gondii cysts with a confocal microscope immediately (Figure 7A-B) then store slides flat and covered from light at 4 °C. NOTE: Alternatively, seal slides completely around all edges to store and imaged at a later time. Use of clear nail polish to seal slide is optional and may make it more difficult to remove coverslip if one were to remove sections from the slide at a later date for further processing.
6. Imaging
NOTE: Any microscope may be used that has the capability of obtaining high-resolution z-stacks.
After initially finding the focal plane at 10X, change to a 20X objective and scan through the section to identify a cyst located inside a GFP+ cell. Center the region of interest (ROI) in the field of view.
Change to a 40X objective. Focus up and down through the entire ROI to make sure that the entire cell and cyst are visible. NOTE: Images were obtained using a confocal microscope with a 40X/1.30 Oil DIC M27 objective.
Using the installed imaging software, obtain a z-stack that includes the entire cell with cyst. Average settings used to obtain images are shown in Figure 6. NOTE: Settings may need to be adjusted depending on each investigators tissue sections and microscope capabilities.
Obtain a maximum projection image of the z-stack by setting the number of projections to 1. Obtain a full rotational view by setting the number of projections to 64. NOTE: Other software packages are available and may be used to obtain maximum projection, rotational as well as other views of z-stack images.
Analyze image with imaging analysis software.
Representative Results
Figure 7 includes representative images of two cyst-containing GFP+ neurons from two different 160 µm thick sections as well as a representative measurement of the distance from cyst-to-cell-body for Figure 7B. Figures 7 A and B illustrate that this new protocol allows for visualization of the infected neuron in its entirety. Figure 7C shows that with this imaging technique, it is now possible to quantify the distance between the cyst and the cell body (Imaris 7.7). Figure 7D is a full rotational view of Figure 7B. Visualization of the z-stack in this manner allows for verification of the complete capture of the cell and cyst. Figure 7E is a 3D rotational movie showing how the image from 7D can be analyzed by Imaris 7.7 software to measure the distance from the cyst to the cell body either from middle-to-middle or edge-to-edge. Though the shown measurement was done using a straight line between the cyst and the cell body, a measurement following the actual GFP+ process to the cell body can also be generated (data not shown).
 Figure 1: Surgical equipment for removal of mouse brain. (A) Absorbent pad. (B) Foam block. (C) Needles to pin down feet. (D) 25 G Blood collection set. (E) 3-way stopcock. (F) 20 ml syringe for Heparin/Saline. (G) 20 ml syringe for 4% PFA. (H) Thumb forceps. (I) Fine scissors, angled to side, sharp-sharp. (J) Sharp-sharp scissors. (K) Kelly hemostats. (L) Mayo scissors. (M) Micro spatula. (N) Scintillation vial for 4% PFA. Scale bar = 8 cm.
Figure 1: Surgical equipment for removal of mouse brain. (A) Absorbent pad. (B) Foam block. (C) Needles to pin down feet. (D) 25 G Blood collection set. (E) 3-way stopcock. (F) 20 ml syringe for Heparin/Saline. (G) 20 ml syringe for 4% PFA. (H) Thumb forceps. (I) Fine scissors, angled to side, sharp-sharp. (J) Sharp-sharp scissors. (K) Kelly hemostats. (L) Mayo scissors. (M) Micro spatula. (N) Scintillation vial for 4% PFA. Scale bar = 8 cm.
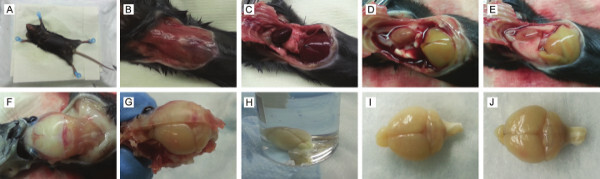 Figure 2: Harvesting mouse brain. (A) Mouse pinned down and chest sprayed with 70% EtOH. (B) Exposed chest. (C) Chest cavity open. (D) Liver after proper perfusion. (E) Liver after improper perfusion. (F) Exposed skull. (G) Brain with half skull removed. (H) Harvested brain in vial filled with 4% PFA. (I) Brain after proper perfusion. (J) Brain after improper perfusion.
Figure 2: Harvesting mouse brain. (A) Mouse pinned down and chest sprayed with 70% EtOH. (B) Exposed chest. (C) Chest cavity open. (D) Liver after proper perfusion. (E) Liver after improper perfusion. (F) Exposed skull. (G) Brain with half skull removed. (H) Harvested brain in vial filled with 4% PFA. (I) Brain after proper perfusion. (J) Brain after improper perfusion.
 Figure 3: Sectioning mouse brain. (A) Mouse brain matrix. (B) Mouse brain and agarose gel affixed to mounting block with cyanoacrylate inside Vibratome chamber filled with chilled 1x PBS.
Figure 3: Sectioning mouse brain. (A) Mouse brain matrix. (B) Mouse brain and agarose gel affixed to mounting block with cyanoacrylate inside Vibratome chamber filled with chilled 1x PBS.
 Figure 4: Clearing process. Representative images of a mouse brain section during each step of the clearing process.
Figure 4: Clearing process. Representative images of a mouse brain section during each step of the clearing process.
 Figure 5: Cutting and fixing spacer to slide. A. No. 1.5 coverslip with representative marks for cutting coverslip into 5 pieces. B. Plain glass slide with each piece of cut coverslip tacked into place with clear nail polish to create a window.
Figure 5: Cutting and fixing spacer to slide. A. No. 1.5 coverslip with representative marks for cutting coverslip into 5 pieces. B. Plain glass slide with each piece of cut coverslip tacked into place with clear nail polish to create a window.
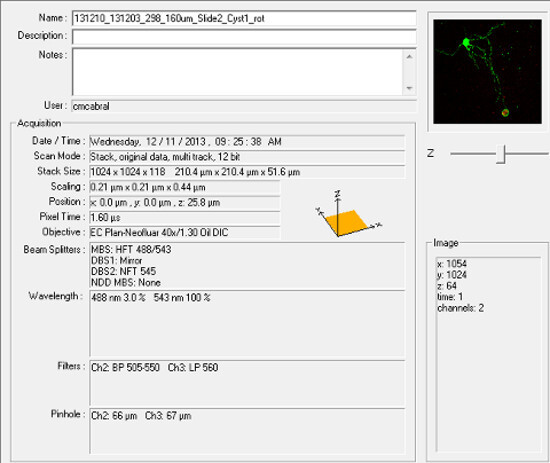 Figure 6: Screen shot of general parameters used for image acquisition.
Figure 6: Screen shot of general parameters used for image acquisition.
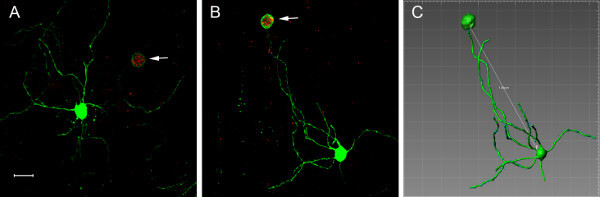 Figure 7: Toxoplasma gondii cysts can reside within distant neuronal processes.(A-B) Maximum projection images of GFP+ neurons containing RFP+ cysts (white arrows). Scale bar = 20 µm. (C-E) provide additional visualization and analysis of the cyst-neuron relationship shown in B. (C) Imaris-generated direct line measurement of distance from the edge of the cyst to the edge of the cell body (149µm). (D)
3-D rotational movie. (E) Imaris-generated 3-D movie showing cyst-to-cell-body measurement from both middle-to-middle (163 µm) and edge-to-edge (149 µm).
Figure 7: Toxoplasma gondii cysts can reside within distant neuronal processes.(A-B) Maximum projection images of GFP+ neurons containing RFP+ cysts (white arrows). Scale bar = 20 µm. (C-E) provide additional visualization and analysis of the cyst-neuron relationship shown in B. (C) Imaris-generated direct line measurement of distance from the edge of the cyst to the edge of the cell body (149µm). (D)
3-D rotational movie. (E) Imaris-generated 3-D movie showing cyst-to-cell-body measurement from both middle-to-middle (163 µm) and edge-to-edge (149 µm).
Discussion
Given that cellular changes in infected host cells have been linked to disease outcomes in infections with other intracellular organisms such as HIV, Rabies, and Chlamydia18,19, we developed a technique that would allow us to study the intimate interactions that occur between the CNS host cell and Toxoplasma. The method described here accomplishes this goal by enabling efficient imaging of chronically infected neurons. Prior to the development of this method, such imaging was time consuming, costly, or not possible.
We can now use this technique to answer basic questions regarding the Toxoplasma-neuron interaction, such as are specific neuronal areas targeted by the parasite? Does this differ between Toxoplasma strains? Do the cellular areas in which the cyst resides differ from the rest of cell (i.e. do infected and uninfected dendrites from the same neuron have the same number of spines)? What are the cellular characteristics shared by infected neurons? The ability to answer such questions is essential to understanding how Toxoplasma infection changes the CNS, including at the level of behavior.
To obtain the best images possible, it is very important to make sure the tissue is cut into even sections. If the sections are uneven, the coverslip does not sit properly on the section leading to uneven contact with the objective resulting in sub-optimal imaging. During optimization of this protocol, several modifications were made. The original clearing process was done with fructose in an attempt to reproduce results shown in a technique known as SeeDB20. This method was incompatible with this model as it quenched the RFP (specifically mCherry). Another key step and probably the most important of all is allowing for maximal clearing of tissue sections. If not cleared properly, background light diffraction will be higher and images become noisy. We found that addition of Tween-20 to the glycerol gave a slightly clearer image than glycerol alone. Current technology allows us to obtain z-stack images through 160µm thick brain tissue sections but in the future, we are looking to increase this thickness to 500 µm or more to allow for even more in depth analysis.
It is only by looking at the entire cell and its connections that we can start to develop a mechanistic idea of how the brain is globally affected by Toxoplasma. This new protocol offers an opportunity to study the Toxoplasma-neuron interaction at the single cell level and do so in a rapid, economical manner. We have yet to explore all of the potential uses for this protocol and how it may translate to other systems but we anticipate it will have multiple applications.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank the entire Koshy lab for helpful discussions. We thank Patty Jansma and the University of Arizona Neuroscience Department for advice and help with imaging. We also thank the Porreca lab for the use of their Vibratome. This research was supported by the US National Institutes of Health (NIH NS065116, A.A.K.).
References
- Luft B, Remington J. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. 1992;15(2):211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. 2002;8(10):634–640. doi: 10.1046/j.1469-0691.2002.00485.x. [DOI] [PubMed] [Google Scholar]
- Ingram WM, Goodrich LM, Robey E, Eisen MB. Mice infected with low-virulence strains of Toxoplasma gondii lose their innate aversion to cat urine, even after extensive parasite clearance. PloS one. 2013;8(9):75246. doi: 10.1371/journal.pone.0075246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AK, Strassmann PS, Lee I-P, Sapolsky RM. Patterns of Toxoplasma gondii cyst distribution in the forebrain associate with individual variation in predator odor avoidance and anxiety-related behavior in male Long-Evans rats. Brain Behav Immun. 2013;37:122–133. doi: 10.1016/j.bbi.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DJ, Hutchison WM. The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch A Pathol Anat Histopathol. 1987;411(1):39–43. doi: 10.1007/BF00734512. [DOI] [PubMed] [Google Scholar]
- Ferguson DJ, Graham DI, Hutchison WM. Pathological changes in the brains of mice infected with Toxoplasma gondii: a histological, immunocytochemical and ultrastructural study. Int J Exp Pathol. 1991;72(4):463–474. [PMC free article] [PubMed] [Google Scholar]
- Melzer TC, Cranston HJ, Weiss LM, Halonen SK. Host Cell Preference of Toxoplasma gondii Cysts in Murine Brain: A Confocal Study. J Neuroparasitology. 2010. [DOI] [PMC free article] [PubMed]
- Selever J, Kong J-Q, Arenkiel BR. A rapid approach to high-resolution fluorescence imaging in semi-thick brain slices. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Koshy A, Fouts A, Lodoen M, Alkan O. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat Methods. 2010;7(4):307–309. doi: 10.1038/nmeth.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy Aa, Dietrich HK, et al. Toxoplasma co-opts host cells it does not invade. PLoS pathog. 2012;8(7) doi: 10.1371/journal.ppat.1002825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman Ta, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffaro CE, Koshy As, Liu L, Zeiner GM, Hirschberg CB, Boothroyd JC. A nucleotide sugar transporter involved in glycosylation of the Toxoplasma tissue cyst wall is required for efficient persistence of bradyzoites. PLoS pathog. 2013;9(5) doi: 10.1371/journal.ppat.1003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij JPJ, Boyle JP, Boothroyd JC. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends in parasitology. 2005;21(10):476–481. doi: 10.1016/j.pt.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Dubey JP, Lindsay DS, Speer C. a Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev. 11(2):267–299. doi: 10.1128/cmr.11.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandota J. Possible Link Between Toxoplasma Gondii and the Anosmia Associated With Neurodegenerative Diseases. Am J Alzheimers Dis Other Demen. 2014;29(3):205–214. doi: 10.1177/1533317513517049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenreiterová M, Flegr J, Kuběna A, Němec P. The distribution of Toxoplasma gondii cysts in the brain of a mouse with latent toxoplasmosis: implications for the behavioral manipulation hypothesis. PloS one. 2011;6(12):28925. doi: 10.1371/journal.pone.0028925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DJ, Hutchison WM. An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice. Parasitol Res. 1987;73(6):483–491. doi: 10.1007/BF00535321. [DOI] [PubMed] [Google Scholar]
- De Chiara G, Marcocci ME, et al. Infectious agents and neurodegeneration. Mol Neurobiol. 2012;46(3):614–638. doi: 10.1007/s12035-012-8320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott Ca, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome of experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol. 2008;82(1):513–521. doi: 10.1128/JVI.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M-T, Fujimoto S, Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci. 2013;16(8):1154–1161. doi: 10.1038/nn.3447. [DOI] [PubMed] [Google Scholar]