

Abstract
PAI-1R, a mutant plasminogen activator inhibitor (PAI-1) that reduces endogenous PAI-1 activity has been shown to inhibit albuminuria and reduce glomerulosclerosis in experimental diabetes. The mechanism of albuminuria reduction is unclear. This study sought to determine whether the administration of PAI-1R protected podocyte from injury directly, thereby reducing albuminuria in the db/db mouse, a model of type 2 diabetes. Untreated uninephrectomized db/db mice developed significant mesangial matrix expansion and albuminuria at week 22 of age, associated with segmental podocyte foot process effacement, reduction of renal nephrin, podocin and zonula occludin-1 (ZO-1) production and induction of renal desmin and B7-1 generation. In contrast, treatment with PAI-1R at 0.5 mg/kg BW through peritoneal injection, twice daily from weeks 20 to 22 reduced glomerular matrix accumulation, fibronectin and collagen production and albuminuria by 36%, 62%, 65% and 31% respectively (p<0.05), without affecting blood glucose level or body weight. Podocyte morphology and protein markers were also significantly attenuated by PAI-1R administration. Importantly, recombinant PAI-1 down-regulated nephrin and ZO-1 but increased desmin and B7-1 mRNA expression and protein production by podocytes in vitro, similar to the effects of TGFβ1. These observations provide evidence that PAI-1, in a manner similar to TGFβ, directly induces podocyte injury, particularly in the setting of diabetes where elevated PAI-1 may contribute to the progression of albuminuria. Reducing the increased PAI-1 activity by PAI-1R, in fact, reduces podocyte injury thereby reducing albuminuria. Therefore, PAI-1R provides additional therapeutic effect in slowing the progression of diabetic nephropathy via the protection of podocytes.
Keywords: PAI-1, diabetes, podocyte, albuminuria
INTRODUCTION
Diabetes is the leading cause of end-stage renal disease (ESRD) and almost half of new ESRD patients have diabetes (Molitch, 2004). Despite the use of angiotensin converting enzymes inhibitors or angiotensin receptor blockers (Brenner, 2001; Parving, 2001), progression of diabetic nephropathy is common. Hence, it is important to identify other interventions that might retard the progression of diabetic nephropathy.
Recently, plasminogen activator inhibitor type 1 (PAI-1) has emerged as a powerful fibrogenic mediator in kidney diseases including diabetic nephropathy (Eddy, 2006; Brown, 2010; Ghosh, 2012). Enhanced glomerular expression of PAI-1 has been reported in both human and animal models of diabetes (Paueksakon, 2002; Huang, 2008). Besides diabetes, increased deposition of PAI-1 in the kidney has been demonstrated in most other human kidney diseases (Rondeau E, 1990; Shihab, 1995; Yamamoto, 1996; Yamamoto, 1999; Grandaliano, 2000; Lee, 2001). The elevated expression of PAI-1 is found in podocytes, endothelial and mesangial cells of glomeruli, in proximal tubular epithelial cells and in small arteries and fibroblast cells in the interstitium (Keeton, 1993). Importantly, genetic PAI-1 deficiency has been shown to be protective in experimental diabetic nephropathy, 5/6 nephrectomy, crescentic anti-GBM nephritis, and the spontaneous renal fibrosis that develops in TGFβ-overexpression mice (Oda, 2001; Krag, 2005; Nicholas, 2005; Eddy, 2006). In contrast, the renal fibrosis is worse in PAI-1 overexpressing mice (Matsuo, 2005). Together, these observations strongly implicate tissue PAI-1 in the renal fibrotic process.
The profibrotic actions of PAI-1 have not been completely elucidated. PAI-1 possesses two distinct functions that could mediate its profibrotic effects alone or in combination: an antiprotease activity that inhibits the generation of plasmin, and a vitronectin-binding ability that interferes with cell adhesion to this extracellular matrix protein (Huang, 2007). As the primary physiological inhibitor of urokinase (uPA) and tissue-type plasminogen activator (tPA), PAI-1 controls the formation and activity of plasmin, and consequently promotes fibrin deposits and accumulation of fibrotic matrix. The binding of PAI-1 to vitronectin could also be important in the pathogenesis of fibrosis. When bound to vitronectin, the antiprotease activity of PAI-1 is significantly stabilized (Podor, 2000). The binding of PAI-1 to vitronectin also blocks the interactions of vitronectin with cell-surface integrin required for cellular motility and with the receptor for cell-bound uPA (uPAR). Furthermore, the binding of PAI-1 to vitronectin induces epithelial-mesenchymal transition (EMT) in epithelial cells by modulating their interaction with the extracellular matrix, a process that is involved in tissue fibrosis (Scaffidi, 2001; Vial, 2008; Senoo, 2010). Whether the properties of PAI-1 observed in vitro are also operative in diseased subjects or animal models remains unclear.
To further understand the actions of PAI-1 and to develop a potential therapeutic agent that inhibits PAI-1 in vivo, a non-inhibitory mutant PAI-1 (PAI-1R) was designed (Stefansson, 2001). This PAI-1R mutant does not inhibit uPA or tPA, but it retains the ability to bind vitronectin thereby decreasing the availability of vitronectin for PAI-1 and effectively shortening its half-life. The administration of PAI-1R to nephritic rats and diabetic db/db mice increased glomerular plasmin generation and reduces proteinuria and glomerulosclerosis (Huang, 2003; Huang, 2008). Using the plasminogen/plasmin blocker, tranexamic acid (TA), we further observed that the therapeutic effect of PAI-1R on glomerular ECM accumulation was largely mediated through plasmin since this effect was blocked by co-treatment with TA (Huang, 2006). However, the effect of PAI-1R on the reduction of proteinuria was not affected by blocking plasmin generation, suggesting that PAI-1R has multiple therapeutic actions on the measures of disease severity. Conversely, elevated PAI-1 may have multiple actions on the induction and progression of kidney disease. It is likely that PAI-1 may act as a direct cause of proteinuria independent of plasmin generation, which could be prevented by inhibition of PAI-1. This notion was strengthened by the observations that PAI-1 deletion significantly reduced the severity of proteinuria in diabetes (Collins, 2006; Lassila, 2007; Nicholas, 2005). It has been shown that proteinuria is often the result of podocyte injury in human diseases and animal models, especially in diabetes (Yu, 2005; Shankland, 2006; Ziyadeh, 2008). Thus, we postulate that PAI-1 plays a role in podocyte injury and proteinuria. We further hypothesize that PAI-1R reduces proteinuria by the amelioration of podocyte injury via PAI-1 inhibition.
To test our hypothesis, we examined if PAI-1 directly damaged podocytes in cell culture and determined the therapeutic efficacy of PAI-1R on podocyte injury in a murine model of uninephrectomized diabetic nephropathy.
METHODS
Reagents
Unless otherwise indicated, materials and chemicals were obtained from Sigma-Aldrich (St. Louis, MO). The recombinant human PAI-1R and active stable PAI-1 have been previously described (Huang, 2009) and were purified as described (Kvassman, 1995).
Study 1: Therapeutic effects of PAI-1R on diabetes-associated podocyte injury in db/db mice
Animals
Diabetic male db/db mice (BKS.Cg-Dock7m +/+ Leprdb/J homozygotes) and their littermate male db/m mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and housed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The experimental protocols were approved by the Animal Care Committee at the University of Utah. The db/db mice were determined to be diabetic by the vendor on the basis of appearance of obesity at approximately 5 week of age and were further demonstrated to be hyperglycemic in our laboratory at week 7. Mice were subjected to right uninephrectomy under anesthesia at week 8 to hasten the development of diabetic nephropathy as described previously (Huang, 2008). Db/m mice received uninephrectomy at week 8 served as the operation control. Our previous work on model development showed that uninephrectomized diabetic db/db mice developed overt nephropathy by 18 weeks of age and disease progression was remarkable between 18 and 22 weeks of age, determined by expansion of the mesangium and significant albuminuria (Huang, 2008). In the present study this model was used to determine the impact of PAI-1R on podocyte injury from 20 to 22 weeks of age.
Experimental design
Groups of 8 to 9 uninephrectomized mice were assigned and treated at 20 weeks of age as follows: untreated non-diabetic db/m mice (n=9) as healthy control; diabetic db/db mice treated with PBS through peritoneal injection from week 20 to week 22 (n=8) as disease control; and diabetic db/db mice treated with 0.5 mg/kg BW PAI-1R through peritoneal injection, twice daily from week 20 to week 22 (n=8) (The effective dose of PAI-1R was determined previously (Huang, 2008)). The blood glucose level and glycosylated hemoglobin (HbA1C) level were monitored in tail blood samples by using a blood glucose meter (Glucometer Elite XL; Bayer Healthcare, Elkhart, IN, USA) and the DC 2000+ HbA1C kit (Bayer Healthcare). Respectively. Twenty-four-hour urine was obtained from each mouse after placement in metabolic cages. Urine albumin was measured using the DC2000+ microalbumin reagent kit (Bayer Healthcare). As for the reproducibility of this assay, the coefficients of variance (CV) were less than 3% when the same sample was measured three times consecutively.
Mice were sacrificed under isoflurane anesthesia in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Briefly, anesthesia was induced by inhalation of 2.5% isoflurane through a complete anesthesia system (VetEquip IMPAC6, VetEquip Inc., Pleasanton, CA, USA). When mice were in deep anesthesia as determined by no jerking when their toes were pinched, blood samples were obtained by cardiac puncture and kidneys were perfused through the heart with cold PBS and then excised. Mice died of massive blood loss. Renal cortex was harvested by dissection and saved for further analysis as described previously (Huang, 2008). Briefly, three pieces of cortex were either snap-frozen in 2-methylbutane at −80°C or fixed in 10% neutralized formalin for immunohistological examination or fixed in 2.5% glutaraledhyde and 0.1 M sodium cacodylate buffer at 4°C for electron microscopy examination. Other pieces of cortex were treated with Trizol™ Reagent (GibcoBRL, Gaithersburg, MD, USA) for isolation of RNA or treated with 100 mM NaCl and 20 mM HEPES to be sonicated for 30 seconds, 3 times on ice and centrifuged at a high speed for 15min at 4°C. The supernatant from the sonication was then collected and stored at −80°C for fibronectin (FN) ELISA (Huang, 2008) and protein measurement by a BCA protein assay kit (PIERCE, Rockford IL, USA). Total collagen level in renal cortex was determined by a colorimetric method according to the manufacturer’s instruction (Bio-color Ltd., UK), in which the Sirius red dye binds to the side chains of amino acids found in collagen.
Histological analyses
Formalin-fixed renal cortex tissues were subsequently embedded in paraffin. Three-micrometer sections were cut from the tissue blocks and stained with periodic acid-Schiff (PAS). The PAS-positive glomerular matrix was quantitated in a blinded fashion by a computer-assisted method as previously described (Huang, 2008). At least 20 glomeruli from each mouse were assessed. The matrix surface area in the mesangium was normalized by that of the total glomerular tuft where the percentage of mesangial matrix occupying each glomerulus was rated on a 0–4 scale (0=0, 1=25, 2=50, 3=75, or 4=100%), as described previously (Huang, 2008).
Immunofluorescent staining for nephrin and podocin was performed on frozen sections. The polyclonal goat anti-nephrin IgG and the polyclonal goat anti-podocin IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were used as the primary antibodies. The rhodamin (TRITC)-conjugated donkey anti-goat IgG (H+L) (Jackson ImmunoResearch, Laboratories Inc., West Grove, PA, USA) was used as the secondary antibody. Intra-glomerular positive staining of both molecules was also quantified separately in a blinded fashion by a computer-assisted method. Briefly, images (x400 magnification) of 20 random glomeruli per slide were captured using a Nikon D50 digital camera (Inkle’s-Ritz Camera, www.ritzcamera.com, Salt Lake City, UT; Nikon Capture 4 Ver. 4.3, Nikon Inc., Melville, NY, USA), and the total staining intensity in each glomerulus was quantified using a computer-assisted color image analysis system (Image J 1.38 for Windows; National Institutes of Health http://rsb.info.nih.gov) (Abramoff, 2004). The results were expressed as the ratio of the staining intensity in the experimental groups to that in the normal glomeruli.
Transmission electron microscopy examination
Glutaraledhyde-fixed renal cortex tissues were further processed with OsO4 and Epon 812. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. Specimens were placed on copper grids, and examined in an electron microscope (JEOL JEM-1010, Tokyo, Japan). Foot process density was calculated by dividing the number of foot process by the GBM length (Zheng, 2008).
Western blot analysis
Equal amounts of renal cortex tissue (15 mg) from each mouse of each group were homogenized in lysis buffer (Cell Signaling Technology, Inc., Beverly, MA, USA) with 1% NP40, 1 mM PMSF and 1 tablet/ 5 ml protease inhibitor mix (Complete, Mini; Roche Diagnostics Corporation, Indianapolis, IN). Protein sample from 8 to 9 mice of each group was pooled for further examination. For western blot analysis, protein samples (20 μg each) were subjected to SDS-PAGE in 4–12% gradient gels (Invitrogen, Carlsbad, CA, USA) and immunblotted onto immobilon-P transfer membranes (Millipore Corporation, Bedford, MA, USA). Desmin and B7-1 were assessed on the Western blots using mouse monoclonal anti-desmin IgG2a (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and rabbit monoclonal anti-B7-1/CD80 IgG (OriGene Technologies, Inc., Rockville, MD, USA). The immunostaining band was visualized and quantified as described previously (Huang, 2008). Desmin and B7-1 protein levels were corrected by the densitometric intensity of the GAPDH (using goat anti-GAPDH IgG antibody, GenScript, Piscataway, NJ, USA) or β-actin (using mouse monoclonal anti-β-actin IgG) in each loading. For comparison, this ratio was set at unity for normal control samples and other lanes on the same gel were expressed as fold-change over this value. Three blots were performed for each primary antibody.
RNA preparation and real-time RT-PCR
Total RNA was extracted from renal cortex tissue using Trizol™ Reagent (GibcoBRL, Gaithersburg, MD, USA) according to the manufacturer’s instructions. Two micrograms of total RNA were reverse-transcribed using the superscript III first-strand synthesis system for RT-PCR kit (Invitrogen). Real-time RT-PCR was performed using the SYBR green dye I (Applied Biosystems, Foster City, CA, USA) and the ABI 7900 Sequence Detection System (PE Applied Biosystems) as described previously (Huang, 2008). Samples were run in triplicates in separate tubes to permit quantification of the target gene normalized to β-actin. Sequences of primers used are listed in Table 1. The specificity of the PCR products was confirmed on a 1.5% agarose gel by showing a specific single band with the expected size.
Table 1.
Primers used for real time reverse transcriptase-PCR
| Gene | Primer | Location (complementary to nucleotides) | Sequence 5′–3′ |
|---|---|---|---|
| Mouse nephrin (NM_019459) | Forward | 4344–4363 | AGATTTTGGGTTGCAGGTTG |
| Reverse | 4476–4495 | TGACCCATCTTTCCAGTTCC | |
| Mouse ZO-1 (NM_009386.1) | Forward | 5975–5994 | GGGGTGCATTTGAGTTCTGT |
| Reverse | 6219–6238 | ACATGCTTTGGTGTCCTTCC | |
| Mouse desmin (NM_010043.1) | Forward | 1374–1393 | CAAAGGGGTTCTGAAGTCCA |
| Reverse | 1552–1571 | GAAAAGTGGCTGGGTGTGAT | |
| Mouse B7-1 (NM_009855.2) | Forward | 297–316 | CCATGTCCAAGGCTCATTCT |
| Reverse | 480–499 | TTCCCAGCAATGACAGACAG | |
| Mouse β-actin (NM_007393.2) | Forward | 857–876 | GCT CTT TTC CAG CCT TCC TT |
| Reverse | 1132–1151 | TGA TCC ACA TCT GCT GGA AG |
Study 2: Effect of wild-type PAI-I on cultured podocytes
Cell culture
Conditionally immortalized mouse podocytes were kindly provided by Dr. Peter Mundel at Massachusetts General Hospital and Harvard Medical School. These cells were cultured and induced to differentiate as described previously (Reiser, 2004). Briefly, podocytes were cultured under permissive conditions (33°C) in RPMI-1640 supplemented with 10% FCS, penicillin/streptomycin, and interferon- (all from Invitrogen). Differentiation was induced by thermoshift to 37°C without interferon- in type I collagen-coated 6-well-plates (Fisher Science Education, Hanover Park, IL, USA) before experimental treatment. All experiments were performed with cells that had been allowed to differentiate for at least 14 days. Podocytes between passages 10 and 15 were used for all experiments.
Effect of PAI-1 on podocyte injury
To examine the effect of PAI-1 on podocyte injury, differentiated podocytes were rendered quiescence in serum-free medium for 24h, then incubated with 150 nM of recombinant active stable human PAI-1 for various time points or incubated for 24h in incremental concentrations of PAI-1 (Huang, 2009). Podocytes treated with serum-free medium only or 2 ng/ml TGF-β1 (R&D, Minneapolis, MN, USA) for 24h served as normal and positive controls respectively. After incubation, the podocytes were washed with ice-cold PBS. Total cellular RNA or protein was isolated from the podocytes as described previously for the analyses of podocyte-associated molecules’ mRNA and protein expression by real time RT/PCR and western blotting respectively (Huang, 2007). The rabbit polyclonal anti-nephrin antibody (TA306037) and rabbit polyclonal anti-zonula occludin-1 (ZO-1) antibody (61-7300) were obtained from OriGene Technologies and Invitrogen respectively. All experiments were performed at least three times in duplicated wells.
Statistical Analyses
Data are expressed as mean ± SD. The disease-induced increase in a variable was defined as the mean value for the disease control group minus the mean value of normal control group. The percentage of reduction in disease severity in a treated group was calculated as described previously (Huang, 2008). Statistical analyses of differences between the groups were performed by one-way ANOVA and subsequent Student-Newman-Keuls or Dunnett testing for multiple comparisons. P< 0.05 was considered statistically significant.
RESULTS
Study 1. Therapeutic efficacy of PAI-1R
Systemic parameters
There was no significant difference in survival rate among the non-diabetic db/m mouse, diabetic db/db mouse treated with PBS and diabetic db/db mouse treated with PAI-1R groups. The baseline and final parameters of the three groups of mice are presented in Table 2. Twenty-week body weights of db/db mice were significantly greater than the corresponding values in db/m controls. There was no significantly change in body weight from weeks 20 to 22 in db/db mice. Diabetic mice remained hyperglycemic throughout the experimental period as indicated by blood glucose and glycosylated hemoglobin levels. PAI-1R treatment had no effect on body weight and blood glucose levels in the db/db mice.
Table 2.
Parameters of the experimental groups of mice
| db/m (n=9) | db/db (n=8) | db/db-PAlR (n=8) | |
|---|---|---|---|
| body wt. initial, g | 24.5±0.8 | 43.1±6.0* | 46.0±3.3* |
| body wt. final, g | 25.1±1.3 | 43.1±5.6* | 42.4±3.5* |
| plasma glucose, initial, mg/dl | 106.2±16.8 | 512.0±96.8* | 553.0±59.1* |
| plasma glucose, final, mg/dl | 98.1±43.7 | 560.0±65.9* | 595.4±5.9* |
| HbA1c, % | 3.8±0.2 | 9.8±2.4* | 10.2±2.2* |
| UAE, initial, μg/d | 7.4±0.5 | 345.8 ±0.6* | 359.5±45.2* |
| UAE, final, μg/d | 6.1±1.3 | 356.9±3.5* | 247.4±19.1*#§ |
Nondiabetic (db/m) or diabetic (db/db) mice were treated for 2 weeks with either PBS or PAI-1R between 20 and 22 weeks of age.
P<0.05 vs. db/m;
P<0.05 vs. db/db;
P<0.05 vs. db/db-PAI-R at 20 weeks of age.
At the beginning of intervention (20 weeks of age), urinary albumin excretion (UAE) of db/db mice in both groups treated with or without PAI-1R was similar and was significantly higher compared with db/m mice. Albumin excretion stayed high between weeks 20 and 22 in PBS-treated db/db mice. PAI-1R treatment, in fact, lowered the UAE by 31.4% (359.5 ± 45.3 μg/d vs. 247.4 ± 19.1 μg/d; P<0.05).
Glomerular histology
At the end of the study (22 weeks), the glomeruli of db/db mice demonstrated increased accumulation of PAS-positive extracellular matrix (ECM) proteins in the mesangium, compared with glomeruli of db/m mice (Figure 1A–B). Treatment with PAI-1R ameliorated ECM deposition from the diabetic glomeruli by 36.3% (P<0.05).
Figure 1. In vivo effect of PAI-1R on glomerular matrix protein accumulation.

A. Representative photomicrographs (at original magnification × 400) of glomeruli at 22 weeks from normal control mice (db/m), diabetic db/db mice treated with PBS (db/db) and diabetic db/db mice treated with PAI-1R (db/db-PAI-1R). Graphic representation of the mean PAS staining scores (B). FN (C) and total collagen (D) of each group are also shown. *P<0.05, vs. db/m. #P<0.05, vs. db/db.
Collagen and FN levels in renal cortex
The levels of both collagen and FN in renal cortex, two major components of glomerular ECM, in mice were quantified. Consistent with the accumulation of glomerular PAS-positive ECM, levels of FN and collagen were significantly increased in db/db mice at week 22. PAI-1R treatment reduced the disease-induced increases in protein levels of FN and collagen by 62.5% and 67.7% respectively (P<0.05, Figure 1C–D).
Electronic microscopy of podocyte ultrastructure
As shown in figure 2A to C, segmental podocyte foot process effacement and decrease in foot process density were evident in the glomeruli from diabetic mice at 22 weeks, compared to non-diabetic glomeruli (db/db vs. db/m, 1.56 ± 0.35 vs. 3.42 ± 0.34, P<0.05). PAI-1R treatment effectively restored foot process density (PAI-1R vs. db/db, 2.83 ± 0.67 vs. 1.56 ± 0.35, p<0.05). The changes of podocyte foot process were clearly observed in the zoom images (Figure 2D to F).
Figure 2. In vivo effect of PAI-1R on podocytes assessed by transmission electron microscope (TEM).

Representative photomicrographs (at original magnification × 5000) of glomerular podocytes obtained at 22 weeks from a normal control mouse (db/m) (A), a diabetic db/db mouse treated with PBS (db/db) (B) and a diabetic mouse treated with PAI-1R (db/db-PAI-1R) (C). Podocyte foot process was point out by an arrow symbol. Their zoom images (D to F) were listed below the original photos.
Expression of podocyte slit diaphragm-associated proteins
To further confirm the protective effects of PAI-1R on podocyte injuries observed under electronic microscopy, podocyte slit diaphragm-associated proteins nephrin and podocin were determined by immunofluorescent staining. As shown in figure 3, non-diabetic db/m mice showed intense and linear staining for both nephrin and podocin along the capillary in glomeruli. In contrast, diabetes resulted in an attenuation of staining by 48.4% for nephrin and 36.9% for podocin (P<0.05) in the glomeruli in db/db mice at 22 weeks of age. Conversely, PAI-1R administration reduced the loss of podocyte specific nephrin and podocin staining by 58.6% and 73.7% respectively (P<0.05).
Figure 3. Effect of PAI-1R on immunofluorescent staining for glomerular nephrin and podocin at week 22.
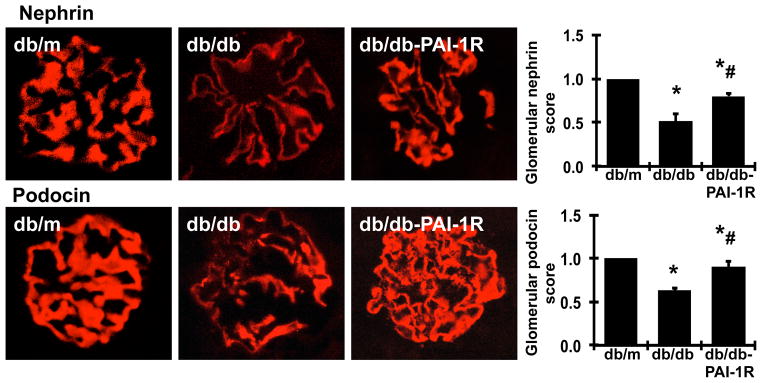
Representative photomicrographs (original magnification × 400) of the glomeruli obtained at 22 weeks from a normal control mouse (db/m), a diabetic db/db mouse treated with PBS (db/db) and a diabetic db/db mouse treated with PAI-1R (db/db-PAI-1R). Graphic representation of the mean nephrin and podocin staining scores of each group is shown on the right. *P<0.05, vs. db/m. #P<0.05, vs. db/db.
Expression of markers of podocyte injury
By western blot, we observed that the protein levels of desmin in renal cortex tissues, a conventional marker for podocyte injury, were up regulated by 190% in diabetic db/db mice (P<0.05), as shown in figure 4. PAI-1R treatment reversed the disease-induced increase of desmin protein. In addition, it has been shown that B7-1 is synthesized in podocytes in response to split-diaphragm protein rearrangement and foot process effacement, served as another injury marker (Reiser, 2004). As shown in figure 4, B7-1 protein levels were markedly increased in diabetic renal tissue in db/db mice. Although we could not confirm whether the elevated productions of desmin and B7-1 were from podocytes, the changes of both desmin and B7-1 were consistent with the changes of the two slit-diaphragm-associated proteins nephrin and podocin shown in figure 3, which may indicate that increased renal desminin and B7-1 productions somehow could be related to podocyte injury in diabetic db/db mice. Consistent with the results on nephrin and podocin, PAI-1R treatment also successfully reduced the elevated B7-1 protein expression in db/db mice by 58.3% (P<0.05).
Figure 4. Effect of PAI-1R on desmin and B7-1 production in renal cortical tissues.

A. Representative western blots showing desmin and B7-1 protein expression. GAPDH was included in the blot for normalization. B & C. Graphic representation of the mean band intensity of desmin and B7-1, normalized against the band intensity of GAPDH. *P<0.05, vs. db/m. #P<0.05, vs. db/db.
In agreement with protein levels, over 50% mRNA expression of nephrin was lost in diabetic db/db mice at week 22, compared to that of db/m controls. PAI-1R treatment rescued 71.5% of the loss in nephrin mRNA levels in diabetic renal tissue (Figure 5A). In contrast, mRNA expression of desmin and B7-1 was markedly increased in diabetic renal tissue by 14-fold and 20-fold, respectively (P<0.05), compared to that found in normal renal tissue (P<0.05). PAI-1R treatment reversed the increased mRNA expression of desmin and B7-1 (Figure 5C–5D), similar to the response of the corresponding proteins. Additionally, we also measured mRNA expression of the tight junction protein, zonula occludin-1 (ZO-1) in podocytes. Not surprisingly, ZO-1 mRNA was 68.6% lower in diabetic renal tissues than in normal controls (P<0.05). This loss in ZO-1 expression was completely reversed by PAI-1R treatment (P<0.05, Figure 5B).
Figure 5. Effect of PAI-1R on nephrin, ZO-1, desmin and B7-1 mRNAs expression in renal cortical tissues.

Expression of mRNA in renal cortical tissues obtained from various mouse groups at 22 weeks was determined by real-time RT/PCR and corrected for β-actin for each samples. Db/m: non-diabetic normal control; db/db: diabetic db/db mouse treated with PBS; and db/db-PAI-R: diabetic db/db mouse treated with PAI-1R. A. Expression of nephrin mRNA, B. Expression of ZO-1 mRNA, C. Expression of desmin mRNA, D. Expression of B7-1 mRNA, *P<0.05, vs. db/m. #P<0.05, vs. db/db.
Collectively, these data suggest that PAI-1R treatment protects podocyte integrity in diabetic db/db mice by preserving the expression of slit-diagram proteins and counter-regulating pathogenic molecules involved in podocyte injury.
Study 2. Direct effect of PAI-1 on podocyte injury in vitro
Effect of PAI-1 on nephrin, ZO-1, desmin and B7-1 mRNA expression by podocytes
It has been shown that the PAI-1R’s therapeutic efficacy is largely due to its ability to reduce endogenous PAI-1’s activity through competing for binding to vitronectin at the site of injury but its lack of protease inhibitory activity (Huang, 2006; Huang, 2009). To further delineate the mechanism of action of PAI-1R, we examined the direct effect of PAI-1 in a cultured podocyte model. Based on our previous studies on cultured mesangial cells (Huang, 2006), a recombinant active stable human PAI-1 at the concentration of 150 nM was chosen. As shown in figure 6, addition of PAI-1 to podocytes up to 48h caused a time-dependent inhibition of both nephrin and ZO-1 mRNA expression. The inhibition at 6h was 13.4% and 27.2% for nephrin and ZO-1 respectively and more than 80% (P<0.05) at 48h (Figure 6A & B). Importantly, these changes seen in PAI-1-treated podocytes were similar to those observed in podocytes exposed to 2 ng/ml TGF-β1 for 24h. The 24h-time point was thus used for further experiments as described below.
Figure 6. Time effects of wild-type PAI-1 on nephrin (A) and ZO-1 (B) mRNAs expression by mouse podocytes.

Quiescent mouse podocytes were incubated in the absence (normal control, Con) or the presence of TGFβ1 (2 ng/ml) or wild-type PAI-1 (150 nmol) for the indicated durations, and real-time RT/PCR was performed. The mRNA expression in the normal controls was used as the reference. *P<0.05 vs. normal control.
A dose-dependent decrease in both nephrin and ZO-1 mRNA levels (P<0.05, Figure 7A–B) and a dose-dependent increase in B7-1 mRNA levels (P<0.05, Figure 7D) in podocytes were seen after 24h of treatment with PAI-1. While there was also an increase in desmin mRNA levels with PAI-1 treatment, the dose-dependency of this phenomenon was less apparent (Figure 7C), In comparison, the addition of 2 ng/ml TGF-β1 to podocytes for 24h resulted in dramatic decreases in both nephrin and ZO-1 mRNA expression and dramatic increases in both desmin and B7-1 mRNA expression (P<0.05 vs. normal control for each comparison). These data indicate that PAI-1 directly alters gene expression of podocyte molecules in a manner similar to TGF-β1.
Figure 7. Dose effect of wild-type PAI-1 on nephrin (A), ZO-1 (B), desmin (C) and B7-1 (D) mRNAs expression by mouse podocytes.


Quiescent mouse podocytes were incubated in the absence (normal control, Con) or the presence of various doses of wild-type PAI-1 or TGFβ1 (2 ng/ml) for 24h, and real-time RT/PCR was performed. The mRNA expression in the normal controls was used as the reference. *P <0.05 vs. normal control.
Effect of PAI-1 on nephrin, ZO-1, and B7-1 protein production by podocytes
Consistent with the results on mRNA expression, 150 nM PAI-1 and 2 ng/ml TGFβ1 decreased the protein levels of nephrin and ZO-1 (by 39.4% and 45.4% for nephrin; 18.7% and 32.0% for ZO-1 respectively; all P<0.05). Meanwhile, both PAI-1 and TGFβ1 significantly increased B7-1 protein production by 230% and 246% respectively. These results indicate that PAI-1, similar to TGF-β1, induces podocyte injury by altering protein production of normal slit-diagram molecules and B7-1.
DISCUSSION
In the present studies, we repeatedly observed that uninephrectomized, diabetic db/db mice developed overt nephropathy associated with progressive albuminuria and glomerular mesangial matrix expansion and increased renal production of fibronectin and collagen at 22 weeks of age. Treatment with human PAI-1R from week 20 to week 22 had no effect on body weight, plasma glucose and Hb1Ac levels but reduced albuminuria and markers of renal fibrosis seen in db/db mice. Although we have not dissected the plasmin-dependent and plasmin-independent actions of PAI-R in the db/db mouse model, we have previously shown that the effects of PAI-1R on pathological matrix accumulation in the model of anti-thy-1 nephritis were completely reversed by TA treatment (Huang, 2003; Huang, 2006), indicating the plasmin dependence of these therapeutic actions. In contrast, TA had no effect on other disease measures, including proteinuria, in anti-thy-1 nephritis model treated with PAI-1R (Huang, 2006). This latter observation suggests that PAI-1R has other actions in addition to enhancing plasmin generation.
Increasing evidence shows that the development of proteinuria is typically associated with podocyte foot-process effacement and/or slit-diaphragm disruption in proteinuric kidney diseases including diabetic nephropathy (Fornoni, 2010). Consistent with this notion, untreated diabetic db/db mice in the present study developed significant segmental podocyte foot-process effacement associated with reduced renal production of nephrin, podocin and ZO-1 and increased renal production of B7-1 and desmin. Importantly, these changes in podocyte morphology and protein molecules that are indicative of podocyte injury were all ameliorated by the treatment with PAI-1R. Nephrin and podocin are critical for maintaining the barrier function of the podocyte slit diaphragm (Liu, 2003; Kawachi, 2006). Dysfunction or loss of nephrin and podocin may cause podocyte shape changes, such as foot process effacement and attenuate the contractile function of podocytes, leading to detachment from the underlying GBM (Liu, 2003; Kawachi, 2006). Therefore, PAI-1R treatment may reduce podocyte injury by preserving the normal structure of podocyte foot processes and slit diaphragms.
B7-1 is a transmembrane protein normally expressed on the surface of B lymphocytes and antigen-presenting cells (Abbas,1999; Chambers, 1999). It has been demonstrated to be synthesized de novo in podocytes and involved in the disruption of the split-diaphgram protein complex and the induction of foot-process effacement in response to a variety of stress conditions (Reiser, 2004). Thus, B7-1 also acts as an autocrine factor to modulate podocyte integrity beyond its role in immune cells. It has been shown that the mRNA expression of B7-1 in urinary sediments indeed correlates with the severity of proteinuria in patients with kidney diseases (Navarro-Munoz, 2011). We observed that the mRNA expression and protein production of B7-1 were significantly increased in diabetes in the present study (Figure 4 & 5). However, PAI-1R treatment substantially reversed the diabetes-induced elevated B7-1, which was accompanied by the recovery of podocyte morphology and slit-diagram protein production and reduction in albuminuria.
In addition, podocyte epithelial-mesenchymal transition (EMT), characterized by the loss of epithelial markers such as ZO-1 and gain of mesenchymal markers such as desmin, has been involved in podocyte foot process effacement (Li, 2008; Yamaguchi, 2009). In the present study, we also observed a reduction in ZO-1 and an increase in desmin in diabetic renal tissue in db/db mice at 22 weeks of age. Treatment with PAI-1R largely reversed these changes in the signaling molecules of podocyte EMT. Together, these results provide evidence that PAI-1R protects podocyte from injury through counter-regulating the expression profile of slit-diagram proteins and pathogenic molecules in db/db mice. Consequently, podocyte protection may be quantified as a marked reduction in albuminuria in the db/db mice (Table 2).
In concordance with the protective effect of PAI-1R on podocyte injury is the notion that PAI-1 directly regulates podocyte protein expression resulting in cellular injury and proteinuria. Notably, PAI-1 deletion prevented diabetic renal complications including albuminuria in both db/db diabetic mice and STZ-induced diabetic mice (Nicholas, 2005; Collins, 2006; Lassila, 2007) while transgenic over-expression of PAI-1 exaggerated proteinuria in an immune nephritis model (Kitching, 2003) although podocyte structures were not examined in those animals. In a mouse model of Ang II-mediated hypertensive kidney disease (Knier, 2011), the knockout of PAI-1 resulted in less glomerulosclerosis. Coincidentally, higher nephrin immunostaining was found in the podocytes. Our present observation in cultured podocytes provides the first evidence that PAI-1 directly induces podocyte injury by altering the expression of nephrin and B7-1, similar to the actions of TGFβ1 (Figure 6–8), a well-known mediator of podocyte injury (Reiser, 2004). In addition, PAI-1 appeared to be sufficient to induce podocyte EMT in cell culture by changing the cellular expression of ZO-1 and desmin, similar to the effects of TGF-β1. Collectively, these observations elucidate a new paradigm of PAI-1’s role in the pathogenesis of kidney disease via induction of podocyte injury, in addition to the known injurious effects of PAI-1 on the matrix degradation (Huang, 2006).
Figure 8. Effect of PAI-1 on nephrin, ZO-1 and B7-1 protein production by mouse podocytes.

Quiescent mouse podocytes were incubated in the absence (normal control, Con) or the presence of TGFβ1 (TGFβ1, 2 ng/ml) or PAI-1R (150 nmol) for 24h, and western blot analysis was performed in cell lysates. A. Representative western blots showing nephrin, ZO-1 and B7-1 protein expression. β-actin was included in the blot as a protein loading control. The graphs (B-D) summarize the results of the respective band density measurements, normalized against the band intensity of β-actin. *P <0.05 vs. normal control.
The signaling pathway by which PAI-1 directly modulates podocyte-associated molecules is unclear. A new signaling pathway in podoctyes that involves vitronectin, uPAR and αvβ3 integrin has recently been identified, which leads to podocyte foot-process effacement and urinary protein loss (Wei, 2008). These findings, together with the ability of PAI-1 to bind to vitronectin and complex with uPAR through PAI-1 complex formation with uPA, have led to the exciting possibility that PAI-1 induces podocyte injury through activation of this uPAR-signaling pathway. This further suggests that the plasmin independent benefit of PAI-1R may stem from its inability to interact stably with uPAR, and thus not activate this signaling pathway.
In conclusion, the present study provides evidence that, similar to TGFβ1, PAI-1 directly injuries podocytes in diabetes. Reducing the increased PAI-1 activity by PAI-1R may, in fact, reduce podocyte injury thereby reducing albuminuria. Therefore, PAI-1R may provide additional therapeutic effect in slowing progression of diabetic nephropathy by maintaining podocyte integrity.
NEW FINDINGS.
The mechanism by which PAI-1R reduces proteinuria has not been previously explored. Using cultured podocytes and diabetic db/db mice, we show that similar to the effects of TGFβ1, PAI-1 directly injuries podocytes. Reducing the increased PAI-1 activity by PAI-1R in diabetic db/db mice, in fact, reduce podocyte injury thereby reducing albuminuria. Therefore, PAI-1R may provide additional therapeutic effect in slowing progression of diabetic nephropathy by maintaining podocyte integrity.
Acknowledgments
FUNDING
This work was supported by grants from National Institutes of Health, DK077955 (Y.H.) and DK081815 (Y.H.).
We wish to thank Linda Hoge for her excellent technical assistance with animal studies.
Footnotes
COMPETING INTERESTS
All the authors declared no competing interests.
AUTHOR CONTRIBUTIONS
Both Jiandong Zhang and Chunyan Gu performed the experiments, collected, analyzed and interpreted the data and drafted the article. Daniel A. Lawrence produced the recombinant human PAI-1R and active stable PAI-1 and edited the article. Alfred K, Cheung edited the paper. Yufeng Huang designed and supervised the experiments, performed the uninephrectomy in mice, interpreted the data and revised the article critically for important intellectual content.
References
- Abbas AK, Sharpe AH. T-cell stimulation: an abundance of B7s. Nat Med. 1999;5:1345–1346. doi: 10.1038/70905. [DOI] [PubMed] [Google Scholar]
- Abramoff WS, et al. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Brenner BM, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- Brown NJ. Therapeutic potential of plasminogen activator inhibitor-1 inhibitors. Ther Adv Cardiovasc Dis. 2010;4:315–324. doi: 10.1177/1753944710379126. [DOI] [PubMed] [Google Scholar]
- Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr Opin Cell Biol. 1999;11:203–210. doi: 10.1016/s0955-0674(99)80027-1. [DOI] [PubMed] [Google Scholar]
- Collins SJ, et al. Plasminogen activator inhibitor-1 deficiency has renal benefits but some adverse systemic consequences in diabetic mice. Nephron Exp Nephrol. 2006;104:e23–34. doi: 10.1159/000093673. [DOI] [PubMed] [Google Scholar]
- Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol. 2006;17:2999–3012. doi: 10.1681/ASN.2006050503. [DOI] [PubMed] [Google Scholar]
- Fornoni A. Proteinuria, the podocyte, and insulin resistance. N Engl J Med. 2010;363:2068–2069. doi: 10.1056/NEJMcibr1008395. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandaliano G, et al. Tissue factor, plasminogen activator inhibitor-1, and thrombin receptor expression in human crescentic glomerulonephritis. Am J Kidney Dis. 2000;35:726–738. doi: 10.1016/s0272-6386(00)70022-9. [DOI] [PubMed] [Google Scholar]
- Huang Y, et al. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest. 2003;112:379–388. doi: 10.1172/JCI18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, et al. Noninhibitory PAI-1 enhances plasmin-mediated matrix degradation both in vitro and in experimental nephritis. Kidney Int. 2006;70:515–522. doi: 10.1038/sj.ki.5000353. [DOI] [PubMed] [Google Scholar]
- Huang Y, Noble NA. PAI-1 as a target in kidney disease. Curr Drug Targets. 2007;8:1007–1015. doi: 10.2174/138945007781662373. [DOI] [PubMed] [Google Scholar]
- Huang Y, et al. Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int. 2007;72:45–52. doi: 10.1038/sj.ki.5002243. [DOI] [PubMed] [Google Scholar]
- Huang Y, et al. A PAI-1 mutant, PAI-1R, slows progression of diabetic nephropathy. J Am Soc Nephrol. 2008;19:329–338. doi: 10.1681/ASN.2007040510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, et al. Mechanisms underlying the antifibrotic properties of noninhibitory PAI-1 (PAI-1R) in experimental nephritis. Am J Physiol Renal Physiol. 2009;297:F1045–1054. doi: 10.1152/ajprenal.00024.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawachi H, et al. Role of podocyte slit diaphragm as a filtration barrier. Nephrology. 2006;11:274–281. doi: 10.1111/j.1440-1797.2006.00583.x. [DOI] [PubMed] [Google Scholar]
- Keeton M, et al. Cellular localization of type 1 plasminogen activator inhibitor messenger RNA and protein in murine renal tissue. Am J Pathol. 1993;142:59–70. [PMC free article] [PubMed] [Google Scholar]
- Kitching AR, et al. Plasminogen activator inhibitor-1 is a significant determinant of renal injury in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2003;14:1487–1495. doi: 10.1097/01.asn.0000065550.13931.00. [DOI] [PubMed] [Google Scholar]
- Knier B, et al. Effect of the plasminogen-plasmin system on hypertensive renal and cardiac damage. J Hypertens. 2011;29:1602–1612. doi: 10.1097/HJH.0b013e32834840e8. [DOI] [PubMed] [Google Scholar]
- Krag S, et al. Plasminogen activator inhibitor-1 gene deficiency attenuates TGF-beta1-induced kidney disease. Kidney Int. 2005;68:2651–2666. doi: 10.1111/j.1523-1755.2005.00737.x. [DOI] [PubMed] [Google Scholar]
- Kvassman J-O, Shore JD. Purification of human plasminogen activator inhibitor (PAI-1) from Escherichia coli and separation of its active and latent forms by hydrophobic chromography. Fibrinolysis. 1995;9:215–221. [Google Scholar]
- Lassila M, et al. Plasminogen activator inhibitor-1 production is pathogenetic in experimental murine diabetic renal disease. Diabetologia. 2007;50:1315–1326. doi: 10.1007/s00125-007-0652-x. [DOI] [PubMed] [Google Scholar]
- Lee HS, et al. mRNA expression of urokinase and plasminogen activator inhibitor-1 in human crescentic glomerulonephritis. Histopathology. 2001;39:203–209. doi: 10.1046/j.1365-2559.2001.01195.x. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 2008;172:299–308. doi: 10.2353/ajpath.2008.070057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, et al. Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J Clin Invest. 2003;112:209–221. doi: 10.1172/JCI18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo S, et al. Multifunctionality of PAI-1 in fibrogenesis: evidence from obstructive nephropathy in PAI-1-overexpressing mice. Kidney Int. 2005;67:2221–2238. doi: 10.1111/j.1523-1755.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- Molitch ME, et al. Nephropathy in diabetes. Diabetes care. 2004;27(Suppl 1):S79–83. doi: 10.2337/diacare.27.2007.s79. [DOI] [PubMed] [Google Scholar]
- Navarro-Munoz M, et al. Messenger RNA expression of B7-1 and NPHS1 in urinary sediment could be useful to differentiate between minimal-change disease and focal segmental glomerulosclerosis in adult patients. Nephrol Dial Transpant. 2011;26:3914–3923. doi: 10.1093/ndt/gfr128. [DOI] [PubMed] [Google Scholar]
- Nicholas SB, et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005;67:1297–1307. doi: 10.1111/j.1523-1755.2005.00207.x. [DOI] [PubMed] [Google Scholar]
- Oda T, et al. PAI-1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int. 2001;60:587–596. doi: 10.1046/j.1523-1755.2001.030002587.x. [DOI] [PubMed] [Google Scholar]
- Parving HH, et al. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345:870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- Paueksakon P, et al. Microangiopathic injury and augmented PAI-1 in human diabetic nephropathy. Kidney Int. 2002;61:2142–2148. doi: 10.1046/j.1523-1755.2002.00384.x. [DOI] [PubMed] [Google Scholar]
- Podor TJ, et al. Type 1 plasminogen activator inhibitor binds to fibrin via vitronectin. J Biol Chem. 2000;275:19788–19794. doi: 10.1074/jbc.M908079199. [DOI] [PubMed] [Google Scholar]
- Reiser J, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondeau E, et al. Plasminogen activator inhibitor 1 in renal fibrin deposits of human nephropathies. Clin Nephrol. 1990;33:55–60. [PubMed] [Google Scholar]
- Scaffidi AK, et al. Regulation of human lung fibroblast phenotype and function by vitronectin and vitronectin integrins. J Cell Sci. 2001;114:3507–3516. doi: 10.1242/jcs.114.19.3507. [DOI] [PubMed] [Google Scholar]
- Senoo T, et al. Suppression of plasminogen activator inhibitor-1 by RNA interference attenuates pulmonary fibrosis. Thorax. 2010;65:334–340. doi: 10.1136/thx.2009.119974. [DOI] [PubMed] [Google Scholar]
- Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- Shihab FS, et al. Transforming growth factor-beta and matrix protein expression in acute and chronic rejection of human renal allografts. J Am Soc Nephrol. 1995;6:286–294. doi: 10.1681/ASN.V62286. [DOI] [PubMed] [Google Scholar]
- Stefansson S, et al. Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J Biol Chem. 2001;276:8135–8141. doi: 10.1074/jbc.M007609200. [DOI] [PubMed] [Google Scholar]
- Vial D, McKeown-Longo PJ. PAI1 stimulates assembly of the fibronectin matrix in osteosarcoma cells through crosstalk between the alphavbeta5 and alpha5beta1 integrins. J Cell Sci. 2008;121:1661–1670. doi: 10.1242/jcs.020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54:653–664. doi: 10.1053/j.ajkd.2009.05.009. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, et al. Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int. 1996;49:461–469. doi: 10.1038/ki.1996.65. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, et al. Increased levels of transforming growth factor-beta in HIV-associated nephropathy. Kidney Int. 1999;55:579–592. doi: 10.1046/j.1523-1755.1999.00296.x. [DOI] [PubMed] [Google Scholar]
- Yu D, et al. Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J Am Soc Nephrol. 2005;16:1733–1741. doi: 10.1681/ASN.2005020159. [DOI] [PubMed] [Google Scholar]
- Zheng S, et al. Podocyte-specific overexpression of the antioxidant metallothionein reduces diabetic nephropathy. J Am Soc Nephrol. 2008;19:2077–2085. doi: 10.1681/ASN.2007080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyadeh FN, Wolf G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr Diabetes Rev. 2008;4:39–45. doi: 10.2174/157339908783502370. [DOI] [PubMed] [Google Scholar]