

Graphical abstract
Abbreviations: ALS, amyotrophic lateral sclerosis; ANOVA, analysis of variance; CME, confocal microendoscopy; DL, deep lumbrical muscle; EPP, endplate potential; FDB, flexor digitorum brevis muscle; MEPP, miniature endplate potential; MPS, mammalian physiological saline; NAD, nicotinamide adenine dinucleotide; NMJ, neuromuscular junction; NMN, nicotinamide mononucleotide; Nmnat, nicotinamide mononucleotide adenyl transferase; QC, quantal content; Sarm1, sterile alpha and TIR motif-containing protein-1; SOD, superoxide dismutase; TTX, tetrodotoxin; WD, Wallerian degeneration; WldS, Wallerian degeneration-Slow mutant; WT, wild-type; YFP, yellow fluorescent protein
Key words: neuromuscular junction, axotomy, synaptic plasticity, activity
Highlights
-
•
Use and disuse may influence synaptic maintenance but so far evidence for this has been indirect.
-
•
We tested whether stimulation or disuse of neuromuscular junctions in adult WldS mice altered vulnerability to axotomy.
-
•
Moderate activity optimized resistance to axotomy while disuse or stimulation increased the rate of synaptic degeneration.
Abstract
Activity and disuse of synapses are thought to influence progression of several neurodegenerative diseases in which synaptic degeneration is an early sign. Here we tested whether stimulation or disuse renders neuromuscular synapses more or less vulnerable to degeneration, using axotomy as a robust trigger. We took advantage of the slow synaptic degeneration phenotype of axotomized neuromuscular junctions in flexor digitorum brevis (FDB) and deep lumbrical (DL) muscles of Wallerian degeneration-Slow (WldS) mutant mice. First, we maintained ex vivo FDB and DL nerve-muscle explants at 32 °C for up to 48 h. About 90% of fibers from WldS mice remained innervated, compared with about 36% in wild-type muscles at the 24-h checkpoint. Periodic high-frequency nerve stimulation (100 Hz: 1 s/100 s) reduced synaptic protection in WldS preparations by about 50%. This effect was abolished in reduced Ca2+ solutions. Next, we assayed FDB and DL innervation after 7 days of complete tetrodotoxin (TTX)-block of sciatic nerve conduction in vivo, followed by tibial nerve axotomy. Five days later, only about 9% of motor endplates remained innervated in the paralyzed muscles, compared with about 50% in 5 day-axotomized muscles from saline-control-treated WldS mice with no conditioning nerve block. Finally, we gave mice access to running wheels for up to 4 weeks prior to axotomy. Surprisingly, exercising WldS mice ad libitum for 4 weeks increased about twofold the amount of subsequent axotomy-induced synaptic degeneration. Together, the data suggest that vulnerability of mature neuromuscular synapses to axotomy, a potent neurodegenerative trigger, may be enhanced bimodally, either by disuse or by hyperactivity.
Introduction
Maintenance and degeneration of synapses are thought to depend on their use or disuse. For instance, activity may influence the rates of synaptic and neuronal degeneration both during normal aging and in neurodegenerative conditions in which cognitive decline or progressive impairment of muscle function is associated with early signs of synaptic dysfunction and demise (Frey et al., 2000; Selkoe, 2002; Swaab et al., 2002; Fischer et al., 2004; Frick and Benoit, 2010; Power et al., 2010; Valdez et al., 2010; Li et al., 2011; Punga and Ruegg, 2012; Stern, 2012; Shors et al., 2012). Altering some forms of neuromuscular activity may also influence progression of disease. For example, imposing moderate levels of activity in mouse models of amyotrophic lateral sclerosis (ALS) delays onset and slow progression of disease signs, reducing premature mortality (Kirkinezos et al., 2003; Veldink et al., 2003; Liebetanz et al., 2004; Deforges et al., 2009; Gordon et al., 2010; de Almeida et al., 2012). By contrast, intensive activity has also been reported to accelerate disease progression, both in mouse models and in sporadic forms of human ALS, perhaps through excitotoxicity or enhancing vulnerability to reactive oxygen species (Carri et al., 2003; Bruijn et al., 2004; Carrasco et al., 2004; Mahoney et al., 2004; Al-Chalabi and Leigh, 2005; Chio et al., 2005; Boillee et al., 2006; Pun et al., 2006; David et al., 2007; Bell and Hardingham, 2011; Alvarez et al., 2013; Mehta et al., 2013).
Despite speculation and debate on the influence of use or disuse of synapses on synaptic degeneration, there is as yet no compelling, direct evidence that connects normal axonal activity to synaptic maintenance, or abnormal axonal activity to the vulnerability of synapses to neurodegenerative triggers. By contrast, there is compelling evidence that some forms of synaptic remodeling or withdrawal are highly sensitive to activity. For instance, the rate of postnatal synapse elimination, a controlled process of presynaptic withdrawal that has been well characterized at developing neuromuscular junctions (NMJs) or following nerve regeneration in adults, is readily modifiable and strongly influenced by activity (Brown et al., 1976; Thompson et al., 1979; Betz et al., 1980a; Ribchester and Taxt, 1983; Taxt, 1983; Thompson, 1983; Barry and Ribchester, 1995; Costanzo et al., 2000; Keller-Peck et al., 2001; Gillingwater and Ribchester, 2003; Walsh and Lichtman, 2003; Personius et al., 2007; Leslie and Nedivi, 2011; Favero et al., 2012; Turney and Lichtman, 2012; Caroni et al., 2014). Reactive growth (sprouting) and enhancement of synaptic function also occurs in adults in response to imposed inactivity or activity (Betz et al., 1980b; Ribchester and Taxt, 1984; Tsujimoto et al., 1990; Dorlochter et al., 1991; Ribchester, 1993; Fahim, 1997). We therefore asked in the present study whether variations in axonal or synaptic activity either before or after a neurodegenerative trigger might influence the resistance of neuromuscular synapses to pathological stimuli for synaptic degeneration in adult muscles as well.
An accessible, reliable and readily controllable model of synaptic degeneration is the Wallerian-like breakdown of motor nerve terminal structure and function that occurs after nerve injury (axotomy) or following functional disruption of axonal transport (Slater, 1966; Miledi and Slater, 1970; Winlow and Usherwood, 1975, 1976; Hudson et al., 1984; Wang et al., 2000; Gillingwater and Ribchester, 2001; Coleman and Freeman, 2010). This process is now thought to have mechanisms in common with several forms of neurodegenerative disease (Conforti et al., 2014). Considerable insight into axotomy-induced degeneration of axons and synapses has been obtained through study of the Wallerian degeneration-Slow mutant (WldS) mouse mutant, in which axonal and synaptic degeneration are profoundly retarded by overexpression of a stable, aberrant isoform of the nicotinamide adenine dinucleotide (NAD)-synthetic enzyme nicotinamide mononucleotide adenyl transferase (Nmnat)-1 (Lunn et al., 1989; Ribchester et al., 1995; Mack et al., 2001; Wang et al., 2001; Gillingwater et al., 2002; Gillingwater et al., 2004; Gillingwater et al., 2006; Coleman and Freeman, 2010). This isoform substitutes for a more labile axoplasmic form, Nmnat-2, whose levels decline steeply after axotomy, an event which normally is sufficient to trigger fragmentation and subsequent degeneration of axons (Beirowski et al., 2005; Beirowski et al., 2009; Babetto et al., 2010; Gilley and Coleman, 2010; Gilley et al., 2013; Milde et al., 2013; Di Stefano et al., 2014).
Interestingly, the protective influence of the WldS protein is modifiable in several ways. For instance, the strength of axonal or neuromuscular synaptic protection in WldS mice is sensitive to: housing environment and husbandry of the mice; neuronal maturity, reduced mutant gene copy-number (“gene dose”); disrupted targeting to nuclei or intracellular organelles; and interaction with other genes, including Sarm-1 (Perry et al., 1990; Mack et al., 2001; Gillingwater et al., 2002; Beirowski et al., 2009; Conforti et al., 2009; Wong et al., 2009; Babetto et al., 2010; Osterloh et al., 2012; Massoll et al., 2013). Moreover, motor axons and their terminals are less well protected from axotomy-induced degeneration in WldS mice than those of sensory neurons (Brown et al., 1994; Oyebode et al., 2012). There are several plausible explanations for this but a key difference is that sensory endings and their axons continue to be active in response to natural orthodromic stimulation, including touch, pressure, nociceptive and proprioceptive stimuli (Oyebode et al., 2012) whereas distal axons and motor nerve endings, while remaining competent to conduct action potentials and evoke transmitter release, require continuity with their initial segments and motor neuron cell bodies for orthodromic activation, which is interrupted upon axotomy (Tsao et al., 1994; Ribchester et al., 1995; Gillingwater et al., 2002; Beirowski et al., 2009; Brown et al., 2014).
Thus, taking together the indirect evidence for an influential role of activity in normal aging or neurodegenerative disease; the strong influence of activity on the rate of natural remodeling processes like synapse elimination; and the enhanced persistence of sensory endings and their axons observed following axotomy in WldS mice, we enquired whether activity also influences Wallerian-like degeneration of axotomized neuromuscular synapses. If this were the case, then we would predict that systematically altering or imposing neuronal activity patterns should change the rate of synaptic degeneration after axotomy.
We tested our hypothesis by controlling axonal and neuromuscular synaptic activity in WldS mice, in three ways. First, we measured the effect of continuous stimulation of isolated and cultured nerve-muscle preparations on the rate of synaptic degeneration ex vivo. Second, we preconditioned axons by chronic nerve conduction block in vivo, then measured the effect of this disuse-priming on synaptic degeneration over several days after axotomy. Finally, we enriched the environment of mice for up to one month, by providing them with running wheels, thus encouraging increased levels of volitional activity, before cutting axons and measuring the subsequent levels of neuromuscular synaptic degeneration. Surprisingly, the data suggest a bimodal response: both inactivity and intense stimulation appear to increase the vulnerability and rate of synaptic degeneration in both WldS and wild-type (WT) mice, while moderate levels of activity were beneficial or without adverse effect. We discuss possible implications of the data for unified views of links between Wallerian-like degeneration of neuromuscular synapses, developmental synapse elimination, and neurodegenerative diseases in which synaptic dysfunction or degeneration are early phenomenological or clinical signs (Gillingwater and Ribchester, 2001; Raff et al., 2002; Gillingwater and Ribchester, 2003).
Experimental procedures
Ethical approval
All experiments reported in the present paper were approved by the University of Edinburgh College of Medicine and Veterinary Medicine Local Ethics Committee and conducted under the terms of a Project Licence and Personal Licences from the UK Home Office, in accordance with requirements of the United Kingdom Animals (Scientific procedures) Act, 1986. At the end of the experiments, mice were swiftly killed by cervical dislocation, in accordance with the UK Home Office regulations, Schedule 1.
Mice
Age-matched (5–10 weeks old) male and female C57Bl6, thy1.2YFP16:C57Bl6, C57BlWldS, thy1.2YFP16:WldS and thy1.2-YFPH:WldS mice generated by cross-breeding to homozygosity were used throughout these studies (Lunn et al., 1989; Feng et al., 2000; Beirowski et al., 2004; Beirowski et al., 2005; Bridge et al., 2009; Wong et al., 2009; Oyebode et al., 2012; Teriakidis et al., 2012; Hirst and Ribchester, 2013). All mice used were housed in standard light (12-h light 12-h darkness) and temperature conditions, in cages of 6 or fewer. Mice had access to food and water ad libitum. The thy1.2YFP16 transgenic line expresses yellow fluorescent protein (YFP) in all motor neurons, serving as a useful endogenous and non-toxic reporter of axonal and synaptic structure and integrity (Feng et al., 2000; Bridge et al., 2009; Wong et al., 2009). Expression of YFP also does not interfere significantly with Wallerian degeneration (WD) or the WldS-protective phenotype (Beirowski et al., 2004; Beirowski et al., 2005; Bridge et al., 2009; Comley et al., 2011). However, WldS mice older than 10 weeks were not used in the present study since the protection of synapses by the chimeric Nmnat1-Ube4b protein responsible for the WldS phenotype weakens with age (Perry et al., 1992; Tsao et al., 1994; Tsao et al., 1999; Gillingwater et al., 2002; Beirowski et al., 2009; Adalbert et al., 2012).
Experimental design
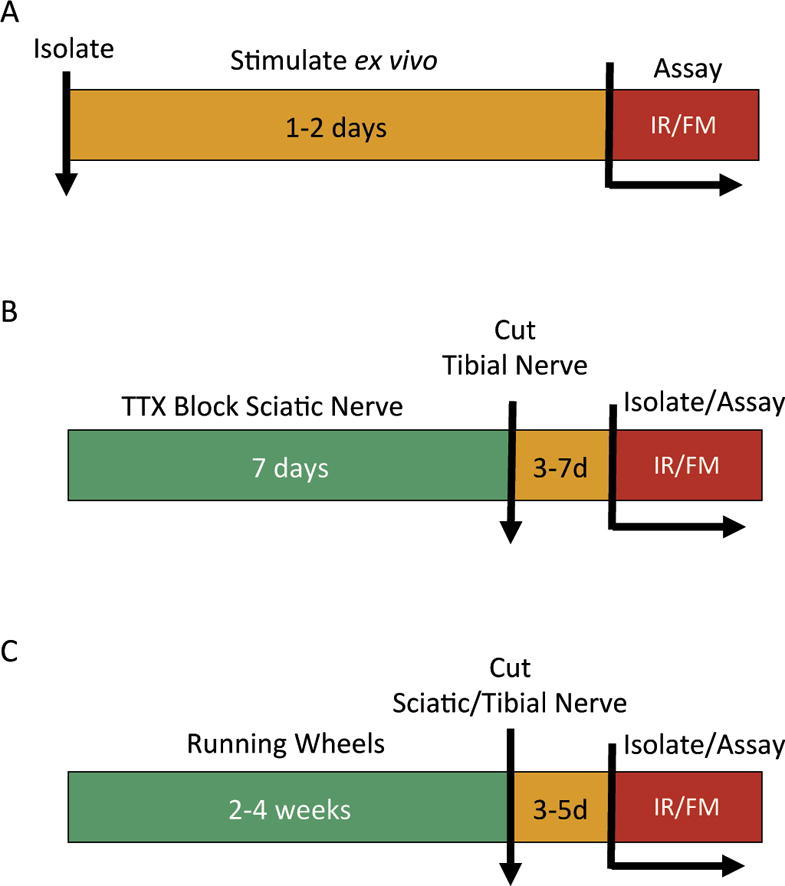
The main principle of our experimental design was to use the response of NMJs to axotomy in WldS mice as an assay for the effects of either preconditioned or coincident experimental alterations in neuromuscular activity (Fig. 1). Our basic strategy was to use a condition-test approach, in a combination of in vivo and ex vivo paradigms and in vitro physiological and morphological measurements, which has proved successful in exploring modifiers and their mechanisms in other contexts, for instance, axonal degeneration and regeneration (McQuarrie et al., 1977; Boegman et al., 1980; Deshpande et al., 1981; Hudson et al., 1984; Lund et al., 2002; Beirowski et al., 2004; Beirowski et al., 2005; Wong et al., 2009; Oyebode et al., 2012). Our assays were conducted on isolated nerve-muscle preparations of hind toe muscles: specifically flexor digitorum brevis (FDB) or one of the four deep lumbrical muscles (1–4DL). FDB is advantageous for electrophysiological analysis because its muscle fibers are isopotential, obviating the need for precise intracellular positioning of a recording microelectrode (Bekoff and Betz, 1977a,b; Gillingwater et al., 2002; Ribchester et al., 2004). The lumbrical muscles are advantageous for synaptic morphology, because they are thin and the NMJs may thereby readily be prepared and scored rapidly in unsectioned, whole-mounted preparations (Betz et al., 1980b; Gillingwater et al., 2002; Ribchester et al., 2004; Oyebode et al., 2012; Teriakidis et al., 2012; Hirst and Ribchester, 2013). We isolated FDB or 1–4DL nerve-muscle preparations and maintained them for up to 2 days ex vivo (that is, in organ culture) at 32 °C, either in the presence or absence of patterned, supramaximal electrical stimulation of their tibial nerve supply (Fig. 1A). In other experiments, we preconditioned the levels of neuromuscular activity in these muscles, supplied by axons in the sciatic nerve, for 1–4 weeks in vivo. Nerve conduction was either blocked completely using implants impregnated with tetrodotoxin (TTX; Fig. 1B); or voluntary, self-motivated activity was facilitated by providing mice free access to running wheels fitted in their cages (Fig. 1C). Mice typically elected to run between 1 and 15 km per night with this provision. Neuromuscular synaptic structure and function were evaluated at the end of this period.
Fig. 1.
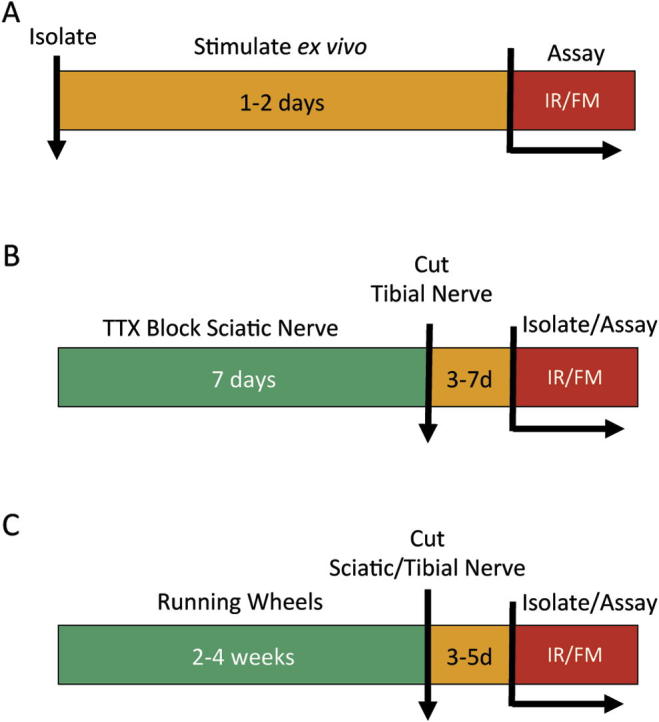
Experimental design. Neuromuscular synapses in WldS mouse FDB muscles were challenged by activity and axotomy in three ways. (A) Isolation and ex vivo organ culture for 1–2 days, with or without exogenous patterned stimulation to the distal tibial nerve stump, followed by assay using intracellular recording (IR) and/or fluorescence microscopy (FM). (B) Preconditioning sciatic nerve conduction block with tetrodotoxin (TTX) for 1 week, followed by tibial nerve section, then assay of residual innervation. (C) Preconditioned self-motivated activity, by providing mice access to running wheels, for 2–4 weeks, followed by either sciatic or tibial nerve section, then assay 3–5 days later.
Ex vivo assay of synaptic degeneration
We developed a novel ex vivo assay for scoring synaptic degeneration during a 48-h period after axotomy (see also (Di Stefano et al., 2014)). We measured the levels of degeneration at different time points in the FDB and lumbrical muscles of four different genotypes of mice: WldS (N = 27 mice), thy1.2YFP16:WldS (N = 15), thy1.2YFP16:Bl6 (N = 10) and C57Bl6 (N = 12). Mice were sacrificed by cervical dislocation and both hind limbs were removed. The hairy and glabrous skin of both hind feet was stripped. The limbs were then placed in fresh oxygenated mammalian physiological saline (MPS; 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 0.4 mM NaH2PO4, 23.8 mM NaHCO3, 5.6 mM d-glucose) and equilibrated by bubbling with 95% oxygen/ 5% CO2. FDB and lumbrical muscles were dissected, and pinned onto small sheets of Sylgard. They were then placed in sterile 20-ml tubes containing filtered and equilibrated MPS (as above), and maintained at 32 °C in a water bath for 8–48 h. Preliminary experiments established that WT and WldS phenotypes could clearly be distinguished after overnight incubation at this temperature. The muscles were bubbled in fresh MPS for 20 min prior to electrophysiological classification of Responsive, Innervated and Unresponsive muscle fibers, as described below. To test for effects of electrical stimulation, FDB muscles were pinned to a Sylgard-lined Petri dish and the nerve was placed in contact with silver wire electrodes integrated into the dish and connected to an external stimulator. Stimulus pulse durations and amplitudes were set to levels that gave visibly strong muscle contractions via neuromuscular transmission. In all cases the stimulus pulses were between 0.1 and 0.5 ms and less than 10V and thus below threshold for direct stimulation of muscle fibers or intramuscular axons. Confirming this, muscles only contracted in response to stimulation when the nerve was placed over the stimulating electrodes. We used three stimulation protocols: 1 Hz continuously, 10 Hz for 1 s every 10 s, and 100 Hz for 1 s every 100 s. Physiological effects were assayed by scoring responsive, innervated and unresponsive muscle fibers after 30 h of stimulation. The effect of varying Ca2+ ionic concentration was investigated in MPS containing reduced Ca2+ (1 mM) and increased Mg2+ (3 mM) in both unstimulated and stimulated muscles. Previous experiments showed that this ratio of Ca2+/Mg2+ was sufficient to abolish muscle contractions in response to supramaximal stimulation and to reduce mean quantal content (QC) of endplate potential (EPPs) in most FDB muscle fibers to fewer than five quanta (D. Thomson, R. Brown and R.R. Ribchester, unpublished observations.)
Electrophysiology
FDB nerve-muscle preparations were dissected and maintained in equilibrated MPS until used for electrophysiological analysis as described above, and previously (Gillingwater et al., 2002; Ribchester et al., 2004). The muscles were pinned in a Sylgard-lined recording chamber. The nerve was stimulated using a glass suction electrode whose inner and outer silver wires were connected to a Digitimer DS2 stimulator (Digitimer, Welwyn Garden City, UK), controlled by a Digitimer D4030 programer. Muscle fiber action potentials were blocked by bathing the isolated preparations in MPS containing 2.5 μM μ-conotoxin (GIIIB μ-CTX; Bachem, Bubendorf, Switzerland) for 20 min, or until muscle twitches in response to nerve stimulation became absent. Glass microelectrodes (1.5 mm o.d. × 0.86 mm i.d., Harvard Apparatus, UK) pulled on a P-87 Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA, USA) were backfilled with 4 M potassium acetate. Microelectrode tip resistances were 30–70 MΩ. Muscle fibers were impaled and EPPs were recorded using an Axoclamp-2B amplifier (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA), low-pass filtered at 3 kHz (Neurolog, Digitimer) and digitized using a CED micro1401 Mk II interface connected to a personal computer (Dell). EPP recordings were analyzed using WinWCP software (Strathclyde electrophysiological software, University of Strathclyde, UK). Spike-2 software (Cambridge Electronic Design, Cambridge, UK) was used to record spontaneous miniature EPPs (MEPPs) and the records were then analyzed using Minianalysis (Synaptosoft, Atlanta, GA, USA). MEPP recordings were only taken from fibers with a resting membrane potential between −60 and −70 mV. Trains of 30 EPPs evoked by supramaximal nerve stimulation at 0.5–2 Hz were recorded from each muscle fiber. From a total of 30 fibers impaled in each muscle, the innervation of fibers was classified by scoring the number of fibers (a) giving an evoked response upon stimulation (“Responsive Fibers”); (b) showing spontaneous MEPPs within a 20–100-s recording period but no evoked response (“Innervated Fibers”); or (c) failing to respond to stimulation and showing no evidence of either evoked EPPs or spontaneous MEPPs in a 20s–100s period of observation (“Unresponsive Fibers”). WinWCP software was used to calculate QC using the Variance Method. The McLachlan–Martin equation: v′ = v/(1 − fv/E) (p. 322 in (McLachlan and Martin, 1981) in WinWCP was used to correct EPP amplitudes for non-linear summation, assuming the muscle-specific correction factor f equal to 0.3; and reversal potential equal to −10 mV.
NMJ staining
Lumbrical muscles were normally dissected from the same foot as the FDB preparations, pinned in Sylgard-lined Petri dishes and immersed in MPS containing 5 μg ml−1 tetramethylrhodamine-isothiocyanate conjugated with α-bungarotoxin (TRITC-α-BTX; Invitrogen, USA) to label postsynaptic acetylcholine receptors (AChRs). The preparations were then placed on a rocking platform (Stuart Scientific, Chelmsford, UK) for 10 min, washed twice with MPS for 10 min, fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Hatfield, PA, USA) for 15 min then washed twice with MPS for 10 min. Once fixed and stained, the muscles were mounted on slides using Vectashield (Vector Laboratories, Peterborough, UK) and imaged on an Olympus BX50WI upright fluorescence microscope fitted with a Hamamatsu Orca-ER camera, captured and processed using OpenLab (Improvision/Perkin-Elmer, Coventry, UK) and Adobe Photoshop (USA) software running on an Apple Mac G5 computer. Confocal images were obtained using a BioRad Radiance 2000 confocal microscope via a Nikon Eclipse EFN600 upright microscope, captured and processed using Zeiss Lasersharp software. Motor endplates were scored as “Occupied” when the TRITC-α-bungarotoxin-labeled endplate showed any coverage with a YFP-labeled axon. Endplates with no overlying, YFP-positive axonal input were scored as “Vacant” (that is, denervated).
Effects of inactivity
These experiments were performed on the following groups of mice: WldS (N = 11 mice), double-homozygous thy1.2YFP16:WldS (N = 32), and thy1.2YFP16:C57Bl6 (N = 1) and C57Bl6 (N = 4).
TTX microcapsule manufacture and implantation
TTX-impregnated microcapsules were manufactured essentially by the method described previously (Martinov and Nja, 2005). Briefly, electrode glass (1.2 mm × 0.94 mm Harvard apparatus, UK) was pulled on a P87 Flaming/Brown micropipette puller. The tip was then broken and polished to a smooth tip using a micro forge (Narishige, Japan) to produce a tip with an external diameter of approximately 200 μm. The other end of the electrode was then scored with a diamond pencil and broken to obtain an overall length of 5 mm, then smoothed using an ethanol lamp. TTX powder in citrate buffer (Alomone Labs, Jerusalem, Israel) was dissolved in sterile saline (Hameln Pharmaceuticals, Gloucester, UK) containing 200 μg/ml Ampicillin (Sigma Aldrich, Dorset, UK) to a final concentration of 15 mM. The microcapsules were filled by capillary action with solutions containing either this solution or the vehicle solution, without TTX, alone. The larger end was then sealed with a small amount of dental wax (Kemdent, Swindon, UK). The tip was protected by inserting it into a small length of polythene tubing (i.d. 2 mm, SF Medical, Hudson, MA, USA).
Prior to surgery, WldS, thy1.2 YFP16: WldS or C57Bl6 mice aged 5–8 weeks, were prepared with a subcutaneous injection of Vetergesic (0.05 mg kg−1). Animals were anesthetized via isoflurane and oxygen inhalation (Merial Animal Health, UK, 2–5%, 0.4–1 l min−1). A skin incision was made at the level of the sciatic notch. The underlying muscle was parted by blunt dissection to reveal the sciatic nerve, which was then enclosed within a small cuff made from polythene tubing (i.d. 3 mm, SF Medical, USA). A TTX-filled microcapsule was dipped in sterile saline to remove any excess TTX from the exterior and the tip was then secured inside the cuff. In initial experiments, the tip was inserted into the epineurium but subsequently we found that the block was as effective when the tip of the micropipette was merely lodged inside the cuff. Since this procedure also reduced the risk of nerve damage, we adopted it for most of the experiments.
Nerve conduction block was monitored daily in four ways: (i) by observing the animals weakened grip strength on cage bars, (ii) abnormal walking gait; (iii) absence of a toe-spreading reflex on lifting by the tail; and (iv) absence of a nociceptive reflex response to firm, brief pinching of the foot pad with blunt-tipped forceps. Absence of the toe-spreading reflex and unresponsiveness to firm toe-pinches have been previously shown to be a reliable indicator of effective and complete nerve conduction block (Brown and Ironton, 1977; Lavoie et al., 1977; Betz et al., 1980b; Ribchester, 1993; Barry and Ribchester, 1995; Costanzo et al., 2000). We confirmed this in one anesthetized WldS mouse in which the sciatic nerve was stimulated above and below the level of application of the TTX-capillary. Foot muscle twitching was observed upon stimulating distal to the block and there was no response to nerve stimulation above the location of the implant.
Tibial nerve axotomy
Animals were anesthetized and the skin of either one or both hindlimbs was swabbed with sterilizing solution as described above. The tibial nerve was exposed by blunt dissection and a small section was removed to delay regeneration. Wounds were sutured using 7-0 Mersilk suture (Ethicon Products, Livingston, UK). Mice were killed 3–7 days later by cervical dislocation and electrophysiological and morphological studies were performed using the methods described above. In C57Bl6 mice the level of degeneration was assessed 12 h after nerve cut. In some cases the contralateral side was used as an unoperated control.
Live imaging of axonal and synaptic degeneration
A handheld fiber-optic confocal microendoscope (Cellvizio, Mauna Kea Technologies, Paris) was used to monitor axonal and synaptic degeneration in vivo in YFP expressing transgenic mice (Wong et al., 2009; Brown et al., 2014). A small incision was made in the hindlimb of the anesthetized mouse, permitting access to a Proflex S-1500 probe, with a tip diameter of 1500 μm. The inbuilt diode laser provided fluorescence excitation at 488 nm. Emitted light of 505–700 nm was collected through the same optical fibers. Tibial nerves and surrounding muscles were exposed and visualized at up to 7 days post-axotomy in mice with and without preconditioning TTX-block of the sciatic nerve. Stills were obtained from the videos via ImageCell software, and nerve fragment length measured using ImageJ (downloadable from http://rsb.info.nih.gov/ij).
Effects of running wheel activity
To facilitate activity in vivo, heterozygous or homozygous WldS mice were individually housed in polycarbonate cages fitted with running wheels, either for 2 weeks (N = 10) or 4 weeks (N = 7). The wheels were connected to revolution counters and average daily running distance was calculated from the number of complete wheel revolutions. In additional WldS mice (N = 8) and C57Bl6 mice (N = 8) mice, circadian wheel-running activity was monitored using Clocklab software (Actimetrics, Wilmette, IL, USA) as described previously (Shen et al., 2000; Harmar et al., 2002). Control WldS mice (N = 6) were individually housed with no additional environmental enrichment. Following the defined self-motivated running period, mice were anesthetized with isoflurane and the sciatic nerve sectioned on one side, as described above. A small (ca. 2 mm) piece of sciatic nerve was removed to inhibit regeneration. Five days later electrophysiological analysis in isolated FDB nerve-muscle preparations was used to score responsive and innervated fibers as described above. The animals were matched for age at the time of sacrifice.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (GraphPad Software Inc, San Diego, CA, USA), using unpaired t-tests for comparing two groups of continuous data and an analysis of variance (ANOVA, with post hoc tests as indicated) for multiple groups. All data are represented as mean ± SEM, unless otherwise stated. ‘N’ refers to the number of animals, whereas ‘n’ represents the number of muscles.
Results
We tested for influences of activity on synaptic degeneration at WldS mouse NMJ in three ways. First, we isolated nerve-muscle preparations and stimulated them at different frequencies for up to 48 h ex vivo (Fig. 1A). Second, we tested the influence of inactivity on synaptic degeneration, by conditioning sciatic nerve axons with chronic conduction block for 7 days in vivo, then we measured the rate of synaptic degeneration 3–7 days after cutting the inactivated axons (Fig. 1B). Finally, we tested whether enriching the behavioral environment of the mice for 2–4 weeks would influence the protection of their neuromuscular synapses from axotomy-induced degeneration, by providing cage access to running wheels before triggering synaptic degeneration by axotomy (Fig. 1C).
As our initial benchmark, we first established the pattern of innervation of toe muscles in WldS mice 5 days after axotomy in vivo, utilizing co-expression of the thy1.2-YFP transgene as a reporter. Whole-mounts of DL muscles showed a pattern of fully occupied, partially occupied and vacant motor endplates, suggesting that degeneration occurs asynchronously within the population of NMJs (Fig. 2). Quantitative scoring of endplate occupancy and functional responses in thy1.2YFP16:WldS mice showed that about 25–50% of endplates were denervated by 5 days post axotomy, which is a similar incidence at that time point to immunostained material (Gillingwater et al., 2002; Bridge et al., 2009). Inspection of single axotomized motor units in thy1.2YFPH:WldS mice, in which only about 5% of motor neurons express YFP, revealed occupied and partially vacated endplates within the same motor unit (Fig. 3), suggesting that synaptic retraction occurs asynchronously even within motor units and may therefore be locally regulated (Keller-Peck et al., 2001; Gillingwater and Ribchester, 2003).
Fig. 2.

Synaptic degeneration in axotomized WldS mouse muscles is asynchronous. (A) Whole-mount montage of a thy1.2YFP16:WldS mouse DL muscle, 5 days after cutting the tibial nerve. YFP fluorescence (green) and TRITC-α-BTX (red) counterstaining of AChR at motor endplates shows many NMJ were still fully occupied (B). Partially occupied (C, D) and vacant (denervated) endplates (E) were also readily discernible. Scale bar in E applies to images B–E. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.

Synaptic degeneration within axotomized motor units of WldS mouse muscle is also asynchronous. (A) Whole-mount montage of a single fluorescent motor unit in a thy1.2YFPH:WldS mouse DL muscle, 5 days after tibial nerve section. Only about 5% of motor units express YFP in the YFP-H transgenic line. In this, fortuitous instance, only a single unit (axon indicated by yellow arrow) was fluorescent. In this unit, some NMJs were still fully occupied (B, B′); others were partially occupied (C, C′) or almost vacant (D, D′). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Synaptic degeneration ex vivo is accelerated by high-frequency stimulation
We tested directly the effects of activity on synaptic degeneration, by isolating tibial nerve/FDB and DL muscle preparations and stimulating them via their nerve supply, for up to 48 h, while they were bathed in MPS (see Fig. 1A).
In preliminary experiments we found that persistence of synaptic function over 12–48h ex vivo was strongly temperature dependent. Surprisingly, and by contrast with the rates of synaptic degeneration observed in vivo, incubating preparations in MPS at 37 °C rendered synaptic degeneration as rapid in preparations from WldS mice as in WT preparations. Conversely, culturing the explanted muscles at 25 °C slowed degeneration of synaptic terminals in WT mice to such an extent that the effect of WldS could not be distinguished at this temperature either (data not shown, but see also (Tsao et al., 1999)). However, when muscles from homozygous WldS mice were incubated at 32 °C, many NMJ’s were still functional 24 h later, compared with WT muscles in which most did not respond to nerve stimulation (Fig. 4B, C, F). Specifically, in preparations from WldS mice, 89.61 ± 2.93% of fibers (n = 11 muscles) responded with nerve-evoked EPPs at 5–15 h. After 15–25 h, there was no significant change (95.88 ± 1.15% responsive fibers; n = 17) and most NMJs (70.42 ± 7.06%, n = 8) still showed significant persistence of synaptic function after 25–30 h ex vivo (Fig. 4F). The number responding at 35–45 h was reduced to 45.72 ± 11.90% (n = 4). By 45–50 h, substantial numbers of fibers had become denervated but 2.50 ± 1.71% (n = 5) still remained functional. By contrast, in WT muscles neuromuscular synapses degenerated much more rapidly: only 36.39 ± 16.02% of NMJs responded to stimulation at 15 h (n = 6 muscles), and only 10.77 ± 5.30% at 15–25 h (n = 13 muscles). By 25 h, only 1.25 ± 1.25% of fibers produced evoked synaptic responses (n = 4 muscles). Thus, 32-h incubation of these nerve muscle preparations ex vivo constituted a suitable checkpoint for determining the rate of synaptic degeneration following stimulation in WldS muscles, as approximately 50% of synapses were innervated, meaning that potential positive or detrimental effects of stimulation could be distinguished.
Fig. 4.

Culturing FDB muscles ex vivo resolves WldS and control phenotypes. (A) Tibial nerve (TibN)/FDB muscle preparation pinned to a Sylgard chamber, used for the ex-vivo culture and subsequent electrophysiological analysis. (B, C) Representative electrophysiological recordings from wild-type (B) and WldS (C) FDB muscle fibers, after 24 h culture ex vivo. (D, E) Confocal images of motor nerve terminals from thy1.2YFP16C57:Bl6 (D) and thy1.2YFP16:WldS (E) lumbrical muscles, 24 h after ex vivo culture at 32 °C. Images digitally adjusted for overall brightness and contrast only. (F) Time course of loss of innervation as measured electrophysiologically from the incidence of FDB muscle fibers responding with EPPs to nerve stimulation in WldS (filled bars) and wild-type mouse muscles. (G) Comparable incidences of occupied NMJ assayed in DL muscles morphologically using fluorescence microscopy. Both graphs show mean ± S.E.M., in n = 2–17 muscles.
Morphological data obtained from thy1.2YFP16:WldS and thy1.2YFP1C57Bl6 mouse DL nerve-muscle preparations corroborated the physiological data (Fig. 4D, E, G). Most NMJ remained occupied by motor nerve terminals up to 24 h after explant of DL nerve-muscle preparations from the thy1.2YFP16:WldS line, whereas most in the control thy1.2YFP1C57Bl6 line had degenerated (Fig. 4G). Specifically, 99.68 ± 0.83% (n = 4 muscles) of NMJs in DL muscles from thy1.2YFP16 WldS mice were innervated for at least 8 h in MPS. By 16 h, innervation of motor endplates declined slightly, to 87.31 ± 4.13% of NMJs (n = 4), then to 54.45 ± 6.50% (n = 5) by 24 h. By 32 h, 22.33 ± 19.38% of motor endplates still remained occupied (n = 3). By 48 h, only 4.25% of NMJs remained (n = 2 muscles). By comparison, NMJs degenerated at least twice as rapidly in explants from the control mice (Fig. 4G). Although almost all intramuscular axons and NMJs in preparations from thy1.2YFP16:Bl6 mice remained intact for about 8 h (92.46 ± 11.62%, n = 7), by 16 h the number of occupied endplates had declined steeply, to 28.51 ± 25.56% (n = 6). By 24 h, innervation had further decreased to only 12.09 ± 12.67% (n = 7) of motor endplates: less than one-fourth the number in WldS preparations. Thus, while the rate of synaptic degeneration ex vivo in preparations excised from WldS mice appears to be more rapid compared with the rate in vivo (Ribchester et al., 1995; Tsao et al., 1999; Gillingwater et al., 2002; Parson et al., 2004), incubation in MPS for 24 h at 32 °C was sufficient to distinguish very clearly the WldS from the WT phenotype.
To test whether activity might alter the rate of synaptic degeneration in these ex-vivo explants, we stimulated the NMJs at frequencies between 1 and 100 Hz before assaying residual functional innervation (Figs. 1A, 5). We used stimulus patterns that applied an equal number of stimuli to the tibial nerve over this period (either 1 Hz continuously; or 10 Hz for 1 s every 10 s; or 100 Hz for 1 s every 100 s). Examples of EPPs recorded after these stimulation protocols are shown in Fig. 5A. Stimulation at a frequency of 1 Hz appeared to delay synaptic degeneration but the difference was not quite statistically significant (78.75 ± 11.21% vs. 53.75 ± 10.28%, n = 4, t = 1.76, P > 0.05: ANOVA; Fig 5B). Stimulation at 10 Hz for 1 s every 10 s produced no significant difference in the number of responsive fibers compared with unstimulated controls (52.50 ± 11.17%, vs. 53.75 ± 10.28%, n = 4, P > 0.05: ANOVA). However, stimulation at 100 Hz for 1 s every 100 s accelerated synaptic degeneration. After 32 h, the mean number of responsive fibers was reduced by about one-half, to 32.67 ± 6.86% (n = 5 muscles; P = 0.056, ANOVA; and Dunnett’s post hoc test: P < 0.05 compared with 1 Hz stimulation; F = 2.93, df = 19; Fig. 5B). When fibers with MEPPs only were included in the tally, the difference comparing unstimulated controls with patterned 100-Hz stimulation was highly significant (93.33 ± 3.12%, vs. 68.67 ± 6.96%; t = 2.953; df = 7; P = 0.011; one-tailed t-test). We observed a similar effect of stimulation when muscles were assessed morphologically (Fig. 4C, E). In DL muscles from thy1.2YFP16:WldS mice maintained in normal MPS and scored after 24 h in culture, 54.45 ± 6.50% of NMJ (n = 5 muscles) were occupied by YFP-positive terminals. Muscles stimulated at 100 Hz in normal MPS showed a significant, fivefold reduction in occupancy (9.95 ± 3.33% of endplates, n = 4) compared to the unstimulated group (P < 0.001; ANOVA/Bonferroni).
Fig. 5.

High-frequency patterned stimulation accelerates synaptic degeneration. (A) Representative recordings of EPPs from FDB muscle fibers in (a) unstimulated control; (b) 1 Hz continuously stimulated; (c) 100 Hz stimulated (1s/100s for 32 h); (d) 100 Hz stimulated, same pattern, but with Ca2+ reduced to 1 mM and Mg2+ increased to 3 mM during the period of stimulation. The assays were all performed in normal MPS. (B) Summary data for responsive fibers, assayed 32 h after axotomy in FDB muscles. The difference between 1 Hz and patterned 100-Hz stimulation is significant (P < 0.05; ANOVA/Bonferroni). The differences between the other groups, including the low Ca2+ medium, were not significant. (C) NMJ in DL muscles from thy1.2YFP16:WldS mice cultured at 32 °C for 24 h, with the tibial nerve supply stimulated at 100 Hz (1 s/100 s) in normal MPS and (D) in MPS with reduced Ca2+ and increased Mg2+. Images digitally enhanced for overall brightness and contrast only. (E) Summary of morphological analysis of occupied NMJ in control muscles without stimulation in normal MPS, compared with MPS containing reduced Ca2+ and increased Mg2+, compared with patterned 100 Hz stimulation, in normal versus low Ca2+ MPS. The reduced incidence of occupied endplates in the group receiving 100-Hz stimulation was highly significantly different from the other groups, and this effect was abolished in the reduced Ca2+/increased Mg2+ group (P < 0.01 ANOVA/Bonferroni. Graphs in B and E show mean ± S.E.M., n = 3–9 muscles. Single asterisks, P < 0.05; triple asterisks: P < 0.001).
The activity-dependent sensitization of synapses to degeneration was inhibited by reducing [Ca2+] in the bathing medium. We reduced extracellular [Ca2+] for 32 h to 1 mM and increased [Mg2+] to 3 mM, then measured physiological responses in FDB muscles after returning them to normal MPS. In unstimulated muscles, the number of responsive fibers was not altered by this change in divalent cation ratio: 53.75 ± 10.28% (n = 4 muscles) of fibers responded with EPPs, compared with 52.11 ± 9.11% (n = 9) in normal MPS (P > 0.05, ANOVA, Fig. 5B). Stimulation at 100 Hz for 32 h in the low [Ca2+] medium reduced the subsequent mean incidence of unresponsive fibers compared with that in stimulated muscles incubated in normal MPS but the difference was not quite statistically significant (32.67 ± 6.86% responsive, n = 5 vs. 56.67 ± 12.02%, n = 3, t = 1.67; P > 0.05, t-test, Fig. 5B). However, the morphological data from cultured DL muscles were more compelling (Fig. 5C–E). As in the FDB muscles assayed physiologically, incubation in low-Ca2+ MPS alone, without stimulation, had no effect on degeneration: 56.60 ± 17.85% (n = 5) of the motor endplates were occupied after 24 h ex vivo, which was not significantly different compared to controls in normal MPS. Incubating muscles in the reduced Ca2+/elevated Mg2+ medium significantly protected NMJs from the effects of stimulation, leaving 40 ± 6.63% of endplates occupied (n = 3; P < 0.001; ANOVA/Bonferroni) about four times the number in muscle stimulated at 100 Hz in normal MPS, and not significantly different from the unstimulated controls (Fig. 5E).
These data suggest prolonged and intensive synaptic activity accelerates synaptic degeneration and the mechanism is sensitive to [Ca2+]. Thus, the enhanced protection of sensory axons and terminals compared with motor axons and their terminals cannot be simply explained by differences in their ongoing activity post axotomy (Brown et al., 1994; Ribchester et al., 1995; Gillingwater et al., 2002; Oyebode et al., 2012).
In the following two groups of experiments we turned to a different but related issue: that is, whether conditioning axonal and neuromuscular activity influences the vulnerability of synapses to subsequent axotomy-induced degeneration. To address this, we reverted to intact animals and either blocked or enhanced activity levels experimentally in vivo.
Conditioning TTX block accelerated axotomy-induced synaptic degeneration in vivo
In this second group of experiments we conditioned axons and NMJ in WldS mice with 7 days of inactivity, achieved by continuous local instillation of TTX from a microcapsule secured to the sciatic nerve (Martinov and Nja, 2005) (see Fig. 6A). We then cut the tibial nerve and assayed synaptic function 3–7 days later. Sample records of evoked EPP responses are shown in Fig. 6B and the time course of degeneration is shown in Fig. 6C. Axotomy triggered synaptic degeneration following 7 days of conditioned inactivity at least twice as rapidly as in muscles that were axotomized but not TTX-blocked beforehand. Three days post axotomy, only 38.34 ± 18.13% of prior TTX-blocked NMJs showed evidence of persistent synaptic transmission, compared with 88.75 ± 3.15% in saline vehicle controls, (n = 4 per group). By 7 days, no NMJs in the TTX-blocked group showed evidence of either spontaneous or evoked neurotransmitter release, compared with 34.58 ± 14.74% in the controls (n = 4 per group; Fig. 6C; compare for example with (Gillingwater et al., 2002).
Fig. 6.

TTX block sensitizes synapses to axotomy. (A) TTX-impregnated capillary (arrow) in situ, with its opening secured to the exposed sciatic nerve of a WldS mouse, via silicone rubber cuff. (B) Representative intracellular recordings obtained 5 days after axotomy from WldS mice: (a) no axotomy (contralateral control); (b) saline implant; (c) TTX block only; (d) TTX-block then axotomy. (C) Time course of loss of functional innervation in axotomized, saline vehicle control muscles (open bars), compared with preconditioning TTX block (filled bars); mean ± S.E.M., n = 4–7 muscles per group. (D) Complete data from six groups of mice showing the incidence of responsive fibers, 5 days after axotomy in those indicated as cut. From left to right, the categories were as follows; TTX block + cut: sciatic nerve block for 7 days followed by section of the tibial nerve; No block + cut: tibial nerve section only; Saline + cut: saline vehicle infusion of the sciatic nerve for 7 days, followed by section of the tibial nerve; TTX block no cut: sciatic nerve block for 7 days only; Saline – saline vehicle infusion of the sciatic nerve only; Contralateral – data from muscles on unoperated contralateral sides of the TTX block + cut group. (E) As in C, but complete data including fibers that showed only MEPPs on intracellular recording. TTX block + cut groups are highly significantly different from the other groups (P < 0.01, ANOVA/Bonferroni).
In order to rule out influences on degeneration unrelated to activity in these experiments, we compared the percentage of muscle fibers with spontaneous or evoked responses to nerve stimulation with that in the following control groups: vehicle (saline-only) with or without nerve section; TTX-block with no nerve section; and nerve section-only (Fig. 6D, E). About five times fewer fibers produced evoked responses in TTX-conditioning blocked preparations 5 days post axotomy, compared with saline-treated axotomized controls, or axotomized muscles with no nerve implants. In the TTX-blocked and axotomized group: 8.57 ± 2.51% (n = 7) of NMJs produced evoked EPPs compared to 55.56 ± 5.88% (n = 3) in saline controls and 43.34 ± 10.11% (n = 6) in axotomized muscles with no implant (P < 0.05, ANOVA, Fig. 6D). When fibers showing only MEPPs were combined with those responding with EPPs, the contrast between TTX pre-conditioned and the other groups was even greater (Fig. 6E).
To test whether microcapsules containing TTX themselves triggered any axonal or synaptic degeneration alone during the conditioning period we implanted microcapsules containing either saline or 15 mM TTX, then FDB muscles were isolated and assayed electrophysiologically 7 days later, without intervening tibial nerve section. There was no evidence of significant synaptic degeneration in these muscles (Fig. 6D, E). The percentage of responsive fibers compared to those in contralateral controls, or those treated with saline were, respectively: TTX-blocked: 87.78 ± 7.78% (n = 3 muscles); contralateral controls: 91.88 ± 2.83% (n = 6); and saline controls: 85.00 ± 2.15% (n = 4; P > 0.05, ANOVA).
Morphological analysis of synaptic occupancy corroborated the electrophysiological data. For these experiments we studied axotomized DL muscles in thy1.2YFP16:WldS mice, in which all motor neurons and their axons and terminals were fluorescent (Feng et al., 2000; Wong et al., 2009; Teriakidis et al., 2012).
First, we obtained live images of NMJ in 7 days TTX-blocked/5 days-axotomized muscles of anesthetized thy1.2YFP16:WldS in vivo, using a hand-held confocal microendoscope (CME; (Wong et al., 2009; Brown et al., 2014). In accordance with the physiological data, terminals in control muscles innervated by unsectioned, TTX-blocked nerves were abundant (Fig. 7A), confirming that nerve block alone did not trigger synaptic degeneration. In muscles with nerve section only and no TTX-block, terminals were still readily discernible (Fig. 7C). However, in muscles supplied by sectioned, TTX-blocked nerves, most intramuscular axons were fragmented and motor nerve terminals had degenerated (Fig. 7E). We quantified synaptic degeneration in isolated DL muscle preparations 5 days post axotomy, from images obtained using conventional fluorescence microscopy (Fig. 7B, D, F). In DL muscles from mice with conditioning nerve block and axotomy, significantly fewer motor endplates (TRITC-α-bungarotoxin positive) were either completely or fractionally occupied compared with those following nerve section without preconditioning block. There were about three times as many vacant (denervated) endplates in TTX-blocked muscles: 75.75 ± 3.65% (n = 4 mice) of NMJs, compared with only 23.58 ± 7.58% in axotomized-only WldS muscles (n = 6; P < 0.001, ANOVA, Bonferroni post hoc test). The number of vacant (denervated) endplates in contralateral unoperated muscles was negligible (0.07 ± 0.05%, n = 7 muscles, Fig. 7G).
Fig. 7.

TTX block selectively sensitizes motor nerve terminals. Images obtained in vivo using confocal microendoscopy (CME; panels A, C, E) and conventional fluorescence microscopy (B, D, F) from WldS mice after TTX block alone (A, B), 5 days after axotomy alone (C, D), or 5 days post axotomy with seven preceding days of complete nerve conduction block with TTX (E, F). Triggering degeneration after 7 days of TTX left most intramuscular axons denuded of their nerve terminals. Images digitally adjusted for overall brightness and contrast only. (G) The number of occupied endplates was significantly reduced in lumbrical muscles 5 days post axotomy from mice preconditioned with 7 days TTX block, compared to untreated axotomized (No block + cut), and unoperated controls (mean ± S.E.M., n = 4–7 muscles; P < 0.001, ANOVA/Bonferroni).
TTX-block selectively sensitized motor nerve terminals
Next we asked to what extent motor nerve terminals were selectively affected by axonal conduction block or whether TTX block also sensitized the axons that supplied them. We found that conditioning block did not affect the rate of degeneration of axons. The evidence for this was based on the appearance of axons in tibial nerves of thy1.2YFP16:WldS double-mutant mice visualized using CME (Fig. 8A–F). Five days after axotomy there was no discernible difference in the lengths of axon fragments comparing the level of axonal degeneration in saline control-treated WldS mice (253 ± 20.46 μm; mean ± SEM; N = 4 mice, n = 127 axons) and those preconditioned with TTX instillation (238.5 ± 15.49 μm, N = 4 mice, n = 130 axons respectively, P > 0.05; Fig. 8I). Neither was different from the apparent length of axon “segments” imaged in unoperated animals. By contrast, as expected, the distal tibial nerve of one WT (thy1.2YFP16:C57Bl6) mouse, axotomized 5 days previously, was copiously populated with short axonal fragments (Fig. 8G–I). Together, these CME imaging data suggest that the sensitizing effects of TTX block on synaptic degeneration occur by a mechanism that is confined to motor nerve terminals.
Fig. 8.

TTX block does not affect axonal integrity. In vivo, CME images of tibial nerve axons in thy1.2YFP16:WldS mice (A–F) and thy1.2YFP16:C57Bl6 mice (G, H), up to 7 days post axotomy. (I) there was no difference in axon preservation 5 days post axotomy in TTX-blocked compared with control mice but, as expected, axons in wild-type mice were extensively fragmented. Mean ± S.E.M., N = 4 mice in each group, except thy1.2YFP16:C57Bl6, wild-type for WldS (YFP16Bl6), showing data from a single mouse; (P < 0.001; ANOVA/Bonferroni on the other three groups). Images digitally adjusted for overall brightness and contrast only.
TTX-block enhanced neurotransmitter release
Several previous studies have shown that neuromuscular paralysis feeds signals back onto motor nerve terminals, leading to enhanced neurotransmitter release (Snider and Harris, 1979; Tsujimoto et al., 1990; Plomp et al., 1992; Ribchester, 1993; Plomp et al., 1994; Barry and Ribchester, 1995; Davis, 2013). In addition, axotomy sensitizes motor endplates to acetylcholine in WldS mouse muscle fibers, enhancing postsynaptic responses to spontaneous transmitter release, most likely due to an activity-dependent change in membrane resistance (Barry and Ribchester, 1995; Ribchester et al., 1995). We therefore asked whether TTX-block administered via the capillary implant method we employed here would also affect neurotransmitter release and other physiological properties of synaptic potentials.
We found no significant effect of TTX block on FDB muscle fiber resting membrane potential (TTX-blocked; −67.33 ± 2.38 mV, n = 5; saline control: −71.59 ± 2.38 mV, n = 3; P > 0.05, t-test). FDB muscles from WldS mice without conditioning nerve block showed a mean MEPP amplitude of 1.08 ± 0.12 mV (n = 5; Fig. 9A). Seven days of nerve block caused a significant, approximately 50% increase in MEPP amplitude (1.58 ± 0.12 mV, n = 9). However, this increase was less than that observed 7 days after complete axotomy (2.55 ± 0.47 mV, n = 3 respectively, P < 0.01 ANOVA; Bonferroni post hoc; compared with Ribchester et al., 1995).
Fig. 9.

TTX block enhances synaptic transmission. (A) Inset, representative recordings of MEPPs from an FDB muscle fiber of a WldS mouse, after 7 days preconditioning, unilateral sciatic nerve block. MEPP amplitude was significantly increased but, interestingly, not as substantially as in remaining innervated fibers 7 days after axotomy, rather than nerve block (P < 0.01; ANOVA/Bonferroni comparing all three groups). Neither MEPP frequency (B) nor EPP rise time (C) was significantly affected by nerve block. However, EPP half-decay time (D), EPP amplitude (E), and quantal content (F) were all significantly increased (P < 0.05; t-tests in each case). Graphs show mean ± S.E.M., n = 3–9 muscles in each group. Single asterisks, P < 0.05; double asterisks: P < 0.01).
By contrast, there was no statistically significant change in MEPP frequency after nerve block (23.68 ± 4.47 MEPPs min−1, n = 6, compared with controls: 30.18 ± 4.97 MEPPs min−1, n = 4, P > 0.05, t-test; Fig. 9B). These data were perhaps surprising given the reported increase in spontaneous MEPP frequency within 1–3 days of chronic paralysis in the mouse extensor digitorum longus (EDL) muscle (Tsujimoto et al., 1990), although the magnitude of the difference in that previous study was quite small and the variability quite large, even after 6 days of TTX block.
TTX block for 7 days also had no significant effect on EPP latency (2.57 ± 0.06 ms n = 7 muscles, versus 2.43 ± 0.12 ms, n = 5, in controls) or rise time (1.47 ± 0.09 ms, n = 5, versus 1.22 ± 0.09 ms, n = 3; Fig. 9C). Half-decay time was significantly increased, however (4.47 ± 0.36 ms, n = 5, versus 2.96 ± 0.23 ms, n = 3: P < 0.05, t-test; Fig. 9D). There was also a significant increase in peak amplitude of EPPs (26.10 ± 1.25 mV, n = 5, vs. 16.51 ± 0.14 mV, n = 3: P < 0.01, Fig. 9E) and a significant increase in QC, calculated using the variance method (40.67 ± 5.32 quanta, n = 6), compared with controls (25.42 ± 3.26, n = 7, P < 0.05, t-test, Fig. 9F).
These observations support previous studies that demonstrated an effect of chronic nerve conduction block on spontaneous and evoked neurotransmitter release at NMJ under other conditions (Snider and Harris, 1979; Tsujimoto et al., 1990; Barry and Ribchester, 1995). In the context of the present study the data further indicate the sensitizing effect of paralysis on properties of motor nerve terminals.
TTX-block primed synaptic degeneration in WT mice
Finally, we asked whether paralysis would also affect Wallerian-like degeneration in motor nerve terminals in WT mice; or whether it was a unique effect in mice expressing the chimeric mutant WldS gene. Synaptic degeneration is one of the earliest signs of WD and is normally complete by 18 h post axotomy in mice (Slater, 1966; Winlow and Usherwood, 1975; Gillingwater et al., 2002; Wong et al., 2009), whereas the earliest signs of axon fragmentation in the distal stump of an axotomized peripheral nerve are normally discernible only after about 36 h (Beirowski et al., 2004; Beirowski et al., 2005; Wong et al., 2009). In light of this aggressive synaptic pathology, there was only a short time window for obtaining definitive data. Thus, we assayed the amount of synaptic degeneration in FDB muscles of WT (C57Bl6) mice, exactly12 h after cutting the tibial nerve.
Seven days of preconditioning TTX-block in WT mice significantly increased the amount of synaptic degeneration observed 12 h after nerve section. In control (no block) FDB muscles, 61.67 ± 17.56% of fibers were responsive, compared to 6.25 ± 3.75% in those primed with a conditioning TTX block (P = 0.022, t-test; n = 4 in each group, Fig. 10). Thus, a mechanism by which disuse sensitizes motor nerve terminals to axotomy in WldS mice seems likely also to be affected in non-mutant mice.
Fig. 10.
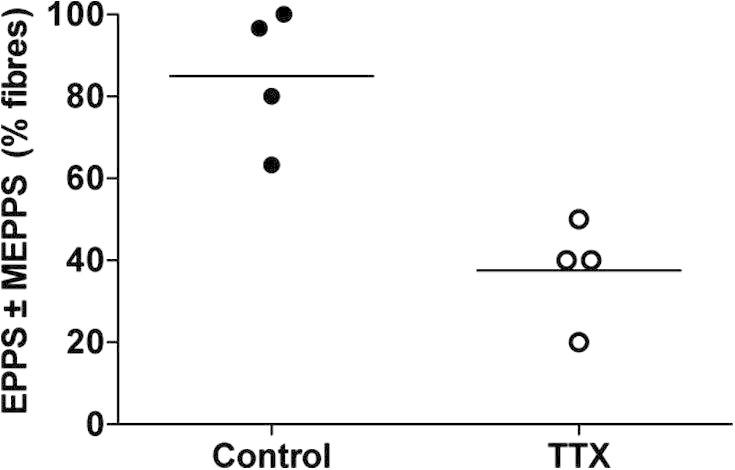
Effect of TTX block on loss of innervation, 12 h after sectioning the sciatic nerve in wild-type mice. Those receiving a preconditioned 7 days of sciatic nerve block showed significantly fewer innervated or responsive NMJs than unblocked controls (P < 0.01; t-test).
Synaptic degeneration was enhanced in mice given prolonged access to running wheels
In the third and final group of experiments, we asked whether facilitating endogenous activity might influence subsequent axotomy-induced synaptic degeneration. To test this, we housed WldS mice individually in cages fitted with running wheels for either 2 weeks or 4 weeks, before cutting the sciatic nerve unilaterally. Innervation of the FDB muscles was assessed 5 days later. WldS and WT C57Bl6 mice showed similar circadian patterns of activity (Fig. 11A, B). During the conditioning period, mice ran 6225 ± 1759 m/day, based on counts of wheel revolutions, but the range of activity varied more than 10-fold: from less than 1.5 km/day to more than 15 km/day. Most of this activity occurred during the controlled 12-h period of darkness.
Fig. 11.

Prolonged self-motivated exercise sensitizes NMJ to axotomy-induced degeneration. (A) Representative circadian activity pattern in a WldS mouse. Each horizontal trace is a record of activity over a 24 h period, divided into 12 h of light and 12 h of darkness. Each vertical line is proportional to the number of wheel revolutions. (B) Integrated activity of WldS mice (red) and C57Bl6 mice (blue) (n = 6 mice in each case) shows no discernible difference in the pattern or amount of circadian activity in the two strains of mice. As expected, most activity was in the dark. (C) Box-whisker plots of incidence (median and interquartile range; outliers >90 percentile shown as dots) of unresponsive FDB muscle fibers (neither MEPPs, nor evoked EPPs observable on tibial nerve stimulation in isolated preparations) in four groups of mice: controls with no nerve section; WldS controls with no running wheels, 5 day post-axotomy; WldS mice with provision of running wheels for 2 weeks, then 5 days post axotomy; WldS mice with provision of running wheels for 4 weeks, then 5 days post axotomy. n = 7–11 muscles per group. (D) Plot of incidence of Responsive fibers (evoked EPPs) versus recorded mean diurnal running wheel activity, averaged over 4 weeks and assayed 5 days post axotomy. While there was no clear evidence of regression or correlation, mice running the least distance had stronger levels of synaptic protection than those running the most. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
First, we tested for the possibility that voluntary activity might enhance, rather than reduce synaptic protection. Protection of axons and synapses by WldS is strongly “gene-dose” dependent and heterozygotes show strong axon protection but weak or absent synaptic protection (Mack et al., 2001; Wong et al., 2009). Thus, WldS heterozygotes constitute a sensitized genetic background for detecting additive protective effects on synaptic degeneration. However, we found that after one-month of conditioning access to running wheels, synaptic transmission was lost from 100% of muscle fibers within 3 days of axotomy in muscles of WldS/+ mice, the same as in mice without wheel access (N = 18 mice, n > 7 muscles in each group; data not shown).
Next we asked whether voluntary activity might enhance degeneration. For these experiments we utilized WldS homozygotes (Fig. 1C). As expected, there were very few unresponsive fibers in the uncut control group (6.78 ± 2.32%, n = 8, P < 0.05, ANOVA/Bonferroni; Fig. 11C). After 2 weeks of activity, the mean number of unresponsive fibers, 5 days after axotomy was not significantly different from that in age-matched homozygous WldS mice housed in cages without wheels (40.33 ± 8.81%, n = 10 muscles vs. 35.15 ± 5.46%, n = 11; P > 0.05, ANOVA). However, in mice with 4 weeks of preconditioning activity, the number of unresponsive fibers 5 days post axotomy was almost twice as great as in the control group (75.14 ± 6.79%, n = 7, P < 0.01, ANOVA/Bonferroni). There was no overt correlation or regression between distance run and the number of responsive fibers after axotomy (Pearson r −0.2313, P > 0.05; Fig. 11D). However, it is perhaps noteworthy that the mice that ran the least average distance per night showed the highest levels of synaptic protection after axotomy.
In sum, the results of all three groups of experiments showed that either: (a) intensive stimulation ex vivo; (b) complete nerve conduction block in vivo; or (c) voluntary wheel running over an extended period, all reduced synaptic protection mediated by WldS gene expression. Normal levels of endogenous activity may therefore be an obligatory moderating influence on synaptic maintenance and degeneration in this paradigm.
Discussion
We have presented here new and direct evidence that the rate of neuromuscular synaptic degeneration in response to axotomy is sensitive to activity. The level of axonal or synaptic activity, either before or after axotomy, exerted clear influences on the persistence of axotomized synapses both in WT mice (see Fig. 10) and in the slow degeneration (WldS) strain in which axons and synapses are normally protected by expression of a chimeric protein containing the NAD synthetic enzyme Nmnat-1 (Coleman and Freeman, 2010; Gilley and Coleman, 2010; Conforti et al., 2014). Thus, we may infer from the present study that moderate levels of activity serve an important function in neuromuscular synaptic maintenance; but extremes – either complete disuse or sustained high-frequency activation – render motor nerve terminals more vulnerable to potent triggers of degeneration, like axotomy. In light of emerging similarities at a molecular level between synaptic degeneration induced by axotomy or in disease (Gillingwater and Ribchester, 2003; Conforti et al., 2014), our findings are potentially of interest in a disease context as well, since the data are consistent with the notion that activity contributes to synaptic maintenance in general and, conversely, synaptic vulnerability to potent neurodegenerative triggers or risk factors.
Our first group of experiments was motivated in part by our previous study, in which we observed that axotomized sensory terminals innervating muscle spindles, which continued to respond to natural stimulation, were more robustly protected from degeneration in WldS mice than motor nerve terminals (Oyebode et al., 2012). We hypothesized that differences in ongoing, endogenous activity in the axotomized sensory and motor axons contributed to the difference in protection. However, the results of stimulation of motor axons in isolated preparations did not wholly support this hypothesis. There was a liminal protective effect of low-frequency stimulation in these experiments but more striking was the fourfold enhancement of degeneration when axons and synapses were activated using patterned high-frequency (100 Hz) stimulation. Reducing the Ca2+/Mg2+ ratio counteracted the effects of stimulation, consistent with a mechanism that depends on activity-dependent increases in cytoplasmic Ca2+. We discuss this further in consideration of the possible mechanisms of synaptic vulnerability to neurodegenerative triggers below.
Our second and third groups of experiments were designed to explore in more detail the relationship between endogenous activity and synaptic protection. The data show that prior levels of axonal or synaptic activity also influence the subsequent rate of synaptic degeneration initiated by a potent trigger: axotomy. The most compelling data were those showing that a sustained, conditioning period of nerve conduction block, either in WldS mice or WT mice led to accelerated synaptic degeneration after nerve section, reducing residual innervation to about one-fifth the level in saline vehicle controls. These data are consistent with the popular aphorism “use it, or lose it”, often quoted in the context of activity-dependent mitigation of cognitive or motor decline associated with either natural aging or triggered by risk factors for neurodegenerative disease (Swaab et al., 2002; Kirkinezos et al., 2003; Veldink et al., 2003; Frick and Benoit, 2010; Gordon et al., 2010; Power et al., 2010). Conversely, and unexpectedly, we found that prolonged (4 weeks) volitional activity (wheel running) also sensitized synapses to degeneration induced by axotomy, consistent with some reports, but not others, that hyperactivity sensitizes vulnerable motor neurons and their connections to degeneration (Carrasco et al., 2004; Liebetanz et al., 2004; Mahoney et al., 2004; Lepore et al., 2010; Carrasco et al., 2012; Tran et al., 2014). Interestingly, chronic nerve conduction block appears not to accelerate progression of disease signs or loss of muscle innervation in the SOD1 mouse model of ALS (Carrasco et al., 2012). The disadvantage of this model is the highly variable time course both for onset of signs, progression and survival (Benatar, 2007; Scott et al., 2008), whereas the response of axons or NMJs to axotomy in either WT or WldS mice is synchronously initiated in motor units by nerve injury and progresses over a time course that is highly consistent between animals with low variance from onset to conclusion. Nevertheless, further studies are required to establish whether the selective autotomy of motor nerve terminals that evidently occurs at early stages in the progression of some familiar or sporadic forms of ALS is a consequence of focal disease or a trigger for Wallerian-like degeneration of motor nerve terminals, or whether there are molecular mechanisms in common (Frey et al., 2000; Fischer et al., 2004; Schaefer et al., 2005; Conforti et al., 2014).
Previous studies have demonstrated that the axonal- and synaptic-protection phenotype of WldS mice is modifiable in several ways. In addition to the sensory/motor differences that prompted the present study (Oyebode et al., 2012), other modifiers of the WldS phenotype include the WldS gene copy-number (“gene-dose”), age, localization of the protective protein to organelles in the axoplasm or synaptic terminals, and interaction with other proteins of known and unknown identity (Mack et al., 2001; Gillingwater et al., 2002; Beirowski et al., 2009; Conforti et al., 2009; Wong et al., 2009; Babetto et al., 2010; Osterloh et al., 2012; Massoll et al., 2013).
The mechanism of activity-dependent sensitization of synaptic degeneration remains a matter of conjecture. There are several differences in the proteomic fingerprints of synaptic terminals in WldS mice compared with WT mice, any of which could be of crucial importance in the mechanism of protection or its modulation, either upstream or downstream of the Nmnat isoforms that insure axonal or synaptic integrity (Wishart et al., 2007). However, several lines of evidence suggest that association of WldS protein with specific intracellular organelles is crucial for its protective functions. Mitochondria and endoplasmic reticulum have been implicated, on the basis of experiments that alter targeting of the protein and mimicry of its effects (Beirowski et al., 2009; Conforti et al., 2009; Babetto et al., 2010; Beirowski et al., 2010; Barrientos et al., 2011; Avery et al., 2012; Gillingwater and Wishart, 2013; Milde et al., 2013; Villegas et al., 2014). Recent data suggest a strong connection between the enzymic activity of WldS protein and sterile alpha and TIR motif-containing protein-1 (Sarm1), and their association with mitochondria may prove to crucial to the mechanism of synaptic maintenance (Osterloh et al., 2012; Massoll et al., 2013; Summers et al., 2014). It has also been shown recently that accumulation of nicotinamide mononucleotide (NMN), the substrate for Nmnat isoforms (and WldS enzymic activity), may be a toxic agent that precipitates axonal fragmentation at the onset of WD, following a steep decline in axonal Nmnat-2 levels (Gilley and Coleman, 2010; Gilley et al., 2013; Milde et al., 2013; Conforti et al., 2014; Di Stefano et al., 2014). It is perhaps of interest to note in this context that aging mitochondria appear to accumulate in motor nerve terminals, because retrograde transport of mitochondria out of motor terminals is slower than their anterograde transport (Misgeld et al., 2007); and that regeneration of motor nerve terminals, or forced localization of WldS protein to the cytoplasm in aged WldS mice, evidently counteracts the age-dependent weakening of synaptic protection in these mice (Gillingwater et al., 2002; Beirowski et al., 2009). It may be of interest and importance to establish whether activity influences the levels or activity of Nmnat, Nampt, or Sarm-1 in axons and synapses, both in WT and WldS mice, as potential explanations for the sensitizing effects of activity shown in the present study.
The disparity between the rate of degeneration of synaptic terminals that we observed in ex-vivo nerve-muscle preparations from WldS mice compared with the rate routinely observed in vivo also begs a number of questions about mechanism (Tsao et al., 1994; Ribchester et al., 1995; Tsao et al., 1999; Gillingwater et al., 2002; Parson et al., 2004; Wishart et al., 2007; Bridge et al., 2009). All the same cell types that could contribute to synaptic degeneration are present in the ex-vivo dissections (Court et al., 2008). So why do the NMJs not persist or continue to function in oxygenated bathing medium, even at a lower temperature (32 °C) than normal mammalian core temperature, for as long as they do when naturally perfused by oxygenated blood in vivo? Perhaps the trauma of isolation itself constitutes a metabolic challenge to motor nerve terminals that WldS protein expression in native WldS mutant mice is insufficient to resist. The energetics of maintaining a functional synapse evidently place considerable demands on the metabolism of motor nerve terminals (Zhu et al., 2012; Schwarz, 2013). Synaptic mitochondria are particularly sensitive to transient hypoxia, ischemia and Ca2+ overload (Brown et al., 2006; David et al., 2007; Misgeld et al., 2007; Nguyen et al., 2009; Nguyen et al., 2011; Nguyen et al., 2012; Talbot et al., 2012). This transient ischemic/hypoxic response is also sensitive to Ca2+ (Barrett et al., 2014). It would be interesting to determine to what extent the effects of activity or isolation and ex-vivo culture converge on a common mechanism, perhaps involving mitochondria, ROS and succinate levels (Chouchani et al., 2013; Chouchani et al., 2014). Intraterminal Ca2+ measurements and their sensitivity to mitochondrial inhibitors, ROS and succinate could be fruitful avenues to explore in seeking resolution of these phenotypic characteristics and their modifiers.
Finally, it is of interest to compare the effects of activity on synaptic degeneration induced by axotomy, shown here, with neuromuscular synapse elimination: the natural process of synaptic remodeling that occurs during normal postnatal development in rodents, or in adults after nerve regeneration (Gillingwater and Ribchester, 2001, 2003). Paralysis retards, and high-frequency stimulation accelerates synapse elimination; and this effect is partly dependent on Ca-activation of proteases (O’Brien et al., 1978; Thompson et al., 1979; Taxt, 1983; Thompson, 1983; Connold et al., 1986). Complete paralysis thus prolongs the period when muscle fibers are polyneuronally innervated, sometimes even after activity has resumed (Brown et al., 1982; Barry and Ribchester, 1995; Costanzo et al., 1999, 2000). Paralysis also promotes reactive growth, rather than degeneration, of nerve terminal sprouts (Brown and Ironton, 1977; Betz et al., 1980b; Tsujimoto et al., 1990; Costanzo et al., 2000). Both characteristics, synapse elimination and terminal sprouting, therefore are modulated by activity in a fashion that appears to be opposite to its effects on axotomy-induced degeneration that we have described in the present study. However, this interpretation is complicated by findings that differences in activity brought about by selective nerve conduction block, or by differential stimulation, promotes withdrawal of the disused or less active axons (Ribchester and Taxt, 1983, 1984; Ridge and Betz, 1984; Favero et al., 2010; Favero et al., 2012). Thus, while the triggers for synaptic degeneration after injury, in disease, or during development may differ, the present findings do not rule out that activity-dependent mechanisms that remove synaptic terminals from NMJs, by degeneration or controlled withdrawal, may ultimately converge (Raff et al., 2002; Gillingwater and Ribchester, 2003; Conforti et al., 2014).
Conclusion
Hitherto, use or disuse was thought to influence synaptic maintenance and mitigate synaptic degeneration but the evidence for this had been indirect. Here we have newly shown, by applying direct methods, that patterned stimulation, chronic disuse, or natural usage of NMJs in adult WldS mice strongly influences their vulnerability to axotomy: a potent trigger of synaptic degeneration. We found that moderate levels of activity either before or after disrupting the functional integrity of axons optimized synaptic maintenance and resistance to degeneration, while complete disuse or intensive stimulation increases the rate of synaptic degeneration. The ex-vivo preparation that we utilized in the first part of this report enables control over both the activity of NMJs and the composition of the cellular environment. This could prove crucial for further analysis of the mechanisms of activity dependence, or other influences, on synaptic degeneration.
Author contributions
RRR, RB, AH-A: Designed the studies; performed experiments and analyzed the data shown in Figs. 1, 4 and 6–11; co-wrote the manuscript.
AS: Conducted experiments and analyzed the data shown in Fig. 5.
KD: Carried out experiments and analyzed some of the data included in Fig. 4.
THG: Performed surgery and obtained and analyzed data included in Figs. 2 and 3.
Acknowledgments
This research was supported by grants from the following UK sources: Medical Research Council (G0401091), Motor Neurone Disease Association; MND Scotland; the Euan MacDonald Centre for MND Research; and the RS MacDonald Trust. We are grateful to Mr Derek Thomson for expert technical assistance; and Dr John Sheward and the late Professor Antony J. Harmar for assistance in measuring levels of behavioural activity in mice.
References
- Adalbert R., Morreale G., Paizs M., Conforti L., Walker S.A., Roderick H.L., Bootman M.D., Siklos L., Coleman M.P. Intra-axonal calcium changes after axotomy in wild-type and slow Wallerian degeneration axons. Neuroscience. 2012;225:44–54. doi: 10.1016/j.neuroscience.2012.08.056. [DOI] [PubMed] [Google Scholar]
- Al-Chalabi A., Leigh P.N. Trouble on the pitch: are professional football players at increased risk of developing amyotrophic lateral sclerosis? Brain. 2005;128:451–453. doi: 10.1093/brain/awh426. [DOI] [PubMed] [Google Scholar]
- Alvarez S., Calin A., Graffmo K.S., Moldovan M., Krarup C. Peripheral motor axons of SOD1(G127X) mutant mice are susceptible to activity-dependent degeneration. Neuroscience. 2013;241:239–249. doi: 10.1016/j.neuroscience.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Avery M.A., Rooney T.M., Pandya J.D., Wishart T.M., Gillingwater T.H., Geddes J.W., Sullivan P.G., Freeman M.R. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babetto E., Beirowski B., Janeckova L., Brown R., Gilley J., Thomson D., Ribchester R.R., Coleman M.P. Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci. 2010;30:13291–13304. doi: 10.1523/JNEUROSCI.1189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett E.F., Barrett J.N., David G. Dysfunctional mitochondrial Ca(2+) handling in mutant SOD1 mouse models of fALS: integration of findings from motor neuron somata and motor terminals. Front Cell Neurosci. 2014;8:184. doi: 10.3389/fncel.2014.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos S.A., Martinez N.W., Yoo S., Jara J.S., Zamorano S., Hetz C., Twiss J.L., Alvarez J., Court F.A. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry J.A., Ribchester R.R. Persistent polyneuronal innervation in partially denervated rat muscle after reinnervation and recovery from prolonged nerve conduction block. J Neurosci. 1995;15:6327–6339. doi: 10.1523/JNEUROSCI.15-10-06327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Adalbert R., Wagner D., Grumme D.S., Addicks K., Ribchester R.R., Coleman M.P. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;6:6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Babetto E., Gilley J., Mazzola F., Conforti L., Janeckova L., Magni G., Ribchester R.R., Coleman M.P. Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J Neurosci. 2009;29:653–668. doi: 10.1523/JNEUROSCI.3814-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Berek L., Adalbert R., Wagner D., Grumme D.S., Addicks K., Ribchester R.R., Coleman M.P. Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP-H mice. J Neurosci Methods. 2004;134:23–35. doi: 10.1016/j.jneumeth.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Beirowski B., Morreale G., Conforti L., Mazzola F., Di Stefano M., Wilbrey A., Babetto E., Janeckova L., Magni G., Coleman M.P. WldS can delay Wallerian degeneration in mice when interaction with valosin-containing protein is weakened. Neuroscience. 2010;166:201–211. doi: 10.1016/j.neuroscience.2009.12.024. [DOI] [PubMed] [Google Scholar]
- Bekoff A., Betz W. Properties of isolated adult rat muscle fibres maintained in tissue culture. J Physiol. 1977;271:537–547. doi: 10.1113/jphysiol.1977.sp012013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekoff A., Betz W.J. Physiological properties of dissociated muscle fibres obtained from innervated and denervated adult rat muscle. J Physiol. 1977;271:25–40. doi: 10.1113/jphysiol.1977.sp011988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell K.F., Hardingham G.E. The influence of synaptic activity on neuronal health. Curr Opin Neurobiol. 2011;21:299–305. doi: 10.1016/j.conb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26:1–13. doi: 10.1016/j.nbd.2006.12.015. [DOI] [PubMed] [Google Scholar]
- Betz W.J., Caldwell J.H., Ribchester R.R. The effects of partial denervation at birth on the development of muscle fibres and motor units in rat lumbrical muscle. J Physiol. 1980;303:265–279. doi: 10.1113/jphysiol.1980.sp013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz W.J., Caldwell J.H., Ribchester R.R. Sprouting of active nerve terminals in partially inactive muscles of the rat. J Physiol. 1980;303:281–297. doi: 10.1113/jphysiol.1980.sp013285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boegman R.J., Deshpande S.S., Albuquerque E.X. Consequences of axonal transport blockade induced by batrachotoxin on mammalian neuromuscular junction I. Early pre- and postsynaptic changes. Brain Res. 1980;187:183–196. doi: 10.1016/0006-8993(80)90503-x. [DOI] [PubMed] [Google Scholar]
- Boillee S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bridge K.E., Berg N., Adalbert R., Babetto E., Dias T., Spillantini M.G., Ribchester R.R., Coleman M.P. Late onset distal axonal swelling in YFP-H transgenic mice. Neurobiol Aging. 2009;30:309–321. doi: 10.1016/j.neurobiolaging.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Brown M.C., Hopkins W.G., Keynes R.J. Short- and long-term effects of paralysis on the motor innervation of two different neonatal mouse muscles. J Physiol. 1982;329:439–450. doi: 10.1113/jphysiol.1982.sp014312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.C., Ironton R. Suppression of motor nerve terminal sprouting in partially denervated mouse muscles [proceedings] J Physiol. 1977;272:70P–71P. [PubMed] [Google Scholar]
- Brown M.C., Jansen J.K., Van Essen D. Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J Physiol. 1976;261:387–422. doi: 10.1113/jphysiol.1976.sp011565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.C., Perry V.H., Hunt S.P., Lapper S.R. Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration. Eur J Neurosci. 1994;6:420–428. doi: 10.1111/j.1460-9568.1994.tb00285.x. [DOI] [PubMed] [Google Scholar]
- Brown M.R., Sullivan P.G., Geddes J.W. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- Brown R., Dissanayake K.N., Skehel P.A., Ribchester R.R. Endomicroscopy and electromyography of neuromuscular junctions in situ. Ann Clin Transl Neurol. 2014;1:867–883. doi: 10.1002/acn3.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn L.I., Miller T.M., Cleveland D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Caroni P., Chowdhury A., Lahr M. Synapse rearrangements upon learning: from divergent-sparse connectivity to dedicated sub-circuits. Trends Neurosci. 2014;37:604–614. doi: 10.1016/j.tins.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Carrasco D.I., Bichler E.K., Rich M.M., Wang X., Seburn K.L., Pinter M.J. Motor terminal degeneration unaffected by activity changes in SOD1(G93A) mice; a possible role for glycolysis. Neurobiol Dis. 2012;48:132–140. doi: 10.1016/j.nbd.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco D.I., Rich M.M., Wang Q., Cope T.C., Pinter M.J. Activity-driven synaptic and axonal degeneration in canine motor neuron disease. J Neurophysiol. 2004;92:1175–1181. doi: 10.1152/jn.00157.2004. [DOI] [PubMed] [Google Scholar]
- Carri M.T., Ferri A., Cozzolino M., Calabrese L., Rotilio G. Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull. 2003;61:365–374. doi: 10.1016/s0361-9230(03)00179-5. [DOI] [PubMed] [Google Scholar]
- Chio A., Benzi G., Dossena M., Mutani R., Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005;128:472–476. doi: 10.1093/brain/awh373. [DOI] [PubMed] [Google Scholar]
- Chouchani E.T., Methner C., Nadtochiy S.M., Logan A., Pell V.R., Ding S., James A.M., Cocheme H.M., Reinhold J., Lilley K.S., Partridge L., Fearnley I.M., Robinson A.J., Hartley R.C., Smith R.A., Krieg T., Brookes P.S., Murphy M.P. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N., Smith A.C., Eyassu F., Shirley R., Hu C.H., Dare A.J., James A.M., Rogatti S., Hartley R.C., Eaton S., Costa A.S., Brookes P.S., Davidson S.M., Duchen M.R., Saeb-Parsy K., Shattock M.J., Robinson A.J., Work L.M., Frezza C., Krieg T., Murphy M.P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014 doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M.P., Freeman M.R. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comley L.H., Wishart T.M., Baxter B., Murray L.M., Nimmo A., Thomson D., Parson S.H., Gillingwater T.H. Induction of cell stress in neurons from transgenic mice expressing yellow fluorescent protein: implications for neurodegeneration research. PLoS One. 2011;6:e17639. doi: 10.1371/journal.pone.0017639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L., Gilley J., Coleman M.P. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci. 2014;15:394–409. doi: 10.1038/nrn3680. [DOI] [PubMed] [Google Scholar]
- Conforti L., Wilbrey A., Morreale G., Janeckova L., Beirowski B., Adalbert R., Mazzola F., Di Stefano M., Hartley R., Babetto E., Smith T., Gilley J., Billington R.A., Genazzani A.A., Ribchester R.R., Magni G., Coleman M. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connold A.L., Evers J.V., Vrbova G. Effect of low calcium and protease inhibitors on synapse elimination during postnatal development in the rat soleus muscle. Brain Res. 1986;393:99–107. doi: 10.1016/0165-3806(86)90069-6. [DOI] [PubMed] [Google Scholar]
- Costanzo E.M., Barry J.A., Ribchester R.R. Co-regulation of synaptic efficacy at stable polyneuronally innervated neuromuscular junctions in reinnervated rat muscle. J Physiol. 1999;521(Pt 2):365–374. doi: 10.1111/j.1469-7793.1999.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo E.M., Barry J.A., Ribchester R.R. Competition at silent synapses in reinnervated skeletal muscle. Nat Neurosci. 2000;3:694–700. doi: 10.1038/76649. [DOI] [PubMed] [Google Scholar]
- Court F.A., Gillingwater T.H., Melrose S., Sherman D.L., Greenshields K.N., Morton A.J., Harris J.B., Willison H.J., Ribchester R.R. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J Cell Sci. 2008;121:3901–3911. doi: 10.1242/jcs.031047. [DOI] [PubMed] [Google Scholar]
- David G., Nguyen K., Barrett E.F. Early vulnerability to ischemia/reperfusion injury in motor terminals innervating fast muscles of SOD1-G93A mice. Exp Neurol. 2007;204:411–420. doi: 10.1016/j.expneurol.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis G.W. Homeostatic signaling and the stabilization of neural function. Neuron. 2013;80:718–728. doi: 10.1016/j.neuron.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida J.P., Silvestre R., Pinto A.C., de Carvalho M. Exercise and amyotrophic lateral sclerosis. Neurol Sci. 2012;33:9–15. doi: 10.1007/s10072-011-0921-9. [DOI] [PubMed] [Google Scholar]
- Deforges S., Branchu J., Biondi O., Grondard C., Pariset C., Lecolle S., Lopes P., Vidal P.P., Chanoine C., Charbonnier F. Motoneuron survival is promoted by specific exercise in a mouse model of amyotrophic lateral sclerosis. J Physiol. 2009;587:3561–3572. doi: 10.1113/jphysiol.2009.169748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande S.S., Boegman R.J., Albuquerque E.X. Consequences of axonal transport blockade by batrachotoxin on mammalian neuromuscular junction. II. Late pre- and postsynaptic changes. Brain Res. 1981;225:115–129. doi: 10.1016/0006-8993(81)90322-x. [DOI] [PubMed] [Google Scholar]
- Di Stefano M., Nascimento-Ferreira I., Orsomando G., Mori V., Gilley J., Brown R., Janeckova L., Vargas M.E., Worrell L.A., Loreto A., Tickle J., Patrick J., Webster J.R., Marangoni M., Carpi F.M., Pucciarelli S., Rossi F., Meng W., Sagasti A., Ribchester R.R., Magni G., Coleman M.P., Conforti L. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlochter M., Irintchev A., Brinkers M., Wernig A. Effects of enhanced activity on synaptic transmission in mouse extensor digitorum longus muscle. J Physiol. 1991;436:283–292. doi: 10.1113/jphysiol.1991.sp018550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahim M.A. Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice. J Appl Physiol (1985) 1997;83:59–66. doi: 10.1152/jappl.1997.83.1.59. [DOI] [PubMed] [Google Scholar]
- Favero M., Buffelli M., Cangiano A., Busetto G. The timing of impulse activity shapes the process of synaptic competition at the neuromuscular junction. Neuroscience. 2010;167:343–353. doi: 10.1016/j.neuroscience.2010.01.055. [DOI] [PubMed] [Google Scholar]
- Favero M., Busetto G., Cangiano A. Spike timing plays a key role in synapse elimination at the neuromuscular junction. Proc Natl Acad Sci U S A. 2012;109:E1667–E1675. doi: 10.1073/pnas.1201147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fischer L.R., Culver D.G., Tennant P., Davis A.A., Wang M., Castellano-Sanchez A., Khan J., Polak M.A., Glass J.D. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D., Schneider C., Xu L., Borg J., Spooren W., Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick K.M., Benoit J.D. Use it or lose it: environmental enrichment as a means to promote successful cognitive aging. ScientificWorldJournal. 2010;10:1129–1141. doi: 10.1100/tsw.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Adalbert R., Yu G., Coleman M.P. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci. 2013;33:13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Coleman M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater T.H., Haley J.E., Ribchester R.R., Horsburgh K. Neuroprotection after transient global cerebral ischemia in Wld(s) mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- Gillingwater T.H., Ingham C.A., Parry K.E., Wright A.K., Haley J.E., Wishart T.M., Arbuthnott G.W., Ribchester R.R. Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain. 2006;129:1546–1556. doi: 10.1093/brain/awl101. [DOI] [PubMed] [Google Scholar]
- Gillingwater T.H., Ribchester R.R. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater T.H., Ribchester R.R. The relationship of neuromuscular synapse elimination to synaptic degeneration and pathology: insights from WldS and other mutant mice. J Neurocytol. 2003;32:863–881. doi: 10.1023/B:NEUR.0000020629.51673.f5. [DOI] [PubMed] [Google Scholar]
- Gillingwater T.H., Thomson D., Mack T.G., Soffin E.M., Mattison R.J., Coleman M.P., Ribchester R.R. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J Physiol. 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater T.H., Wishart T.M. Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol Appl Neurobiol. 2013;39:320–334. doi: 10.1111/nan.12014. [DOI] [PubMed] [Google Scholar]
- Gordon T., Tyreman N., Li S., Putman C.T., Hegedus J. Functional over-load saves motor units in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2010;37:412–422. doi: 10.1016/j.nbd.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Harmar A.J., Marston H.M., Shen S., Spratt C., West K.M., Sheward W.J., Morrison C.F., Dorin J.R., Piggins H.D., Reubi J.C., Kelly J.S., Maywood E.S., Hastings M.H. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- Hirst T.C., Ribchester R.R. Segmentation of the mouse fourth deep lumbrical muscle connectome reveals concentric organisation of motor units. J Physiol. 2013;591:4859–4875. doi: 10.1113/jphysiol.2013.258087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson C.S., Deshpande S.S., Albuquerque E.X. Consequences of axonal transport blockade by batrachotoxin on mammalian neuromuscular junction. III. An ultrastructural study. Brain Res. 1984;296:319–332. doi: 10.1016/0006-8993(84)90068-4. [DOI] [PubMed] [Google Scholar]
- Keller-Peck C.R., Walsh M.K., Gan W.B., Feng G., Sanes J.R., Lichtman J.W. Asynchronous synapse elimination in neonatal motor units: studies using GFP transgenic mice. Neuron. 2001;31:381–394. doi: 10.1016/s0896-6273(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Kirkinezos I.G., Hernandez D., Bradley W.G., Moraes C.T. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann Neurol. 2003;53:804–807. doi: 10.1002/ana.10597. [DOI] [PubMed] [Google Scholar]
- Lavoie P.A., Collier B., Tenenhouse A. Role of skeletal muscle activity in the control of muscle acetylcholine sensitivity. Exp Neurol. 1977;54:148–171. doi: 10.1016/0014-4886(77)90242-4. [DOI] [PubMed] [Google Scholar]
- Lepore A.C., Tolmie C., O’Donnell J., Wright M.C., Dejea C., Rauck B., Hoke A., Ignagni A.R., Onders R.P., Maragakis N.J. Peripheral hyperstimulation alters site of disease onset and course in SOD1 rats. Neurobiol Dis. 2010;39:252–264. doi: 10.1016/j.nbd.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie J.H., Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol. 2011;94:223–237. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Lee Y., Thompson W.J. Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J Neurosci. 2011;31:14910–14919. doi: 10.1523/JNEUROSCI.3590-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebetanz D., Hagemann K., von Lewinski F., Kahler E., Paulus W. Extensive exercise is not harmful in amyotrophic lateral sclerosis. Eur J Neurosci. 2004;20:3115–3120. doi: 10.1111/j.1460-9568.2004.03769.x. [DOI] [PubMed] [Google Scholar]
- Lund L.M., Machado V.M., McQuarrie I.G. Increased beta-actin and tubulin polymerization in regrowing axons: relationship to the conditioning lesion effect. Exp Neurol. 2002;178:306–312. doi: 10.1006/exnr.2002.8034. [DOI] [PubMed] [Google Scholar]
- Lunn E.R., Perry V.H., Brown M.C., Rosen H., Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Mack T.G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L., Fernando F.S., Tarlton A., Andressen C., Addicks K., Magni G., Ribchester R.R., Perry V.H., Coleman M.P. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Mahoney D.J., Rodriguez C., Devries M., Yasuda N., Tarnopolsky M.A. Effects of high-intensity endurance exercise training in the G93A mouse model of amyotrophic lateral sclerosis. Muscle Nerve. 2004;29:656–662. doi: 10.1002/mus.20004. [DOI] [PubMed] [Google Scholar]
- Martinov V.N., Nja A. A microcapsule technique for long-term conduction block of the sciatic nerve by tetrodotoxin. J Neurosci Methods. 2005;141:199–205. doi: 10.1016/j.jneumeth.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Massoll C., Mando W., Chintala S.K. Excitotoxicity upregulates SARM1 protein expression and promotes Wallerian-like degeneration of retinal ganglion cells and their axons. Invest Ophthalmol Vis Sci. 2013;54:2771–2780. doi: 10.1167/iovs.12-10973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan E.M., Martin A.R. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie I.G., Grafstein B., Gershon M.D. Axonal regeneration in the rat sciatic nerve: effect of a conditioning lesion and of dbcAMP. Brain Res. 1977;132:443–453. doi: 10.1016/0006-8993(77)90193-7. [DOI] [PubMed] [Google Scholar]
- Mehta A., Prabhakar M., Kumar P., Deshmukh R., Sharma P.L. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol. 2013;698:6–18. doi: 10.1016/j.ejphar.2012.10.032. [DOI] [PubMed] [Google Scholar]
- Milde S., Gilley J., Coleman M.P. Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 2013;11:e1001539. doi: 10.1371/journal.pbio.1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miledi R., Slater C.R. On the degeneration of rat neuromuscular junctions after nerve section. J Physiol. 1970;207:507–528. doi: 10.1113/jphysiol.1970.sp009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T., Kerschensteiner M., Bareyre F.M., Burgess R.W., Lichtman J.W. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Nguyen K.T., Barrett J.N., Garcia-Chacon L., David G., Barrett E.F. Repetitive nerve stimulation transiently opens the mitochondrial permeability transition pore in motor nerve terminals of symptomatic mutant SOD1 mice. Neurobiol Dis. 2011;42:381–390. doi: 10.1016/j.nbd.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K.T., Garcia-Chacon L.E., Barrett J.N., Barrett E.F., David G. The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc Natl Acad Sci U S A. 2009;106:2007–2011. doi: 10.1073/pnas.0810934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K.T., Zhang Z., Barrett E.F., David G. Morphological and functional changes in innervation of a fast forelimb muscle in SOD1-G85R mice. Neurobiol Dis. 2012;48:399–408. doi: 10.1016/j.nbd.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien R.A., Ostberg A.J., Vrbova G. Observations on the elimination of polyneuronal innervation in developing mammalian skeletal muscle. J Physiol. 1978;282:571–582. doi: 10.1113/jphysiol.1978.sp012482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A., MacDonald J.M., Ziegenfuss J.S., Milde S., Hou Y.J., Nathan C., Ding A., Brown R.H., Jr., Conforti L., Coleman M., Tessier-Lavigne M., Zuchner S., Freeman M.R. DSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyebode O.R., Hartley R., Singhota J., Thomson D., Ribchester R.R. Differential protection of neuromuscular sensory and motor axons and their endings in Wld(S) mutant mice. Neuroscience. 2012;200:142–158. doi: 10.1016/j.neuroscience.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Parson S.H., Ribchester R.R., Davie N., Gandhi N.P., Malik R.Q., Gillingwater T.H., Thomson D. Axotomy-dependent and -independent synapse elimination in organ cultures of Wld(s) mutant mouse skeletal muscle. J Neurosci Res. 2004;76:64–75. doi: 10.1002/jnr.20016. [DOI] [PubMed] [Google Scholar]
- Perry V.H., Brown M.C., Tsao J.W. The effectiveness of the gene which slows the rate of Wallerian degeneration in C57BL/Ola mice declines with age. Eur J Neurosci. 1992;4:1000–1002. doi: 10.1111/j.1460-9568.1992.tb00126.x. [DOI] [PubMed] [Google Scholar]
- Perry V.H., Lunn E.R., Brown M.C., Cahusac S., Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur J Neurosci. 1990;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Personius K.E., Chang Q., Mentis G.Z., O’Donovan M.J., Balice-Gordon R.J. Reduced gap junctional coupling leads to uncorrelated motor neuron firing and precocious neuromuscular synapse elimination. Proc Natl Acad Sci U S A. 2007;104:11808–11813. doi: 10.1073/pnas.0703357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp J.J., van Kempen G.T., Molenaar P.C. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp J.J., van Kempen G.T., Molenaar P.C. The upregulation of acetylcholine release at endplates of alpha-bungarotoxin-treated rats: its dependency on calcium. J Physiol. 1994;478(Pt 1):125–136. doi: 10.1113/jphysiol.1994.sp020236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power G.A., Dalton B.H., Behm D.G., Vandervoort A.A., Doherty T.J., Rice C.L. Motor unit number estimates in masters runners: use it or lose it? Med Sci Sports Exerc. 2010;42:1644–1650. doi: 10.1249/MSS.0b013e3181d6f9e9. [DOI] [PubMed] [Google Scholar]
- Pun S., Santos A.F., Saxena S., Xu L., Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Punga A.R., Ruegg M.A. Signaling and aging at the neuromuscular synapse: lessons learnt from neuromuscular diseases. Curr Opin Pharmacol. 2012;12:340–346. doi: 10.1016/j.coph.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Raff M.C., Whitmore A.V., Finn J.T. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ribchester R.R. Co-existence and elimination of convergent motor nerve terminals in reinnervated and paralysed adult rat skeletal muscle. J Physiol. 1993;466:421–441. [PMC free article] [PubMed] [Google Scholar]
- Ribchester R.R., Taxt T. Motor unit size and synaptic competition in rat lumbrical muscles reinnervated by active and inactive motor axons. J Physiol. 1983;344:89–111. doi: 10.1113/jphysiol.1983.sp014926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribchester R.R., Taxt T. Repression of inactive motor nerve terminals in partially denervated rat muscle after regeneration of active motor axons. J Physiol. 1984;347:497–511. doi: 10.1113/jphysiol.1984.sp015078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribchester R.R., Thomson D., Wood N.I., Hinks T., Gillingwater T.H., Wishart T.M., Court F.A., Morton A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Ribchester R.R., Tsao J.W., Barry J.A., Asgari-Jirhandeh N., Perry V.H., Brown M.C. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS) Eur J Neurosci. 1995;7:1641–1650. doi: 10.1111/j.1460-9568.1995.tb01159.x. [DOI] [PubMed] [Google Scholar]
- Ridge R.M., Betz W.J. The effect of selective, chronic stimulation on motor unit size in developing rat muscle. J Neurosci. 1984;4:2614–2620. doi: 10.1523/JNEUROSCI.04-10-02614.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A.M., Sanes J.R., Lichtman J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- Schwarz T.L. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S., Kranz J.E., Cole J., Lincecum J.M., Thompson K., Kelly N., Bostrom A., Theodoss J., Al-Nakhala B.M., Vieira F.G., Ramasubbu J., Heywood J.A. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- Selkoe D.J. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shen S., Spratt C., Sheward W.J., Kallo I., West K., Morrison C.F., Coen C.W., Marston H.M., Harmar A.J. Overexpression of the human VPAC2 receptor in the suprachiasmatic nucleus alters the circadian phenotype of mice. Proc Natl Acad Sci U S A. 2000;97:11575–11580. doi: 10.1073/pnas.97.21.11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors T.J., Anderson M.L., Curlik D.M., 2nd, Nokia M.S. Use it or lose it: how neurogenesis keeps the brain fit for learning. Behav Brain Res. 2012;227:450–458. doi: 10.1016/j.bbr.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater C.R. Time course of failure of neuromuscular transmission after motor nerve section. Nature. 1966;209:305–306. doi: 10.1038/209305b0. [DOI] [PubMed] [Google Scholar]
- Snider W.D., Harris G.L. A physiological correlate of disuse-induced sprouting at the neuromuscular junction. Nature. 1979;281:69–71. doi: 10.1038/281069a0. [DOI] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012;11:1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers D.W., DiAntonio A., Milbrandt J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci. 2014;34:9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab D.F., Dubelaar E.J., Hofman M.A., Scherder E.J., van Someren E.J., Verwer R.W. Brain aging and Alzheimer’s disease; use it or lose it. Prog Brain Res. 2002;138:343–373. doi: 10.1016/S0079-6123(02)38086-5. [DOI] [PubMed] [Google Scholar]
- Talbot J.D., David G., Barrett E.F., Barrett J.N. Calcium dependence of damage to mouse motor nerve terminals following oxygen/glucose deprivation. Exp Neurol. 2012;234:95–104. doi: 10.1016/j.expneurol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxt T. Local and systemic effects of tetrodotoxin on the formation and elimination of synapses in reinnervated adult rat muscle. J Physiol. 1983;340:175–194. doi: 10.1113/jphysiol.1983.sp014757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teriakidis A., Willshaw D.J., Ribchester R.R. Prevalence and elimination of sibling neurite convergence in motor units supplying neonatal and adult mouse skeletal muscle. J Comp Neurol. 2012;520:3203–3216. doi: 10.1002/cne.23091. [DOI] [PubMed] [Google Scholar]
- Thompson W. Synapse elimination in neonatal rat muscle is sensitive to pattern of muscle use. Nature. 1983;302:614–616. doi: 10.1038/302614a0. [DOI] [PubMed] [Google Scholar]
- Thompson W., Kuffler D.P., Jansen J.K. The effect of prolonged, reversible block of nerve impulses on the elimination of polyneuronal innervation of new-born rat skeletal muscle fibers. Neuroscience. 1979;4:271–281. doi: 10.1016/0306-4522(79)90088-5. [DOI] [PubMed] [Google Scholar]
- Tran L.T., Gentil B.J., Sullivan K.E., Durham H.D. The voltage-gated calcium channel blocker lomerizine is neuroprotective in motor neurons expressing mutant SOD1, but not TDP-43. J Neurochem. 2014;130:455–466. doi: 10.1111/jnc.12738. [DOI] [PubMed] [Google Scholar]
- Tsao J.W., Brown M.C., Carden M.J., McLean W.G., Perry V.H. Loss of the compound action potential: an electrophysiological, biochemical and morphological study of early events in axonal degeneration in the C57BL/Ola mouse. Eur J Neurosci. 1994;6:516–524. doi: 10.1111/j.1460-9568.1994.tb00295.x. [DOI] [PubMed] [Google Scholar]
- Tsao J.W., George E.B., Griffin J.W. Temperature modulation reveals three distinct stages of Wallerian degeneration. J Neurosci. 1999;19:4718–4726. doi: 10.1523/JNEUROSCI.19-12-04718.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T., Umemiya M., Kuno M. Terminal sprouting is not responsible for enhanced transmitter release at disused neuromuscular junctions of the rat. J Neurosci. 1990;10:2059–2065. doi: 10.1523/JNEUROSCI.10-07-02059.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turney S.G., Lichtman J.W. Reversing the outcome of synapse elimination at developing neuromuscular junctions in vivo: evidence for synaptic competition and its mechanism. PLoS Biol. 2012;10:e1001352. doi: 10.1371/journal.pbio.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G., Tapia J.C., Kang H., Clemenson G.D., Jr., Gage F.H., Lichtman J.W., Sanes J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci U S A. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldink J.H., Bar P.R., Joosten E.A., Otten M., Wokke J.H., van den Berg L.H. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord. 2003;13:737–743. doi: 10.1016/s0960-8966(03)00104-4. [DOI] [PubMed] [Google Scholar]
- Villegas R., Martinez N.W., Lillo J., Pihan P., Hernandez D., Twiss J.L., Court F.A. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J Neurosci. 2014;34:7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M.K., Lichtman J.W. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Wang M.S., Fang G., Culver D.G., Davis A.A., Rich M.M., Glass J.D. The WldS protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann Neurol. 2001;50:773–779. doi: 10.1002/ana.10039. [DOI] [PubMed] [Google Scholar]
- Wang M.S., Wu Y., Culver D.G., Glass J.D. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincristine neuropathy. J Neuropathol Exp Neurol. 2000;59:599–606. doi: 10.1093/jnen/59.7.599. [DOI] [PubMed] [Google Scholar]
- Winlow W., Usherwood P.N. Ultrastructural studies of normal and degenerating mouse neuromuscular junctions. J Neurocytol. 1975;4:377–394. doi: 10.1007/BF01261371. [DOI] [PubMed] [Google Scholar]
- Winlow W., Usherwood P.N. Electrophysiological studies of normal and degenerating mouse neuromuscular junctions. Brain Res. 1976;110:447–461. doi: 10.1016/0006-8993(76)90857-x. [DOI] [PubMed] [Google Scholar]
- Wishart T.M., Paterson J.M., Short D.M., Meredith S., Robertson K.A., Sutherland C., Cousin M.A., Dutia M.B., Gillingwater T.H. Differential proteomics analysis of synaptic proteins identifies potential cellular targets and protein mediators of synaptic neuroprotection conferred by the slow Wallerian degeneration (Wlds) gene. Mol Cell Proteomics. 2007;6:1318–1330. doi: 10.1074/mcp.M600457-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F., Fan L., Wells S., Hartley R., Mackenzie F.E., Oyebode O., Brown R., Thomson D., Coleman M.P., Blanco G., Ribchester R.R. Axonal and neuromuscular synaptic phenotypes in Wld(S), SOD1(G93A) and ostes mutant mice identified by fiber-optic confocal microendoscopy. Mol Cell Neurosci. 2009;42:296–307. doi: 10.1016/j.mcn.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Zhu X.H., Qiao H., Du F., Xiong Q., Liu X., Zhang X., Ugurbil K., Chen W. Quantitative imaging of energy expenditure in human brain. NeuroImage. 2012;60:2107–2117. doi: 10.1016/j.neuroimage.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]