

Abstract
DNA damage is a deleterious threat, but occurs daily in all types of cells. In response to DNA damage, poly(ADP-ribosyl)ation, a unique posttranslational modification, is immediately catalyzed by poly(ADP-ribose) polymerases (PARPs) at DNA lesions, which facilitates DNA damage repair. Recent studies suggest that poly(ADP-ribosyl)ation is one of the first steps of cellular DNA damage response and governs early DNA damage response pathways. Suppression of DNA damage-induced poly(ADP-ribosyl)ation by PARP inhibitors impairs early DNA damage response events. Moreover, PARP inhibitors are emerging as anti-cancer drugs in phase III clinical trials for BRCA-deficient tumors. In this review, we discuss recent findings on poly(ADP-ribosyl)ation in DNA damage response as well as the molecular mechanism by which PARP inhibitors selectively kill tumor cells with BRCA mutations.
Review
Both environmental and internal hazards induce lesions in genomic DNA1. If not repaired, DNA lesions will induce genomic instability and ultimately cause tumorigenesis. Fortunately, DNA damage response system recognizes and repairs DNA lesions, which protects genomic stability and suppresses tumorigenesis2, 3. Accumulated evidence suggests that poly(ADP-ribosyl)ation is a crucial part of DNA damage response system for sensing of DNA lesions, activation of DNA damage response pathways, and facilitating DNA damage repair4, 5.
Poly(ADP-ribosyl)ation has been identified for 50 years6, 7. The process of poly(ADP-ribosyl)ation is catalyzed by poly(ADP-ribose) polymerases (PARPs)8–10. Using NAD+ as the donor, mono-ADP-ribose is covalently linked to the side chains of arginine, lysine, aspartate, and glutamate residues of target proteins by PARPs. After catalyzing the first ADP-ribose on the proteins, other ADP-ribose can be covalently linked onto the first ADP-ribose and the continuous reactions produce both linear and branched polymers, known as poly(ADP-ribose) (PAR)5, 11. The structure of PAR has been well characterized for many years: the ADP-ribose units in the polymer are linked by glycosidic ribose-ribose 1′–2′ bonds, and the chain length is heterogeneous, which can reach around 200 units, with 20–50 ADP-ribose units in each branch12–14 (Fig. 1). Accumulated evidence shows that DNA damage induces massive synthesis of PAR in a very short period15, 16. In this review, we summarize the recent findings of this dynamic posttranslational modification in DNA damage response, and discuss the possible molecular mechanism of PARP inhibitors in cancer treatment.
Figure 1. Sketch of poly(ADP-ribosyl)ation.

With NAD+ as the donor, PARPs mediate the genotoxic stress-dependent poly(ADP-ribosyl)ation. ADP-ribose residues are covalently linked to the side chains of arginine, lysine, aspartate, or glutamate residues of acceptor proteins. Glycosidic ribose-ribose 1′–2′ bonds between ADP-ribose units generate both linear and branched polymers. The chain length of PAR is heterogeneous, which can reach up to 200 ADP-ribose units, with 20–50 units in each branch.
Metabolism of PAR during DNA damage response
Although the cellular concentration of NAD+ is around 0.3 – 1 mM, the basal level of poly(ADP-ribosyl)ation is very low15, 17. However, following genotoxic stress, level of poly(ADP-ribosyl)ation increases 10- to 1000-fold in a few seconds15–18, which could consume up to 75% of cellular NAD+15, 18. Since NAD+ is a key coenzyme in many biological processes such as glucose and fatty acid metabolism, poly(ADP-ribosyl)ation may transiently suppress these biochemical reactions immediately following DNA damage. The DNA damage-induced poly(ADP-ribosyl)ation is mainly catalyzed by PARP1, 2 and 3, although seventeen PARPs have been identified on the basis of homologous information to the funding member PARP14, 11, 19. With the enzymatic activity significantly higher than the other members in vitro, PARP1 is believed to make the major contribution to DNA damage-induced PAR synthesis in vivo4, 18. PARP1 is a 116-kDa protein, consisting of three functional domains, namely N-terminal DNA-binding domain, automodification domain, and C-terminal catalytic domains5, 20 (Fig. 2). The DNA-binding domain includes three zinc fingers that are required for the interaction with DNA breaks21, 22. Once the DNA-binding domain recognizes DNA breaks, it induces the conformational changes of C-terminal catalytic domain to expose the activation site to NAD+, and activates the enzymatic activity23. Of note, a BRCA1 carboxy-terminal (BRCT) motif is also found in this automodification domain, although its function is still elusive5, 20. The BRCT motif is known as the phospho-group binding domain and is usually involved in DNA damage response24–27. The existence of the BRCT motif may imply an unidentified role of PARP1 especially in DNA damage response. Besides auto-poly(ADP-ribosyl)ation, PARP1 also induces histone poly(ADP-ribosyl)ation at vicinity of DNA lesions, which may facilitate chromatin remodeling in response to DNA damage28, 29.
Figure 2. Domain architecture of human PARP1.
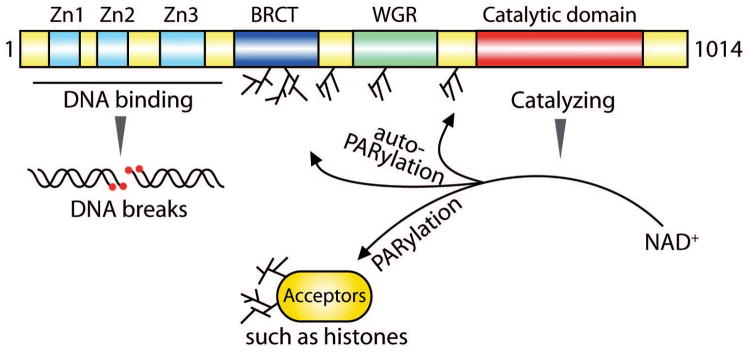
Human PARP1 contains 1014 residues of amino acid with the molecular weight of 116 kDa. N-terminal three zinc finger motifs (Zn1-3) recognize SSBs and DSBs, which induces the conformational changes of the Tryptophan-glycine-arginine rich (WGR) and Catalytic domains and activates the enzymatic activity of PARP1.
The massive DNA damage-induced PAR synthesis consumes huge amount of NAD+15, 18, which is not sustainable for cells if NAD+ is depleted for the prolonged time. It has been shown that the half-life of DNA damage-induced PAR is between 40 s to 6 min15, 30–33. PAR is quickly hydrolyzed into free ADP-ribose (ADPr) that is probably recycled back to NAD+. The best studied PAR-degrading enzyme is poly(ADP-ribose) glycohydrolase (PARG)34, which possesses both endoglycosidic and exoglycosidic activities and therefore generates free ADPr from PAR35, 36. In mammals, several isoforms of PARG from a single PARG gene have been identified4, 11. The full length 110kDa-PARG mainly localizes in nucleus while other short forms of PARG exist in cytoplasm36, 37. Following DNA damage-induced PAR synthesis, PARG is recruited to DNA lesions and breaks 1′–2′ glycosic bonds between two riboses38, 39. However, PARG cannot remove the last ADP-ribose linking to the amino acid residue40, 41. Recent studies suggest that several other enzymes including TARG, Macro D1 and Macro D2 could remove the last ADP-ribose residue42–44. In particular, TARG mainly localizes in nucleus, and is likely to function with PARG to degrade DNA damage-induced poly(ADP-ribosyl)ation44.
PAR-dependent chromatin remodeling during DNA damage response
The major substrates of DNA damage-induced poly(ADP-ribosyl)ation are PARP1 itself and histones including nucleosomal histones and linker histones surrounding DNA lesions11, 28. Over the past few decades, PAR is known to be covalently linked to arginine, glutamate or aspartate residues of acceptor proteins45. The identification of lysine as an acceptor site on PARP2 and histone tails updated the convention concept of poly(ADP-ribosyl)ation by ester linkage46, 47. Recent proteomic analyses with various enrichment approaches further reveal the in vivo poly(ADP-ribosyl)ation sites. For example, Zhang et al. used boronate beads to enrich the substrates and identified novel poly(ADP-ribosyl)ation sites48. Jungmichel et al. dissected poly(ADP-ribosyl)ated targets by affinity purification using a bacterial PAR-binding domain49. Also, using phosphoproteomic approach, two other groups have mapped auto-ADP-ribosylation sites of PARP150 and mono/poly- ADP-ribosylation sites from whole cell lysates51. Interestingly, poly(ADP-ribosyl)ation is a unique chromatin modification as each ADP-ribose residue contains two phosphate groups carrying two negative charges, so that the polymer brings a large amount of negative charges to the damaged chromatin4, 11, 52. Since genomic DNA has the same negative charges, PAR relaxes chromatin by electron repel. Besides its own chemical property, PAR regulates chromatin remodeling through its binding partners. To date, PAR is recognized by several modules including the PBZ, Macro, RRM, BRCT, FHA and OB-fold domains32, 33, 53–59. CHD4, a PBZ domain containing protein, is a subunit in the histone deacetylase NuRD complex60, 61. It has been shown that CHD4 is recruited by PAR in response to DNA damage and facilitates the loading of the NuRD complex for DNA damage-induced chromatin remodeling, which may indirectly impact DNA damage repair62. Another example is ALC1, a DNA helicase with a Macro domain. Upon binding PAR, the helicase activity of ALC1 is activated, which induces nucleosomes sliding away from DNA damage sites63, 64. Thus, PAR mediates chromatin remodeling at DNA lesions through both its own chemical properties and its binding partners.
The role of PAR in DNA single-strand damage repair
Besides the role in DNA damage-induced chromatin remodeling, PARP1 is a major sensor to detect DNA single-strand lesions and participates in DNA damage repair65–68 (Fig. 3). Numerous DNA single strand breaks (SSBs) (breakage in the sugar-phosphate backbone of one strand of a DNA helix), are induced daily by various types of environmental and internal hazards in every cell. If not repaired timely, SSBs can be converted into DNA double-strand breaks (DSBs), a more lethal type of DNA lesion69, 70. SSBs can be generated directly by disintegration of DNA backbone or arise as a result of erroneous activity of cellular enzymes such as DNA topoisomerase 1 (TOP1) 69, 71. SSBs can also be indirectly induced during the base excision repair (BER)72, 73. This process is initiated by DNA glycosylases, which recognize and remove damaged bases, forming apurinic/apyrimidinic sites (AP sites). These sites are then cleaved by an AP endonuclease to generate SSBs for DNA patching74. These lesion-induced SSBs are repaired by a general process including four steps: SSB detection, DNA end processing, DNA gap filling, and DNA ligation70. During the process, SSBs are detected by PARPs (mainly by PARP1), and the interaction between the DNA nicks and PARPs triggers massive synthesis of PAR at the sites of SSBs68, 70. The SSB-induced PAR is recognized by X-ray repair cross-complementing protein 1 (XRCC1), one of the core factors in SSBs repair 75. Previous studies have shown that XRCC1 has high affinity to PAR both in vitro and in vivo, and the interaction is required for the rapid recruitment of XRCC1 to the sites of SSBs32, 76, 77. As a scaffold, XRCC1 interacts with and stabilizes other SSB repair machineries75, 78, 79. More recently, polynucleotide kinase 3′-phosphatase (PNKP), aprataxin (APTX) and aprataxin and PNK-like factor (APLF) were found to recognize PAR too32, 53, 54. All of them are important enzymes for the SSB repair. PNKP has been shown to possess 3′-DNA phosphatase and 5′-DNA kinase activity and can thus restore normal termini from DNA lesions with 3′-phosphate and 5′-hydroxyl end groups80, 81. These enzymatic activities of PNKP facilitate DNA end ligation by DNA ligase III70. APTX catalyzes the nucleophilic release of adenylate groups covalently linked to 5′-phosphate termini at single-strand nicks and gaps, which is generated during aborted ligation. This activity produces 5′-phosphate termini for efficiently rejoining82. For APLF, it has an AP endonuclease activity as well as a 3′–5′ exonuclease activity for DNA end resection83. Thus, PARP1 senses the ends of SSBs and synthesizes PAR at DNA lesions. Damage-induced PAR functions as the earliest alarm and targets at least XRCC1, PNKP, APTX and APLF to SSBs for the repair.
Figure 3. Poly(ADP-ribosyl)ation functions as a sensor for activating DNA damage response.

In response to SSBs and DSBs, massive PAR is rapidly generated by PARPs and jumpstarts DNA damage response. In the NER pathway for SSB repair, PAR mediates the recruitment of DDB2 and ALC1. In the BER pathway, PAR targets PNKP, APTX, XRCC1 and APLF to DNA lesions for the subsequent repair. In NHEJ pathway for DSB repair, PAR mediates the recruitment of the Ligase IV -XRCC4 complex. In HR pathway for DSB repair, PAR is recognized by several repair machineries, such as the BRCA1-BARD1 complex, the MRN complex and the hSSB1-INTS complex.
Besides BER, nucleotide excision repair (NER) also generates SSBs84. Accumulated evidence suggests that PARP1 participates in NER, a repair mechanism for bulky DNA adducts, such as UV-induced thymine dimer and 6,4-photoproducts85. Similar to BER, damaged bases are recognized and removed to expose single-strand ends during NER86. However, compared with BER, a relatively long patch of single-stranded DNA containing the lesions is removed. The undamaged single-stranded DNA in the helix is used as the template for the synthesis of the complementary strand by DNA polymerases86, 87. Although the detailed function of PAR in NER is not clear, it has been shown that UV induces massive poly(ADP-ribosyl)ation, and auto poly(ADP-ribosyl)ated PARP1 is associated with specific NER machineries such as XPA and DDB276, 88, which facilitates the recruitment of the chromatin-remodeling enzyme ALC185.
The role of PAR in DNA double-strand break repair
Beside SSB repair, recent studies suggest that poly(ADP-ribosylation) also plays a key role in DNA double-strand break (DSB) repair. Compared with SSB, DSB is much more deleterious. If not repaired, DSBs instantly induce the loss of partial genomic DNA or chromosomal abnormal rearrangements that alter gene codes. It ultimately causes genomic instability and tumorigenesis2, 89. To avoid genomic instability, cells have a sophisticated DSB response system including checkpoint pathways and DSB repair pathways90, 91. Poly(ADP-ribosylation) has been shown to regulate both DSB-induced checkpoint activation and DSB repair.
PARP1 is one of the first proteins that directly recognize DSB ends22. Due to high expression level of PARP1 in nucleoplasm4, 19, abundant PARP1 is able to scan genomic DNA. Once DSB occurs, PARP1 reaches the sites of DNA damage within milliseconds92, 93. Structural analysis has shown that the N-terminal three zinc fingers coordinate together to recognize one DSB end and induces the conformational changes in the catalytic domain for the activation of massive PAR synthesis22. The abundance and rapid activation of PARP1 suggests that PARP1 is a key DNA damage sensor for DSB response.
DSB-induced PAR is then recognized by PAR-binding proteins for both cell cycle checkpoint activation and DSB repair. Recently, several novel PAR-binding modules, including the BRCT, FHA and OB-fold domains, have been revealed32, 33, 57. Interestingly, the BRCT and FHA domains are known as phospho-protein binding domain24, 25, 94–96. It has been shown that a set of BRCT and FHA domains directly bind PAR and possibly recognize phosphate groups in PAR32. Among these BRCT and FHA domain containing proteins, NBS1 is important for cell cycle checkpoint activation32. NBS1 is a subunit in the MRN complex that activates ATM in response to DSB97–99. ATM is a PI3-like kinase that governs DSB-induced cell cycle checkpoint100, 101. Once DSBs occur, the BRCT domain of NBS1 recognizes PAR at DNA lesions, which rapidly recruits the MRN complex to the sites of DNA damage and facilitates the early activation of ATM-dependent signal transduction pathway as well as cell cycle checkpoints32. Besides NBS1, hSSB1 is another PAR-binding protein that may mediate early checkpoint activation57. hSSB1 is a subunit in the hSSB-INTS complex, and contains a N-terminal OB-fold domain102. The OB-fold domain is known to interact with single-stranded DNA/RNA103, 104. Recent study suggests that a set of OB-fold domains, such as the OB-fold domain of hSSB1, prefer to bind PAR over oligo nucleotide. The interaction between PAR and hSSB1 also facilitates the fast recruitment of hSSB1 to the sites of DSBs57. It has been shown that hSSB1 plays an important role to stabilize the MRN complex at DNA lesions105. Thus, it is possible that the hSSB-INTS complex indirectly regulates ATM-dependent checkpoint activation through the MRN complex.
Besides cell cycle checkpoint activation, poly(ADP-ribosyl)ation also regulates DSB repair. There are two well-studied DSB repair mechanisms, namely non-homologous end joining (NHEJ) and homologous recombination (HR)2, 106. DNA damage-induced poly(ADP-ribosyl)ation regulates both NHEJ and HR. Among the NHEJ pathway, Ligase IV plays a key role to religate DSB ends107. Recent evidence shows that the BRCT domain of Ligase IV is a PAR-binding domain32. The interaction with PAR mediates the fast recruitment of Ligase IV to DNA lesions, which is likely to promote quick NHEJ. Regarding HR, the BRCA1/BARD1 complex is required for loading downstream HR repair machinery such as RAD51 to DSBs108, 109. Like other BRCT domain, the BRCT domain of BARD1 directly binds PAR at DNA lesions and mediates the fast recruitment of the BRCA1/BARD1 complex to DNA lesions33. The early DNA damage response mediates by the BRCA1/BARD1 complex is likely to promote HR repair.
Collectively, poly(ADP-ribosyl)ation regulates both cell cycle checkpoint activation and DSB repair. DNA damage-induced PAR is recognized by a lot of DNA damage response factors, and mediates the fast recruitment of these factors to DNA lesions, which jumpstarts DNA damage response pathways. Since most DNA damage-induced poly(ADP-ribosyl)ation events are mediated by PARP1, PARP1 is a bona fide DSB sensor. After various DNA damage response pathways are activated, the high level of PAR begins to be degraded by PARG, and DNA repair factors are selectively retained at DNA lesions through other mechanisms such as DNA damage-induced phosphorylation and ubiquitination events for fulfilling the DNA damage repair (Fig. 3).
PARP inhibitors in cancer chemotherapies
Since poly(ADP-ribosyl)ation regulates DNA damage response, suppression of DNA damage-induced PAR may sensitize tumor cells, especially DNA damage repair-deficient tumor cells, to genotoxic stress. Thus, PARP inhibitors have been designed and tested for cancer treatment20, 110, 111. Two explicit studies have shown that BRCA1 and BRCA2-deficient tumor cells are hypersensitive to PARP inhibitor treatment112, 113. Germline mutations of BRCA1 and BRCA2 induce hereditary breast and ovarian cancers, and account for 5 ~ 10 % of total breast and ovarian cancers114, 115. Both BRCA1 and BRCA2 are involved in HR repair and facilitate the loading of RAD51, the key enzyme for HR repair, to DNA lesions109. Mutations of BRCA1 and BRCA2 impair RAD51-dependent HR, induce the accumulation of DNA lesions, lead to genomic instability, and eventually cause cell malignant transformation116–118. It has been hypothesized that PARP inhibitors selectively killing BRCA-deficient tumor cells through synthetic lethality approach119, 120. The synthetic lethality concept was first proposed by Hartwell et al. in 1990s during their study of anticancer drugs121. It described the condition in which defects in either one of two genes individually had mild effect, but the lethality ensued when the defects in the two combined122. Previous study on PARP inhibitor mainly focuses on its role in BER repair. It has been shown that DNA lesions induced by endogenous metabolism or replication errors result in SSBs that are repaired by the BER pathway. When PARPs are inhibited, the BER pathway is suppressed. It induces the SSBs degraded to DSBs during replication. In HR proficient cells, these DSBs would be repaired by HR. However, in absence of BRCA1, BRCA2, or other HR machineries, failure of DSB repair induces cell apoptosis 123. However, several questions on this model arise from recent studies: 1) Besides PARPs, other enzymes also participate in SSB repair70. To date, it is unclear whether suppression of other SSB repair machineries could also kill BRCA-deficient tumors. 2) Besides HR repair, NHEJ is an alternative mechanism for DSB repair. Why couldn’t NHEJ compensate the loss of HR during PARP inhibitor treatment? 3) Recent study suggests that poly(ADP-ribosyl)ation is likely to play a much broader role in not only SSB repair but also DSB repair. The BRCA1/BARD1 complex even directly recognize DNA damage-induced PAR33, suggesting that PAR directly participates in BRCA1-dependent HR repair. 4) Phase II clinical trials suggest that PARP inhibitors are effective for around 40 % of BRCA-deficient tumors110. There are still ~ 60 % of patients who did not respond well to PARP inhibitor treatment. Thus, we propose a new model of how PARP inhibitors selectively kill tumor cells with BRCA mutations.
New model
Recent findings in our laboratory show an important function of PAR in HR32, 33. We find that BRCA1 is recruited to the sites of DNA damage by PAR and the PAR-dependent fast recruitment of BRCA1 is required for HR33, 124. It has been shown that BRCA1 and BARD1forms a complex through the Ring-Ring interaction125, 126. The BRCT domain of BARD1 recognizes PAR and target the whole complex to DNA lesions in a few seconds following DSBs. Under normal condition, PAR is degraded by PARG in a few minutes following DSBs. But the BRCA1-BARD1 complex is still able to be retained at DNA lesions because the BRCT domain of BRCA1 is a pSer-binding domain that recognizes pSer406 of Abraxas/CCDC98 at DNA damage sites33, 124, 127, 128. Suppression of PAR synthesis by PARP inhibitors abolishes the fast recruitment of the BRCA1-BARD1 complex. But through Abraxsa/CCDC98 and phosphorylation-dependent events, BRCA1is still recruited to DNA lesions, albeit in very slow kinetics (Fig. 4).
Figure 4. The molecular mechanism of the recruitment of BRCA1 to DNA lesions.

BRCA1 and BARD1 form heterodimer via the interaction between the Ring domains. Upon DNA damage, PAR quickly recruits the BRCA1-BARD1 complex via the interaction with the BARD1 BRCT. The BRCT of BRCA1 is important for the stable retention of BRCA1/BARD1 complex at the sites of DNA damage through the interaction with Abraxas/CCDC98.
Most cancer-associated mutations of BRCA1 occur in the exon 11 of BRCA1 or the C-terminal BRCT domain. These mutations either generate truncated forms of BRCA1 deleting the C-terminal BRCT domain or abolish the tertiary structure of the BRCT domain129–131. These mutations are likely to be hypomorphic mutations because the mutants could still be recruited to DNA lesions transiently via the interaction between the BARD1 BRCT domain and PAR, although they could not be stabilized at DNA lesions because of lacking the BRCA1 BRCT domain. Transient recruitment of these BRCA1 mutants could only repair some of but not all of DNA lesions. Accumulation of lesions in the genome will induce genomic instability and tumorigenesis. However, with the PARP inhibitor treatment, the BRCA1 mutants could not be recruited to the sites of DNA damage since DNA damage-induced PAR synthesis is suppressed. Nor could the mutants slowly accumulate at DNA lesions because the cancer-associated mutations abolish the BRCA1 BRCT domain. Under this condition, cells completely lose BRCA1, which eventually induces apoptosis132. This is likely to be the molecular mechanism by which PARP inhibitors selectively kill tumor cells with BRCA1 mutations (Fig. 5). In agreement with this model, we found that cells bearing mutations in the BRCA1 BRCT domain (e.g. P1749R andM1775R) are hypersensitive to PARP inhibitor treatment33. However, not all the cancer-associated BRCA1 mutations disrupt the BRCA1 BRCT domain. A set of mutations have been identified in the Ring domain of BRCA1 as well as in the BARD BRCT domain133–137. In this case, regardless of the treatment of PARP inhibitors, these BRCA1/BARD1 mutants could not be quickly recruited to the sites of DNA damage. Thus, tumor cells bearing these mutations may not be sensitive to PARP inhibitor treatment (Fig. 5). Consistently, we found that cells bearing mutations in the BRCA1 Ring domain (e.g. C61G) are much less sensitive to PARP inhibitors than cells with the mutations in the BRCA1 BRCT domain33. Thus, different cancer-associated BRCA1 mutations have distinct responses to PARP inhibitors, PARP inhibitors may only be effective to tumors with certain BRCA1 mutations.
Figure 5. A new model of how PARP inhibitors selectively suppress BRCA1-deficient tumors.
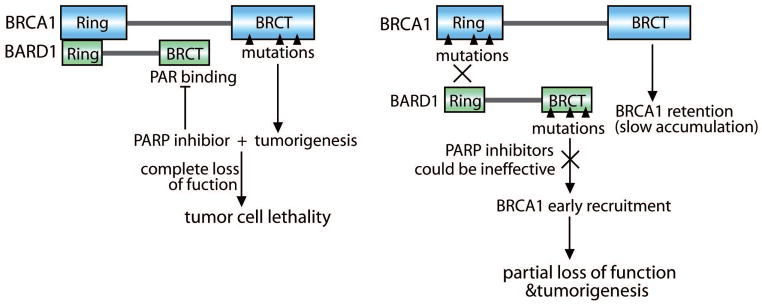
Hypomorphic mutations of BRCA1 abolish the BRCA1 BRCT domain. The mutants can still be transiently recruited to the sites of DNA damage by PAR. BRCA1 mutants fail to repair all the lesions, and induce genomic instability and tumorigenesis. Treating these tumor cells with PARP inhibitors abolishes the transient recruitment of mutant BRCA1. Without BRCA1, tumor cells undergo apoptosis. However, a set of cancer-associated mutations exist in the Ring domain of BRCA1 or the BRCT domain of BARD1. In these cases, PAR does not affect the recruitment of BRCA1. Thus, PARP inhibitors do not selectively kill tumor cells with these mutations.
Recently, we also demonstrate that the Oligonucleotide/oligosaccharide-binding (OB)-fold motif is a novel PAR-binding domain that mediates DNA damage response57. Interestingly, the OB-fold motif also exists in BRCA2, implying a similar mechanism for PARP inhibitors selectively suppressing tumors with BRCA2 mutations.
Conclusion and future direction for PARP inhibitors in cancer chemotherapies
In conclusion, during DNA damage response, PAR serves as an initial sensor and mediates the early recruitment of SSB and DSB repair machineries. Suppression of PAR synthesis by PARP inhibitors abolishes the early recruitment of DNA damage repair machineries such as BRCA1, thus sensitizes tumor cells to DNA damaging agents. This could be a novel molecular mechanism for PARP inhibitors to selectively kill tumor cells, which might be important for personalized cancer chemotherapies. In addition to BRCA1, PARP inhibitors suppress the fast recruitment of many other DNA damage repair machineries, which induces cell lethality or hypersensitive to DNA damaging agents. Thus, it is possible to target other types of cancers with PARP inhibitors. For example, Ligase IV is a key enzyme in NHEJ and is recruited to DNA lesions by PAR32. Mutation of Ligase IV is associated with Ligase IV syndrome with clinical features such as T-cell lymphoma138–140. These cancer-associated mutations do not exist in the PAR-binding domain and are likely to be hypomorphic mutations. PARP inhibitor treatment may abolish the fast recruitment of Ligase IV for NHEJ, thus induces tumor cell lethality or hypersensitive to DNA damaging agents such as etoposide, mitomycin C and cisplatin. Another example is XRCC1, a scaffold protein in SSB repair. Mutation of XRCC1 is associated with non-melanoma skin cancer141, 142. With similar mechanism, it is possible that PARP inhibitor treatment will selectively kill tumor cells with XRCC1 mutation. Thus, PARP inhibitors may have broader clinical implications in cancer chemotherapies.
Acknowledgments
We thank Dr. Chao Liu to proofread the manuscript. This work was supported by the National Institute of Health (CA132755 and CA130899 to X.Y.). X.Y. is a recipient of the Era of Hope Scholar Award from the Department of Defense. M.L. is a recipient of the Ovarian Cancer Research Foundation Award.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 4.Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 5.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- 7.Weill JD, Busch S, Chambon P, Mandel P. The effect of estradiol injections upon chicken liver nuclei ribonucleic acid polymerase. Biochem Biophys Res Commun. 1963;10:122–126. doi: 10.1016/0006-291x(63)90036-6. [DOI] [PubMed] [Google Scholar]
- 8.Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 9.Hottiger MO, Hassa PO, Luscher B, Schuler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35:208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Leung AK. Poly(ADP-ribose): an organizer of cellular architecture. J Cell Biol. 2014;205:613–619. doi: 10.1083/jcb.201402114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 12.Alvarez-Gonzalez R, Jacobson MK. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry. 1987;26:3218–3224. doi: 10.1021/bi00385a042. [DOI] [PubMed] [Google Scholar]
- 13.Juarez-Salinas H, Levi V, Jacobson EL, Jacobson MK. Poly(ADP-ribose) has a branched structure in vivo. J Biol Chem. 1982;257:607–609. [PubMed] [Google Scholar]
- 14.Miwa M, Ishihara M, Takishima S, Takasuka N, Maeda M, Yamaizumi Z, et al. The branching and linear portions of poly(adenosine diphosphate ribose) have the same alpha(1 leads to 2) ribose-ribose linkage. J Biol Chem. 1981;256:2916–2921. [PubMed] [Google Scholar]
- 15.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342 (Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez-Gonzalez R, Pacheco-Rodriguez G, Mendoza-Alvarez H. Enzymology of ADP-ribose polymer synthesis. Mol Cell Biochem. 1994;138:33–37. doi: 10.1007/BF00928440. [DOI] [PubMed] [Google Scholar]
- 17.Gasser A, Guse AH. Determination of intracellular concentrations of the TRPM2 agonist ADP-ribose by reversed-phase HPLC. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;821:181–187. doi: 10.1016/j.jchromb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 20.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gradwohl G, Menissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, et al. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc Natl Acad Sci U S A. 1990;87:2990–2994. doi: 10.1073/pnas.87.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ali AA, Timinszky G, Arribas-Bosacoma R, Kozlowski M, Hassa PO, Hassler M, et al. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol. 2012;19:685–692. doi: 10.1038/nsmb.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336:728–732. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 25.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 26.Glover JN, Williams RS, Lee MS. Interactions between BRCT repeats and phosphoproteins: tangled up in two. Trends Biochem Sci. 2004;29:579–585. doi: 10.1016/j.tibs.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst) 2009;8:1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Messner S, Hottiger MO. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011;21:534–542. doi: 10.1016/j.tcb.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Martinez-Zamudio R, Ha HC. Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol. 2012;32:2490–2502. doi: 10.1128/MCB.06667-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alvarez-Gonzalez R, Althaus FR. Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res. 1989;218:67–74. doi: 10.1016/0921-8777(89)90012-8. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson EL, Antol KM, Juarez-Salinas H, Jacobson MK. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J Biol Chem. 1983;258:103–107. [PubMed] [Google Scholar]
- 32.Li M, Lu LY, Yang CY, Wang S, Yu X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013;27:1752–1768. doi: 10.1101/gad.226357.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M, Yu X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell. 2013;23:693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakura H, Miwa M, Tanaka M, Kanai Y, Shimada T, Matsushima T, et al. Natural occurence of a biopolymer, poly (adenosine diphosphate ribose) Nucleic Acids Res. 1977;4:2903–2915. doi: 10.1093/nar/4.8.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min W, Wang ZQ. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci (Landmark Ed) 2009;14:1619–1626. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 36.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 37.Meyer-Ficca ML, Meyer RG, Jacobson EL, Jacobson MK. Poly(ADP-ribose) polymerases: managing genome stability. Int J Biochem Cell Biol. 2005;37:920–926. doi: 10.1016/j.biocel.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, Schreiber V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011;39:5045–5056. doi: 10.1093/nar/gkr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng X, Koh DW. Roles of poly(ADP-ribose) glycohydrolase in DNA damage and apoptosis. Int Rev Cell Mol Biol. 2013;304:227–281. doi: 10.1016/B978-0-12-407696-9.00005-1. [DOI] [PubMed] [Google Scholar]
- 40.Moss J, Stanley SJ, Nightingale MS, Murtagh JJ, Jr, Monaco L, Mishima K, et al. Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J Biol Chem. 1992;267:10481–10488. [PubMed] [Google Scholar]
- 41.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, et al. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, et al. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat Struct Mol Biol. 2013;20:508–514. doi: 10.1038/nsmb.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol. 2013;20:502–507. doi: 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]
- 44.Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, et al. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013;32:1225–1237. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilz H. ADP-ribosylation of proteins--a multifunctional process. Hoppe Seylers Z Physiol Chem. 1981;362:1415–1425. [PubMed] [Google Scholar]
- 46.Haenni SS, Hassa PO, Altmeyer M, Fey M, Imhof R, Hottiger MO. Identification of lysines 36 and 37 of PARP-2 as targets for acetylation and auto-ADP-ribosylation. Int J Biochem Cell Biol. 2008;40:2274–2283. doi: 10.1016/j.biocel.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 47.Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010;38:6350–6362. doi: 10.1093/nar/gkq463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods. 2013;10:981–984. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- 49.Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013;52:272–285. doi: 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 50.Chapman JD, Gagne JP, Poirier GG, Goodlett DR. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J Proteome Res. 2013;12:1868–1880. doi: 10.1021/pr301219h. [DOI] [PubMed] [Google Scholar]
- 51.Daniels CM, Ong SE, Leung AK. Phosphoproteomic Approach to Characterize Protein Mono- and Poly(ADP-ribosyl)ation Sites from Cells. J Proteome Res. 2014 doi: 10.1021/pr401032q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, et al. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 54.Li GY, McCulloch RD, Fenton AL, Cheung M, Meng L, Ikura M, et al. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc Natl Acad Sci U S A. 2010;107:9129–9134. doi: 10.1073/pnas.1000556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, et al. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, et al. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang F, Chen Y, Li M, Yu X. The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proc Natl Acad Sci U S A. 2014;111:7278–7283. doi: 10.1073/pnas.1318367111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malanga M, Czubaty A, Girstun A, Staron K, Althaus FR. Poly(ADP-ribose) binds to the splicing factor ASF/SF2 and regulates its phosphorylation by DNA topoisomerase I. J Biol Chem. 2008;283:19991–19998. doi: 10.1074/jbc.M709495200. [DOI] [PubMed] [Google Scholar]
- 59.Gagne JP, Hunter JM, Labrecque B, Chabot B, Poirier GG. A proteomic approach to the identification of heterogeneous nuclear ribonucleoproteins as a new family of poly(ADP-ribose)-binding proteins. Biochem J. 2003;371:331–340. doi: 10.1042/BJ20021675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- 61.Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 62.Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325:1240–1243. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci U S A. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 66.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le Page F, Schreiber V, Dherin C, De Murcia G, Boiteux S. Poly(ADP-ribose) polymerase-1 (PARP-1) is required in murine cell lines for base excision repair of oxidative DNA damage in the absence of DNA polymerase beta. J Biol Chem. 2003;278:18471–18477. doi: 10.1074/jbc.M212905200. [DOI] [PubMed] [Google Scholar]
- 68.Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol. 2003;23:3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caldecott KW. Protein-protein interactions during mammalian DNA single-strand break repair. Biochem Soc Trans. 2003;31:247–251. doi: 10.1042/bst0310247. [DOI] [PubMed] [Google Scholar]
- 70.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 71.Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res. 2003;532:173–203. doi: 10.1016/j.mrfmmm.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 72.Demple B, DeMott MS. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene. 2002;21:8926–8934. doi: 10.1038/sj.onc.1206178. [DOI] [PubMed] [Google Scholar]
- 73.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson DM, 3rd, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J Biol Chem. 1995;270:16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 75.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003;2:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 76.Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 77.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brem R, Hall J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005;33:2512–2520. doi: 10.1093/nar/gki543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 80.Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, et al. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem. 1999;274:24176–24186. doi: 10.1074/jbc.274.34.24176. [DOI] [PubMed] [Google Scholar]
- 81.Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJ, Sgouros J, et al. Molecular characterization of a human DNA kinase. J Biol Chem. 1999;274:24187–24194. doi: 10.1074/jbc.274.34.24187. [DOI] [PubMed] [Google Scholar]
- 82.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 83.Li S, Kanno S, Watanabe R, Ogiwara H, Kohno T, Watanabe G, et al. Polynucleotide kinase and aprataxin-like forkhead-associated protein (PALF) acts as both a single-stranded DNA endonuclease and a single-stranded DNA 3′ exonuclease and can participate in DNA end joining in a biochemical system. J Biol Chem. 2011;286:36368–36377. doi: 10.1074/jbc.M111.287797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81:39–44. doi: 10.1016/s0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- 85.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, et al. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199:235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 87.Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer. 2001;1:22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- 88.Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F, Shah GM. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. 2013;110:1658–1663. doi: 10.1073/pnas.1209507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 90.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 91.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 92.Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 93.Tartier L, Spenlehauer C, Newman HC, Folkard M, Prise KM, Michael BD, et al. Local DNA damage by proton microbeam irradiation induces poly(ADP-ribose) synthesis in mammalian cells. Mutagenesis. 2003;18:411–416. doi: 10.1093/mutage/geg015. [DOI] [PubMed] [Google Scholar]
- 94.Durocher D, Henckel J, Fersht AR, Jackson SP. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell. 1999;4:387–394. doi: 10.1016/s1097-2765(00)80340-8. [DOI] [PubMed] [Google Scholar]
- 95.Li J, Williams BL, Haire LF, Goldberg M, Wilker E, Durocher D, et al. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol Cell. 2002;9:1045–1054. doi: 10.1016/s1097-2765(02)00527-0. [DOI] [PubMed] [Google Scholar]
- 96.Mahajan A, Yuan C, Lee H, Chen ES, Wu PY, Tsai MD. Structure and function of the phosphothreonine-specific FHA domain. Sci Signal. 2008;1:re12. doi: 10.1126/scisignal.151re12. [DOI] [PubMed] [Google Scholar]
- 97.Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 99.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 100.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- 101.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 102.Zhang F, Ma T, Yu X. A core hSSB1-INTS complex participates in the DNA damage response. J Cell Sci. 2013;126:4850–4855. doi: 10.1242/jcs.132514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Flynn RL, Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010;45:266–275. doi: 10.3109/10409238.2010.488216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meyer RR, Laine PS. The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev. 1990;54:342–380. doi: 10.1128/mr.54.4.342-380.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Richard DJ, Cubeddu L, Urquhart AJ, Bain A, Bolderson E, Menon D, et al. hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Res. 2011;39:3643–3651. doi: 10.1093/nar/gkq1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23:687–696. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 107.Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 108.Westermark UK, Reyngold M, Olshen AB, Baer R, Jasin M, Moynahan ME. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol Cell Biol. 2003;23:7926–7936. doi: 10.1128/MCB.23.21.7926-7936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22:5784–5791. doi: 10.1038/sj.onc.1206678. [DOI] [PubMed] [Google Scholar]
- 110.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 111.Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2013 doi: 10.1038/onc.2013.352. [DOI] [PubMed] [Google Scholar]
- 112.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 113.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 114.Petrucelli N, Daly MB, Feldman GL. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet Med. 2010;12:245–259. doi: 10.1097/GIM.0b013e3181d38f2f. [DOI] [PubMed] [Google Scholar]
- 115.Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet. 2001;10:705–713. doi: 10.1093/hmg/10.7.705. [DOI] [PubMed] [Google Scholar]
- 116.Xia F, Taghian DG, DeFrank JS, Zeng ZC, Willers H, Iliakis G, et al. Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc Natl Acad Sci U S A. 2001;98:8644–8649. doi: 10.1073/pnas.151253498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Polyak K, Garber J. Targeting the missing links for cancer therapy. Nat Med. 2011;17:283–284. doi: 10.1038/nm0311-283. [DOI] [PubMed] [Google Scholar]
- 120.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 121.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 122.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 123.De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front Oncol. 2013;3:228. doi: 10.3389/fonc.2013.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baer R. Luring BRCA1 to the scene of the crime. Cancer Cell. 2013;23:565–567. doi: 10.1016/j.ccr.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 126.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–837. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 127.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim H, Huang J, Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol. 2007;14:710–715. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- 129.Li ML, Greenberg RA. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem Sci. 2012;37:418–424. doi: 10.1016/j.tibs.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11:519–525. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 131.Lee MS, Green R, Marsillac SM, Coquelle N, Williams RS, Yeung T, et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010;70:4880–4890. doi: 10.1158/0008-5472.CAN-09-4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH. Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. Genes Dev. 1996;10:1835–1843. doi: 10.1101/gad.10.14.1835. [DOI] [PubMed] [Google Scholar]
- 133.Thai TH, Du F, Tsan JT, Jin Y, Phung A, Spillman MA, et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum Mol Genet. 1998;7:195–202. doi: 10.1093/hmg/7.2.195. [DOI] [PubMed] [Google Scholar]
- 134.Ghimenti C, Sensi E, Presciuttini S, Brunetti IM, Conte P, Bevilacqua G, et al. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer. 2002;33:235–242. doi: 10.1002/gcc.1223. [DOI] [PubMed] [Google Scholar]
- 135.Ishitobi M, Miyoshi Y, Hasegawa S, Egawa C, Tamaki Y, Monden M, et al. Mutational analysis of BARD1 in familial breast cancer patients in Japan. Cancer Lett. 2003;200:1–7. doi: 10.1016/s0304-3835(03)00387-2. [DOI] [PubMed] [Google Scholar]
- 136.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci U S A. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brzovic PS, Meza JE, King MC, Klevit RE. BRCA1 RING domain cancer-predisposing mutations. Structural consequences and effects on protein-protein interactions. J Biol Chem. 2001;276:41399–41406. doi: 10.1074/jbc.M106551200. [DOI] [PubMed] [Google Scholar]
- 138.Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. 2006;176:5060–5068. doi: 10.4049/jimmunol.176.8.5060. [DOI] [PubMed] [Google Scholar]
- 139.Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. 2006;36:224–235. doi: 10.1002/eji.200535401. [DOI] [PubMed] [Google Scholar]
- 140.van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116:137–145. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nelson HH, Kelsey KT, Mott LA, Karagas MR. The XRCC1 Arg399Gln polymorphism, sunburn, and non-melanoma skin cancer: evidence of gene-environment interaction. Cancer Res. 2002;62:152–155. [PubMed] [Google Scholar]
- 142.Han J, Hankinson SE, Colditz GA, Hunter DJ. Genetic variation in XRCC1, sun exposure, and risk of skin cancer. Br J Cancer. 2004;91:1604–1609. doi: 10.1038/sj.bjc.6602174. [DOI] [PMC free article] [PubMed] [Google Scholar]