

Abstract
Androgen receptor splicing variants (ARVs) which lack the ligand-binding domain (LBD) are associated with the development of castration-resistant prostate cancer (CRPC), including resistance to the new generation of high affinity anti-androgens. However, the mechanism by which ARVs expression is regulated is not fully understood. In this study, we show that activation of classical NF-κB signaling increases the expression of ARVs in prostate cancer (PCa) cells and converts androgen sensitive PCa cells to become androgen insensitive; while, downregulation of NF-κB signaling inhibits ARVs expression and restores responsiveness of CRPC to anti-androgen therapy. In addition, we demonstrated that combination of anti-androgen with NF-κB targeted therapy inhibits efficiently tumor growth of human CRPC xenografts. These results indicate that induction ARVs by activated NF-κB signaling in PCa cells is a critical mechanism by which the PCa progresses to CRPC. This has important implications since it can prolong the survival of CRPC patients by restoring the tumors to once again respond to conventional androgen-deprivation therapy (ADT).
Keywords: NF-κB, Androgen receptor variants, Prostate cancer
INTRODUCTION
Androgen-deprivation therapy (ADT) in the majority of prostate cancer (PCa) patients results in initial regression of disease and a dramatic decrease in serum prostate-specific antigen (PSA). Eventually, all patients will fail ADT and the regrowing cancer is now commonly referred to as Castrate Resistant Prostate Cancer (CRPC). It is now clear that Androgen Receptor Addiction (ARA) is the driving force behind the development of CRPC.1 The recent development of abiraterone acetate, that blocks the synthesis of androgens by the tumor, and the new high affinity anti-androgens, such as Enzalutamide (formerly MDV3100), once again block androgen receptor (AR) action in CRPC validating that the AR pathway is a central target for drug therapy.2,3 However, in due course, failure to these new drugs occurs. Multiple pathways are proposed that result in continue AR function4,5 but recent studies have reported the elevated expression of AR splice variants (ARVs), which lack the ligand-binding domain (LBD), are associated with rapid progression to CRPC.6-10 The ARVs result in constitutive activation of the AR pathway thereby promoting PCa cell growth at even low concentrations of androgen,9,10 enhance growth of androgen-dependent xenografts in castrated mice8 and the development of Enzalutamide resistant PCa.11 Based on these findings, it has been proposed that ARVs can function as important drivers of CRPC.6-8
ARVs originate from AR transcripts by insertions of cryptic exons downstream of the coding sequences for AR DNA-binding domain (DBD),6-8,10 or by deletions of exons coding for AR-LBD.9,12 These alterations in AR transcripts disrupt the AR open reading frame, leading to truncated AR proteins with an intact N-terminal domain (NTD) and the DNA binding domain (DBD), and in some cases a short variant-specific peptide replacing the LBD.13 AR gene rearrangements have been proposed as one mechanism to explain the appearance of truncated ARVs in PCa cell lines and xenografts.14,15 Most recently, Nadiminty et al reported that activation of the non-canonical Nuclear Factor-kappa B (NF-κB) pathway (NF-κB2/p52) can enhance expression of ARVs thereby contributing to resistance to anti-androgen in PCa.16 However, the detailed mechanism(s) by which the ARVs increase in advanced CRPC is still not fully understood.
NF-κB proteins are an important class of transcriptional regulators in PCa. Many stimuli activate NF-κB, mostly through IκB kinase-dependent (IKK-dependent) phosphorylation and subsequent degradation of inhibitor κB (IκB) protein.17-19 The degradation of IκB occurs by the inhibitor of kappa kinase (IKK) complex20 which has two catalytic subunits, IKKβ and IKKα, that regulate the classical and alternative NF-κB signaling pathways, respectively. The classical NF-κB pathway targets dimers composed of p65 (RelA), c-Rel, and NF-κB1 (p50), which regulate transcription of diverse set of genes encoding cytokines, growth factors, cell adhesion molecules, pro- and anti-apoptotic proteins (see review21,22). The alternative NF-κB pathway depends on IKKα dimers that phosphorylate the NF-κB2 (p100) precursor protein, leading to degradation of its C-terminal half and release of its N-terminal portion, p52, that enters the nucleus as a dimer with RelB.23
Abundant data supports a key role for the NF-κB signaling pathway in controlling the initiation and progression of human cancer.24-27 Many studies indicate that activation of NF-κB signaling correlates with PCa progression, including chemoresistance, advanced stage, PSA recurrence, metastatic spread, and growth of PCa in bone.6,28-35 NF-κB is shown to the third most activated pathway out of >100 pathways in metastatic34 and in primary PCa where it correlates with patient outcome,36 also supported by previous reports.16,33,35,37 Further, multiple NF-κB binding sites in the AR promoter have been shown to increase transcription of AR-FL.38 Previously, we and other researchers have confirmed that classical NF-κB pathway plays a critical role in the progression of PCa to castrate resistant growth and bone metastasis.38-40
However, detailed mechanism(s) by which NF-κB signaling contributes to CRPC progression is still not fully understood. In this study, we report that activation of classical NF-κB signaling induces the transcription of the AR splice variants in PCa cells and by blocking NF-κB signaling we can restore CRPC responsiveness to anti-androgen treatment. These results indicate that ARVs elevation by the activation of NF-κB signaling in PCa cells is an important mechanism whereby tumors progress to CRPC. Further, NF-κB inhibition is sufficient to restore responsiveness of CRPC to androgen blockade.
RESULTS
Activation of NF-kB signaling increases ARVs expression in the PCa cells
Full length AR (AR-FL) contains an N-terminal domain (NTD) encoded by exon 1, the DBD (exon 2 and 3), a short hinge region (exon 4), and the C-terminal LBD encoded by Exon 4-8,41 while ARVs lack the ability to bind androgens due to a lack off or a truncation of the LBD. In order to investigate whether NF-κB signaling affects ARVs expression, we designed two pairs of primers to specifically detect AR Exon 1 (Exon 1) and AR-FL. Exon 1 primers were used to detect both AR-FL and all ARVs; AR-FL primers were used to detect AR-FL only. LNCaP is an androgen-dependent (AD) PCa cell, which has low levels of NF-κB activity.40 NF-κB signaling in LNCaP cells was activated by infecting with IKK2-EE retroviral vector (LNCaP-EE where EE contains constitutive active mutant of IKK2 resulting in activation the classical NF-κB pathway)42,43 (Figure 1A). The cells infected with the empty vector (EV) were used as controls (LNCaP-EV). The results from real time qRT-PCR showed that activation of NF-κB signaling increased both AR-FL and AR exon 1 expression (Figure 1B). However, the change of exon 1 expression is higher than that of AR-FL (Figure 1B). In addition, activation of NF-κB signaling increased AR-V7 mRNA expression significantly, which is one of the well-known ARVs and commonly expressed in CRPC7 (Figure 1B). These result indicated activation of NF-κB signaling increase ARVs expression in PCa cells. Western blot assay further confirmed that activation of NF-κB signaling in LNCaP cells increased both AR-FL and ARVs expression (Figure 1C). This result further confirmed by directly over expression of RelA (p65), a specific transcription factor of the classical NF-κB pathway in LNCaP cells. The result from Western blot assay showed that over expression of RelA increased AR-V7 significantly in LNCaP cells (Figure 1D). In contrast to LNCaP cells, C4-2B and 22RV1 cells are castrate resistant. In order to further confirm our findings, NF-κB signaling in C4-2B and 22RV1 cells was inactivated by infecting with IKK2-KD retroviral vector (which contains the kinase dead, KD, mutant of IKK2).42,43 Consistent with our findings in LNCaP cells, inactivation of NF-κB signaling in C4-2B and 22RV1 cells decreased ARVs expression in both cell lines (Figure 1E and F).
Figure 1. Activation of NF-κB signaling increases ARVs expression in PCa cells.

A) NF-κB signaling was activated in LNCaP cells by infecting with IKK2-EE retroviral vector (LNCaP-EE). LNCaP cells infected with IKK2-EV (empty vector) (LNCaP-EV) were used as controls. NF-κB activity was determined using NGL, a NF-κB reporter vector. B) Three sets of primers were used to amplify wild-type AR (AR-FL), Exon 1 (which amplify both wild-type AR and ARVs) and AR-V7, respectively. AR-FL and ARVs mRNA expression were measured by qRT-PCR. Results are presented as means ± SD of 3 experiments performed in triplicate. C) Proteins expression of AR-FL (molecular weight is 110 kDa) and ARVs (molecular weight is 75 kDa) was measured by Western blot analysis using AR N20 antibody. Blot signals were quantified using ImageJ program. Results were normalized by TBA signals. D) NF-κB signaling was activated in LNCaP cells by infecting with RelA expression vector (LNCaP-RelA). LNCaP cells infected with empty vector (LNCaP-EV) were used as control. Protein expression of AR-V7 was measured by Western blot analysis using AR-V7 antibody. Blot signals were quantified using ImageJ program. Results were normalized by β-actin signals. NF-κB signaling was inactivated by infecting with IKK2-KD retroviral vector in E) C4-2B (C4-2B-KC) and F) 22RV1 (22RV1-KD) cells. Cells infected with IKK2-EV (C4-2B-EV, 22RV1-EV) were used as control. Three sets of primers were used to amplify AR-FL, Exon 1 (which amplify both wild-type AR and ARVs) and AR-V7, respectively. AR-FL and ARVs mRNA expression were measured by qRT-PCR. Results are presented as means ± SD of 3 experiments each performed in triplicate.
BMS345541 is a well-known specific inhibitor of the NF-κB pathway. Treatment of PCa cells with BMS345541 blocks NF-κB signaling efficiently in these cells (Supplemental Figure 1). In order to confirm that NF-κB signaling elevates ARVs expression in PCa cells, C4-2B and 22RV1 cells were treated with BMS345541. The results from real time qRT-PCR showed that blocking of NF-κB signaling efficiently decreased AR-FL, AR exon 1 and AR-V7 mRNA expression (Figure 2A and B). By western blot, detection of AR-FL and ARVs proteins in 22RV1 cells is apparent (AR-FL ~ 110 kDa; ARVs ~ 75 kDa). Consistent with the findings on AR-FL and ARVs mRNA by qRT-PCR, inactivation of NF-κB signaling decreased AR-FL and ARVs expression in 22RV1 cells at the protein level (Figure 2C). This result was further confirmed by Western blot assay using an antibody that only recognizes AR-V7 (Figure 2D). These findings indicate that activation of the classical NF-κB pathway is an important mechanism by which the ARVs levels increase in advanced CRPC. However, BMS345541 alone did not affect the proliferation rate significantly in androgen independent C4-2B and 22RV1 cells in vitro (Figure 2E and F).
Figure 2. BMS345541, a specific NF-κB inhibitor, decreases ARVs expression efficiently in PCa cells.

A) C4-2B and B) 22RV1 cells were treated with BMS345541 (BMS) for 24 hours. AR-FL and ARVs mRNA expression were measured by qRT-PCR. Results are presented as means ± SD of 3 experiments performed in triplicate. Proteins expression of AR-FL (molecular weight is 110 kDa) and ARVs (molecular weight is 75 kDa) in BMS treated (24 hours) 22RV1 cells was measured by Western blot analysis using (C) AR N20 and (D) AR-V7 antibodies. Blot signals were quantified using ImageJ program. Results were normalized by actin signals. E) C4-2B and F) 22RV1 cells were treated with referenced concentration of BMS345541 (BMS). Cell proliferation assay was performed at 48 hours after treatment. Results are presented as means ± SD of 3 experiments performed in triplicate.
Blocking of NF-κB signaling increases the sensitivity of CRPC cells to the anti-androgen
In order to determine if antagonizing NF-κB signaling effects the sensitivity of CRPC cells to an anti-androgen, we generated NF-κB inactivated PCa cell lines by stably infecting with IKK2-KD vectors (C4-2B-KD) (Supplemental Figure 2A). Although NF-κB blockade slightly altered proliferation rates, the engineered cells grew well (survive) in vitro after down regulation of NF-κB activity (Supplemental Figure 2B). NF-κB activated (C4-2B-EV; infected with empty vector) and inactivated (C4-2B-KD) PCa cells were treated with bicalutamide, an anti-androgen. As expected, control cells (C4-2B-EV), which have high levels of NF-κB activity and express the ARVs, had a lower response to bicalutamide (Figure 3A). However, the sensitivity of NF-κB inactivated C4-2B-KD cells to the bicalutamide is increased significantly (Figure 3B). Conversely, activation of NF-κB signaling reversed androgen-dependent LNCaP cells to become androgen-insensitive (Figure 1A and 3C). These results indicate that activation of NF-κB signaling is sufficient to cause progression of androgen dependent PCa cells to become castrate resistant; while, blocking NF-κB signaling increase the sensitivity of CRPC cells to an anti-androgen. Mechanistically, NF-κB controls the expression of the AR-FL and ARVs thereby controlling the response to anti-androgens.
Figure 3. Blocking of NF-κB signaling increases the sensitivity of androgen-independent PCa cells to the anti-androgen.
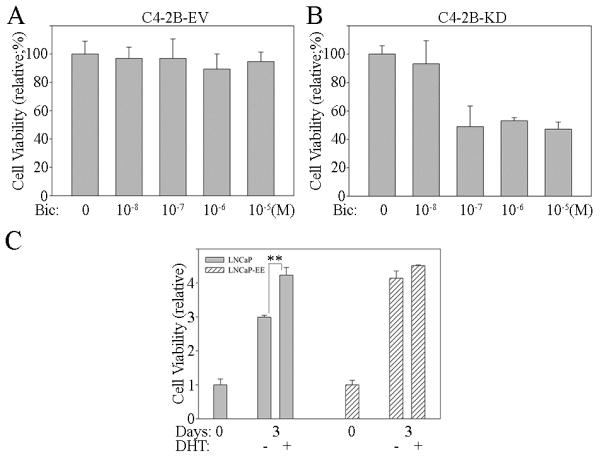
C4-2B cells were stably infected with IKK2-KD retroviral vector (C4-2B-KD), in which NF-κB activity was inhibited with a kinase dead (KD) IKK2 mutant. The cells infected with empty vector (EV) were used as controls leaving NF-κB signaling activated (C4-2B-EV). Both A) C4-2B-EV and B) C4-2B-KD cells were treated with Bicalutamide (Bic). Cell proliferation assay was performed at 48 hours after treatment. Results are presented as means ± SD of 3 experiments performed in triplicate. C) NF-κB signaling activated (LNCaP-EE) and inactivated (LNCaP-EV) LNCaP cells were treated with or without androgen (DHT; 10−8M). Cell proliferation assay was performed at 72 hours after treatment. Results are presented as means ± SD of 3 experiments performed in triplicate. **, P < 0.001 by Student’s t test.
Inhibition of NF-κB signaling restores responsiveness of CRPC cells to anti-androgen treatment
BMS345541, a specific inhibitor of the NF-κB pathway, efficiently blocks NF-κB signaling in PCa cells (Supplemental Figure 1). Unfortunately, BMS345541 is not suitable for clinical use. Bortezomib is a FDA approved drug that is an inhibitor of the 26S proteasome complex and blocks the degradation of IκB. Elevation of IκB, the NF-κB inhibitor, blocks NF-κB signaling.44 Our studies show that bortezomib blocks NF-κB activity efficiently in both C4-2B and 22RV1 CRPC cells (Supplemental Figure 3A and B). Since bortezomib is clinically approved to treat cancer, we selected this agent to test whether inhibition of NF-κB signaling would restore responsiveness of CRPC cells to the anti-androgen treatment. First, we investigated bortezomib effect on AR-FL and ARVs expression in PCa cells. AR-FL and ARVs expression was analyzed by real time qRT-PCR and Western blotting. The results show that bortezomib decreases AR-FL and ARVs expression efficiently in PCa cells at both the mRNA and protein levels (Figure 4A, B, C and D). To test if bortezomib can restore responsiveness of CRPC cells to the anti-androgen treatment, we treated C4-2B and 22RV1 cells with anti-androgen alone or combined it with bortezomib. As expected, the anti-androgen (bicalutamide) or bortezomib alone had no significant effect on the growth rate on CRPC C4-2B and 22RV1 cells (Figure 5A, B, C and D). However, when the cells were treated with anti-androgen plus an antagonist of NF-κB signaling, the growth rate of both C4-2B and 22RV1 cells was significantly inhibited (Figure 5E and F). These results further demonstrate that blocking of NF-κB signaling decreases AR-FL and ARVs expression and restores responsiveness of CRPC cells to the anti-androgen treatment.
Figure 4. Bortezomib decreases ARVs expression efficiently in PCa cells.

A) C4-2B and B) 22RV1 cells were treated with Bortezomib (BZ) for 24 hours. AR-FL and ARVs mRNA expression were measured by qRT-PCR. Results are presented as means ± SD of 3 experiments performed in triplicate. Proteins expression of AR-FL (molecular weight is 110 kDa) and ARVs (molecular weight is 75 kDa) in Bortezomib treated (24 hours) 22RV1 cells was measured by Western blot analysis using C) AR N20 and D) AR-V7 antibodies. Blot signals were quantified using ImageJ program. Results were normalized by Actin signals.
Figure 5. Bortezomib increases the sensitivity of androgen-independent PCa cells to the anti-androgen.

A, C, E) C4-2B and B, D, F) 22RV1 cells were treated with referenced concentration of Bicalutamide (Bic) and/or Bortezomib (BZ). Cell proliferation assay was performed at 48 hours after treatment. Results are presented as means ± SD of 3 experiments performed in triplicate. *, P < 0.05; **, P<0.001, by Student’s t test, compared with control.
Combination of an anti-androgen and NF-κB targeted therapy efficiently inhibits CRPC tumor growth in vivo
In order to extend our in vitro results, we generated CRPC xenograft mouse models of C4-2B or 22RV1 cells in male athymic nude mice. After the primary s.c. tumors reached 3-4 mm diameter, the mice were treated with bicalutamide, bortezomib or bicalutamide plus bortezomib by IP injection. Control mice were treated with vehicle only. Tumor volume was measured weekly, and the xenograft tissues were harvested 2 weeks after treatment for analysis. After treatment with either bicalutamide or bortezomib alone, both of C4-2B and 22RV1 tumors continued to grow and there was no significant difference in the tumor size compared to the control group (treated with vehicle) (Figure 6A and B). However, when the mice were treated with bicalutamide combined with bortezomib, the tumor growth was significantly inhibited in both C4-2B and 22RV1 bearing mice (p<0.05; Figure 6A and B). Proliferation rates, NF-κB activity and ARVs expression of tumor cells in the xenograft model were determined by immunohistochemical staining of Ki67, p65-pho and AR-V7 (Figure 6C and D). Our results showed that the number of the cancer cells stained by Ki67 in the tumor from mice treated with bicalutamide plus bortezomib was significantly lower (p < 0.01) than that the mice treated with bicalutamide, bortezomib or vehicle alone (Figure 6C). In addition, bortezomib significantly inhibits p65-pho (p < 0.05) and AR-V7 (p < 0.001) expression in this xenograft model (Figure 6D). These results indicate that blocking NF-κB signaling increases the sensitivity of CRPC tumor to the anti-androgen treatment. Therefore, combination therapy that attacks both the androgen-AR axis and NF-κB pathway can provide a new therapeutic approach against lethal CRPC.
Figure 6. Blocking of NF-κB signaling increases the sensitivity of CRPC tumor to the anti-androgen.

A) C4-2B and B) 22RV1 cells were subcutaneously injected into the right flank of 7-week-old male athymic nude mice. After the primary tumors reached 3-4 mm diameter, the mice were treated with Bicalutamide (Bic: 20mg/kg in oil; 3 times/wk), Bortezomib (BZ: 1mg/kg in DMSO; 3 times/wk) or Bicalutamide plus Bortezomib (Bic+BZ) by IP injection. Control group mice (Con) were treated with vehicle only. Tumor volume was measured weekly and calculated by the formula: Volume = π/6 × W × H × L (mm3). Results are presented as the mean percentage change in tumor volume; bars, ± SD. *, P < 0.05 by Student’s t test, compared with control. C) Immunohistochemical staining of Ki67 was performed to determine cell proliferation in the tumors. Each tissue section was counted manually in three different areas to assess the Ki67 positive cells index. The data were then presented as number of Ki67 positive cells/400× microscope field. Results are presented as the means ± SD. **, P < 0.001 by Student’s t test, compared with control. D) Immunohistochemical staining of p65-pho and AR-V7 were performed to determine NF-κB activity and ARVs expression in the tumors. Expression of p65-pho and AR-V7 were measured by calculating the percentage of DAB-stained nuclear area over total nuclear area (labeling index) using ImmunoRate program (lower panel). Results are presented as the means ± SD. **, P < 0.001; *, P < 0.05 by Student’s t test, compared with control.
DISCUSSION
It is widely accepted that androgen receptor addition (ARA) can lead to the failure of ADT and the emergence of CRPC. Although multiple mechanisms have been proposed to explain escape from ADT (such as AR amplification, modification of the AR by point mutations or phosphorylation, and changes in AR co-activators), there is evidence that the ARVs are an important driving force in developing CRPC.10,11,13-15 Despite initial success in treating CRPC with the new generation of high affinity anti-androgens such as enzalutamide or inhibitors of androgen synthesis such as abiraterone, patients still fail, within 4-5 months, the drug therapy targeting AR action.2,3,45 Most recently, several studies demonstrated that ARVs play a major role in the resistance to this new generation of inhibitors of AR action.11,16,46 Therefore, understanding the mechanism of failure of therapy is critical to develop new treatments against CRPC.
Neuroendocrine differentiation (NED) of the primary adenocarcinoma (defined as the adenocarcinoma expressing NE markers such as enolase, chromogranin A, and/or synaptophysin) is associated with poor prognosis.47-50 As the patients undergo treatment of metastatic disease, there is an increase in NED and eventually with failure to ADT, small cell carcinoma appears (NE cancer that has low levels of AR or is AR negative).51 We have reported that Wnt-signaling induces NED in adenocarcinoma,52 a pathway that is now recognized to be activated in advanced PCa.53 Previously, we have reported that neuropeptides, such as bombesin (BBS) and gastrin-releasing peptide (GRP), activates NF-κB signaling in PCa cells, and neuropeptides released from NE cells cause LNCaP xenografts to become castrate resistant by activating the classical NF-κB signaling and increasing AR levels.39,54 Most recently, Nadiminty et al. reported that activation of the non-canonical NF-κB pathway (NF-κB2/p52) enhances expression of ARVs thereby contributing to resistance to anti-androgens in PCa cell lines.16 In this study, we demonstrate that activation of NF-κB signaling plays a critical role in the expression of ARVs in PCa cells and that blocking of NF-κB signaling down-regulates AR-FL and ARVs expression to restore responsiveness of CRPC to anti-androgen treatment. Both overexpression of RelA (p65) and constitutively active IKK2 mutants do not result in physiological levels of active the NF-κB. However, this data still provides a fundamental explanation on how the activation of the NF-κB pathway can control progression to CRPC by the induction of ARVs. Our study shows that activation of NF-κB signaling increases ARVs expression at both the mRNA and protein levels. It is reported that NF-κB binds to the AR promoter to transcriptionally regulate gene expression.38 The rapid induction of ARVs expression in LNCaP by NF-κB activation (within 24 hours) would suggest that NF-κB also regulation splicing of the AR pre-mRNA. However, we do not have direct proof that NF-κB is involved in spliceosome function. Our preclinical studies using human CRPC xenograft models show that the combination of an anti-androgen and antagonist of NF-κB signaling is sufficient to inhibit CRPC tumor growth. Taken together, these findings suggest that as NED or small cell carcinoma occurs, signaling by neuropeptides activates the NF-κB pathway in the adenocarcinoma to increase ARVs expression and failure to ADT. These findings have important clinical implications by providing the mechanism whereby the tumor escapes ADT and an approach for a new intervention to treat CRPC.
The results from previous phase I and II trials of bortezomib alone or in combination with docetaxel showed limited antitumor activity in CRPC.55,56 This is consistent with our results since it is the combination of ADT with the NF-κB targeted therapy that was effective against CRPC xenografts. Under this combination, we see that blocking NF-κB signaling down regulates the expression of both the AR-FL and the ARVs, while the anti-androgen blocks the remaining low levels of androgen seen by the AR-FL. AR binds to DNA as a dimer and it has been shown that AR-FL and ARVs dimer can drive CRPC.10 Our current data demonstrates that blocking the AR axis or NF-κB signaling alone cannot inhibit CRPC tumor growth efficiently; while, the combination of anti-androgen with NF-κB targeted therapy inhibits CRPC tumor growth significantly in preclinical CRPC mouse models (Figure 6). These results strongly support that combination of inhibiting both the AR axis and NF-κB pathway can serve as an effective therapy against lethal CRPC. Recently, a small clinical trial to test bortezomib in combination with ADT was reported for early stage disease. Bortezomib plus hormone blockade for three months induced a rapid clinical response that lasted 42 days longer (median time to progression) when compared to hormone blockade alone. Progression was measured as a change in the slope of the PSA rise.57 This clinical response was considered short lived and bortezomib was associated with significant neural toxicity in the patients. Our studies indicate that the maximum benefit to a patient would not occur in early stage disease but patients in the late stage of the disease that are CRPC would benefit by combining the ADT with inhibition of the NF-κB pathway. Our studies suggest that blocking the expression of the NF-κB pathway during the late stages of the disease when ARVs are extensively expressed would provide the maximum benefit in combination with ADT. Clearly, agents that block NF-κB activity with minimal side effects that can be used in combination with ADT for late stage CRPC would benefit the patients.
In summary, the data presented here indicates that activation of NF-κB signaling increases ARVs expression in the PCa cells and blocking NF-κB signaling down-regulates ARVs expression and restores responsiveness of CRPC cells to the anti-androgen treatment. Combination of anti-androgen and NF-κB targeted therapy is a new approach to treat CRPC.
MATERIALS AND METHODS
Cell culture and materials
The human prostate carcinoma cell lines LNCaP and 22RV1 were obtained from ATCC (Manassas, VA). C4-2B cells were gifts of Dr. Leland Chung (Cedars Sinai Medical Center, Los Angeles, CA).58 Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in the air. Cell lines were routinely cultured in RPMI 1640 (Gibco-BRL) medium containing 5% fetal calf serum (FBS) (Hyclone), 0.1% ITS and 0.1% Glutamine (Gibco-BRL). The following reagents were purchased for in vivo and in vitro experiments: Bicalutamide (Selleckchem), BMS345541 (Sigma-Aldrich) and Bortezomib (LC Laboratories, Woburn, MA).
Generation of NF-κB signaling activated/inactivated PCa cells
To generate NF-κB signaling activated/inactivated PCa cells, LNCaP cells were infected with IKK2-EE retroviral vector resulting in LNCaP-EE, in which NF-κB signaling was activated with a constitutively active (EE) mutants of IKK2,42,43 while, C4-2B and 22RV1 cells were infected with IKK2-KD retroviral vector, in which NF-κB signaling was inhibited with a kinase dead (KD) IKK2 mutant (C4-2B-KD and 22RV1-KD).42,43 The cells infected with empty vector were used as controls (LNCaP-EV, C4-2B-EV and 22RV1-EV). All retroviral vectors were a gift from Dr. Martin Leverkus, University of Magdeburg, Germany.
Reverse transcription and real-time PCR
Total RNAs from experimental cells were extracted using Trizol (Gibco-BRL), and residual genomic DNA was removed by DNaseI (Invitrogen) treatment. The RNAs were reverse transcribed using random primers and Superscript II (Gibco-BRL) according to the manufacturer’s protocol. The primers used to amplify wild-type AR (AR-FL) were 5′-TTCGAATGAACTACATCAAGGAACTCGATCG-3′ (forward), 5′-TTGGGCACTTGCACAGAGAT-3′ (reverse); AR Exon 1 (Exon 1) were 5′-CCTGGCACACTCTCTTCACA-3′ (forward), 5′-CCGGAGTAGCTATCCATCCA-3′ (reverse). Primers of AR-V7 were 5′-CCATCTTGTCGTCTTCGGAAATGTTATGAAGC-3′ (forward), 5′-TTTGAATGAGGCAAGTCAGCCTTTCT-3′ (reverse)7. Real-time PCR reactions were carried out in a 20μl volume using a 96-well plate format and fluorescence was detected utilizing the Bio-Rad I-Cycler IQ Real-time detection system. Gene expression was normalized to 18s rRNA by the 2−ΔΔCt method.59 The values plotted represent the mean of at least three individual samples ± SD.
Western blot analysis
Whole cell lysate was extracted from NF-κB activated/inactivated PCa cells. A 20μg aliquot (40-60μg for AR-V7 antibody) of each protein sample was separated on a 4 to 12% Tris-glycine gradient gel (NOVEX™), and then transferred to nitrocellulose membranes (Schleicher & Schuell, Germany). The membranes were blocked with 5% skim milk in TBS-T (Trypsin buffered saline, 1% Tween-20) buffer. The AR (N20, Santa Criuz) and AR-V7 (Precision) antibodies were added (AR, 1:1000; AR-V7, 1:200) and the blots were incubated o/n in 4 C°. After washing three times for 10 minutes each in TBS-T, incubation was performed for 1 hour with the secondary horseradish-peroxidase-conjugated anti-rabbit antibody. The signals were developed by an ECL detection system (Amersham Biosciences, Amersham, USA).
Transient transfection assay
The NGL vector [a NF-κB responsive reporter vector which has Luciferase and Green Fluorescent Protein (GFP) reporter genes]60 was used to measure NF-κB activity in the PCa cancer cells by transient transfection experiments. Experimental cells were plated at an initial density of 2.5 × 104/well in 24-well tissue culture plates. After 24 hours, the cells were transfected with NGL vector using Lipofectamine (Invitrogen) for four hours according to the manufacturer’s protocol. Luciferase activity was determined using the Promega Corp luciferase assay system 24 hours after transfection or further treatment with BMS345541 or bortezomib. The transfection efficiency was determined by co-transfecting pRL-CMV containing the Renilla luciferase reporter gene (Promega). The values plotted represent the mean of at least three individual samples ± SD.
Proliferation assay
Experimental cells were plated in a 96-well plate (1 × 104/well). After 24 h, the cells were treated with or without different combination of drugs (Bicalutamide, BMS345541 or Bortezomib) in different concentration. MTT assay was performed at 24, 48, and 72 hours after the cells had been treated with referenced drugs. All of the measurements were carried out in triplicate.
CRPC xenograft mouse model
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Vanderbilt Institutional Animal Care & Use Committee (Permit Number: M/09/387). CRPC xenograft mouse model was generated by injection of C4-2B or 22RV1 cells subcutaneously into the right flank of 7-week-old male athymic nude mice (BALB/c strain). After the primary tumors reached 3-4 mm diameter, the mice were treated with Bicalutamide (20mg/kg in oil; 3 times/wk), Bortezomib (1mg/kg in DMSO; 3 times/wk) or Bicalutamide plus Bortezomib by IP injection. Control group mice were treated with vehicle only. Tumor volume was measured weekly and calculated by the formula: Volume = π/6 × W × H × L (mm3). After 2 weeks treatment, the xenograft tissues were harvested and fixed in 10% buffered formalin and paraffin embedded for histologic and immunohistochemical analyses. Each group had at least five mice. The results are reported as the mean percent ± SD.
Immunohistochemistry
Paraffin-embedded tissue sections were stained immunohistochemically with antibodies against Ki67 (clone TEC-3, DAKO), p65-pho (Abcam) and AR-V7 (Precision). The primary antibody was incubated at the appropriate concentration (1:1000) for one hour at room temperature. The secondary antibody was incubated for 60 minutes. Slides were rinsed extensively in tap water, counterstained with Mayer’s hematoxylin and mounted. For quantitation of the cell proliferation, the cells were counted as positive for Ki67 when nuclear immunoreactivity was observed. Each tissue section was counted manually in three different areas to assess the Ki67 positive cells index. The data were then presented as number of Ki67 positive cells/400x microscope field. Expression of p65-pho and AR-V7 were measured by calculating the percentage of DAB-stained nuclear area over total nuclear area (labeling index) using ImmunoRate program.61
Statistical and image analysis
Where appropriate, experimental groups were compared using two-sample t-test, with significance defined as P <0.05.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Martin Leverkus (University of Magdeburg, Germany) for providing us with IKK2-EE and IKK2-KD retroviral vectors, and Tom C. Case and Manik Paul for technical assistance. This work was supported to RJ by the Department of Defense (DOD) Prostate Cancer Research Program (PCRP) (W81XWH-10-1-0236); to RJM by the National Cancer Institute (4R01 CA076142-14) and the Frances Preston Laboratories of the T.J. Martell Foundation.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. Urologic Oncology. 2009;27:36–41. doi: 10.1016/j.urolonc.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2013 doi: 10.1038/onc.2013.235. doi:10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Inaugural Article: Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Rre. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate. 2011;71:1656–1667. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–2117. doi: 10.1158/0008-5472.CAN-10-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP, et al. NF-kappaB2/p52 Induces Resistance to Enzalutamide in Prostate Cancer: Role of Androgen Receptor and Its Variants. Molecular Cancer Therapeutics. 2013;12:1629–1637. doi: 10.1158/1535-7163.MCT-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 18.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 19.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 20.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;5:RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 22.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 24.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 25.Karin M. NF-kappaB and cancer: mechanisms and targets. Mol Carcinog. 2006;45:355–361. doi: 10.1002/mc.20217. [DOI] [PubMed] [Google Scholar]
- 26.Inoue J, Gohda J, Akiyama T, Semba K. NF-kappaB activation in development and progression of cancer. Cancer Sci. 2007;98:268–274. doi: 10.1111/j.1349-7006.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pacifico F, Leonardi A. NF-kappaB in solid tumors. Bio Chem Pharmacol. 2006;72:1142–1152. doi: 10.1016/j.bcp.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 28.Lessard L, Begin LR, Gleave ME, Mes-Masson AM, Saad F. Nuclear localisation of nuclear factor-kappaB transcription factors in prostate cancer: an immunohistochemical study. Br J Cancer. 2005;93:1019–1023. doi: 10.1038/sj.bjc.6602796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lessard L, Karakiewicz PI, Bellon-Gagnon P, Alam-Fahmy M, Ismail HA, Mes-Masson AM, et al. Nuclear localization of nuclear factor-kappaB p65 in primary prostate tumors is highly predictive of pelvic lymph node metastases. Clin Cancer Res. 2006;12:5741–5745. doi: 10.1158/1078-0432.CCR-06-0330. [DOI] [PubMed] [Google Scholar]
- 30.Domingo-Domenech J, Mellado B, Ferrer B, Truan D, Codony-Servat J, Sauleda S, et al. Activation of nuclear factor-kappaB in human prostate carcinogenesis and association to biochemical relapse. Br J Cancer. 2005;93:1285–1294. doi: 10.1038/sj.bjc.6602851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingo-Domenech J, Oliva C, Rovira A, Codony-Servat J, Bosch M, Filella X, et al. Interleukin 6, a nuclear factor-kappaB target, predicts resistance to docetaxel in hormone-independent prostate cancer and nuclear factor-kappaB inhibition by PS-1145 enhances docetaxel antitumor activity. Clin Cancer Res. 2006;12:5578–5586. doi: 10.1158/1078-0432.CCR-05-2767. [DOI] [PubMed] [Google Scholar]
- 32.Ismail HA, Lessard L, Mes-Masson AM, Saad F. Expression of NF-kappaB in prostate cancer lymph node metastases. Prostate. 2004;58:308–313. doi: 10.1002/pros.10335. [DOI] [PubMed] [Google Scholar]
- 33.Ross JS, Kallakury BV, Sheehan CE, Fisher HA, Kaufman RP, Jr., Kaur P, et al. Expression of nuclear factor-kappa B and I kappa B alpha proteins in prostatic adenocarcinomas: correlation of nuclear factor-kappa B immunoreactivity with disease recurrence. Clin Cancer Res. 2004;10:466–4672. doi: 10.1158/1078-0432.ccr-0543-3. [DOI] [PubMed] [Google Scholar]
- 34.Setlur SR, Royce TE, Sboner A, Mosquera JM, Demichelis F, Hofer MD, et al. Integrative microarray analysis of pathways dysregulated in metastatic prostate cancer. Cancer Res. 2007;67:10296–10303. doi: 10.1158/0008-5472.CAN-07-2173. [DOI] [PubMed] [Google Scholar]
- 35.McCall P, Bennett L, Ahmad I, Mackenzie LM, Forbes IW, Leung HY, et al. NFkappaB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J of Cancer. 2012;107:1554–1563. doi: 10.1038/bjc.2012.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gannon PO, Lessard L, Stevens LM, Forest V, Begin LR, Minner S, et al. Large-scale independent validation of the nuclear factor-kappa B p65 prognostic biomarker in prostate cancer. European J of Cancer. 2013;49:2441–2448. doi: 10.1016/j.ejca.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 37.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Altuwaijri S, Deng F, Chen L, Lal P, Bhanot UK, et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am J Pathol. 2009;175:489–499. doi: 10.2353/ajpath.2009.080727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008;68:6762–6769. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin R, Sterling JA, Edwards JR, DeGraff DJ, Lee C, Park SI, et al. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PloS One. 2013;8:e60983. doi: 10.1371/journal.pone.0060983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claessens F, Denayer S, Van Tilborgh N, Kerkhofs S, Helsen C, Haelens A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nuclear Receptor Signaling. 2008;6:e008. doi: 10.1621/nrs.06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diessenbacher P, Hupe M, Sprick MR, Kerstan A, Geserick P, Haas TL, et al. NF-kappaB inhibition reveals differential mechanisms of TNF versus TRAIL-induced apoptosis upstream or at the level of caspase-8 activation independent of cIAP2. J Invest Dermatol. 2008;128:1134–1147. doi: 10.1038/sj.jid.5701141. [DOI] [PubMed] [Google Scholar]
- 43.Leverkus M, Sprick MR, Wachter T, Denk A, Brocker EB, Walczak H, et al. TRAIL-induced apoptosis and gene induction in HaCaT keratinocytes: differential contribution of TRAIL receptors 1 and 2. J Invest Dermatol. 2003;121:149–155. doi: 10.1046/j.1523-1747.2003.12332.x. [DOI] [PubMed] [Google Scholar]
- 44.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Investigation. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 45.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.di Sant’Agnese PA. Neuroendocrine differentiation in prostatic carcinoma: an update. Prostate Suppl. 1998;8:74–79. [PubMed] [Google Scholar]
- 48.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd CPPRAADAAV, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 49.Taplin ME, George DJ, Halabi S, Sanford B, Febbo PG, Hennessy KT, et al. Prognostic significance of plasma chromogranin a levels in patients with hormone-refractory prostate cancer treated in Cancer and Leukemia Group B 9480 study. Urology. 2005;66:386–391. doi: 10.1016/j.urology.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 50.Berruti A, Mosca A, Tucci M, Terrone C, Torta M, Tarabuzzi R, et al. Independent prognostic role of circulating chromogranin A in prostate cancer patients with hormone-refractory disease. Endocrine-Related Cancer. 2005;12:109–117. doi: 10.1677/erc.1.00876. [DOI] [PubMed] [Google Scholar]
- 51.Akfirat C, Zhang X, Ventura A, Berel D, Colangelo ME, Miranti CK, et al. Tumour cell survival mechanisms in lethal metastatic prostate cancer differ between bone and soft tissue metastases. J of Pathology. 2013;230:291–297. doi: 10.1002/path.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X, Wang Y, DeGraff DJ, Wills ML, Matusik RJ. Wnt/beta-Catenin activation promotes prostate tumor progression in a mouse model. Oncogene. 2011;30:1868–1879. doi: 10.1038/onc.2010.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin RJ, Wang Y, Masumori N, Ishii K, Tsukamoto T, Shappell SB, et al. NE-10 neuroendocrine cancer promotes the LNCaP xenograft growth in castrated mice. Cancer Res. 2004;64:5489–5495. doi: 10.1158/0008-5472.CAN-03-3117. [DOI] [PubMed] [Google Scholar]
- 55.Morris MJ, Kelly WK, Slovin S, Ryan C, Eicher C, Heller G, et al. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. J of Urology. 2007;178:2378–2383. doi: 10.1016/j.juro.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 56.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J of Clin Oncology. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 57.Kraft AS, Garrett-Mayer E, Wahlquist AE, Golshayan A, Chen CS, Butler W, et al. Combination therapy of recurrent prostate cancer with the proteasome inhibitor bortezomib plus hormone blockade. Cancer Biology & Therapy. 2011;12:119–124. doi: 10.4161/cbt.12.2.15723. [DOI] [PubMed] [Google Scholar]
- 58.Lin DL, Tarnowski CP, Zhang J, Dai J, Rohn E, Patel AH, et al. Bone metastatic LNCaP-derivative C4-2B prostate cancer cell line mineralizes in vitro. Prostate. 2001;47:212–221. doi: 10.1002/pros.1065. [DOI] [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 60.Everhart MB, Han W, Sherrill TP, Arutiunov M, Polosukhin VV, Burke JR, et al. Duration and Intensity of NF-kappaB Activity Determine the Severity of Endotoxin-Induced Acute Lung Injury. J Immunol. 2006;176:4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 61.Tuominen VJ, Ruotoistenmaki S, Viitanen A, Jumppanen M, Isola J. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Br Cancer Res. 2010;12:R56. doi: 10.1186/bcr2615. doi: 10.1186/bcr2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.