

Abstract
Background
Wilms tumor is the most common childhood renal tumor. While the majority of patients with favorable histology Wilms Tumor (FHWT) have good outcomes, some patients still experience recurrence and death from disease. This study’s goal was to determine if tumor-specific chromosome 1q gain is associated with event-free (EFS) and overall survival (OS) in FHWT.
Methods
Unilateral FHWT samples were obtained from patients enrolled on National Wilms Tumor Study-4 and Pediatric Oncology Group 9046, “A Molecular Genetic analysis of Wilms Tumor.” 1q gain, 1p loss, and 16q loss were determined using multiplex ligation-dependent probe amplification (MLPA).
Results
The eight-year EFS was 87% (95% CI 82%, 91%) for the entire cohort of 212 patients. Tumors of 58/212 patients (27%) displayed 1q gain. A strong relationship between 1q gain and 1p/16q loss was observed. The eight-year EFS was 76% (95% CI 63%, 85%) for those with 1q gain and 93% (95% CI 87%, 96%) for those lacking 1q gain (p=0.0024). The eight-year OS was 89% (95% CI 78%, 95%) for those with 1q gain, and 98% (95% CI 94%, 99%) for those lacking 1q gain (p=0.0075). Gain of 1q did not correlate with disease stage (p=0.16). After stratification for stage, 1q gain was associated with significantly increased risk of recurrence (risk ratio estimate: 2.72, p=0.0089).
Conclusions
Gain of 1q may provide a valuable prognostic marker to stratify therapy for patients with FHWT. A confirmatory study is necessary before this biomarker is incorporated into risk stratification schema of future therapeutic studies.
Keywords: Wilms tumor, prognosis, chromosome 1, kidney, pediatrics
Introduction
Wilms tumor is the most common primary renal tumor occurring in childhood, with approximately 500 cases identified each year in the United States.1 The majority of Wilms tumors occur in children younger than five years of age, and are characterized by a triphasic histologic pattern consisting of blastemal, stromal, and epithelial elements. A small minority of Wilms tumors contain anaplastic histology consisting of nuclear enlargement, nuclear atypia, and irregular mitotic figures, which is associated with increased relapse risk 2–3. The absence of anaplastic features identifies patients with favorable histology Wilms tumor (FHWT), which represents the patient population analyzed in this study.
Multidisciplinary collaboration and research through the National Wilms Tumor Study Group (NWTSG), now the Renal Tumor Committee of the Children’s Oncology Group (COG), as well as the European pediatric cooperative groups has led to dramatic improvements in survival for the majority of patients with FHWT.4–10 Multiple patient and tumor factors have been identified as prognostically significant in the NWTSG/COG patient cohorts, including tumor histology,2 disease stage,11 tumor-specific loss of heterozygosity (LOH) for chromosomes 1p and 16q,10,12 and patient age and tumor weight.13–15 While the majority of patients with FHWT have good outcomes, many patients still experience recurrence and death from disease, even among patients with lower disease stages. Also, present therapeutic approaches expose patients to significant risk for both immediate and late morbidity and mortality, including cardiac16 and hepatic toxicity,17 secondary malignancies,18 and pregnancy complications.19 Additional prognostic factors are necessary to prospectively identify those patients at the time of diagnosis who are at greater risk for recurrence. LOH of 1p and 16q in Wilms tumor was first described from a cohort of NWTSG patients enrolled on the third and fourth National Wilms tumor studies (NWTS-3 and -4) which demonstrated inferior outcomes for patients with LOH for 16q, and trends toward inferior outcome for patients with LOH for 1p.12 Further work using prospectively gathered samples as part of the 5th National Wilms tumor study (NWTS-5) provided compelling data that patients with combined LOH for 1p and 16q had inferior event-free and overall survival, regardless of stage of disease. However, only 4.6% (76/1656) of FHWT patients had tumors with combined LOH 1p/16q, and combined LOH for 1p/16q was present in only 9.4% (20/213) of relapses.10
A number of additional studies have identified other genetic changes in FHWT that are associated with outcome. The change that has been consistently reported in all such studies is gain of chromosome 1q.20–31 Despite the strength of the data previously reported, all these studies were performed in convenience samples that were not consistently treated. Further, limited data have been published showing the relationship of 1q gain and outcome within different tumor stages. Importantly, 1q gain and LOH at 1p/16q are not independent events. LOH at 1p and 16q often arises through chromosomal translocations that also result in 1q gain. The goal of the current study was to determine if 1q gain, analyzed using multiplex ligation-dependent probe amplification (MLPA), is associated with event-free and overall survival in a carefully stratified cohort of FHWT patients enrolled on the National Wilms Tumor Study-4 (NWTS-4). The study also aimed to better define the relationship between 1q gain and 1p/16q LOH.
Methods
Clinical Samples
Patients included in this study were drawn from those who registered prospectively after providing informed consent on the Pediatric Oncology Group Study 9046, “A Molecular Genetic analysis of Wilms Tumor.” While some of the PO6 9046 patients were enrolled on NWTS-3, the entire cohort from this study came from patients enrolled on NWTS-4, which represents a slightly different cohort from that reported previously.12 Requirements for inclusion into the current study were the presence of unilateral renal disease, FHWT confirmed by central pathology review, and registration with full eligibility and follow-up on NWTS-4.
Multiplex Ligation-Dependent Probe Amplification (MLPA)
MLPA was performed as previously described using a synthetic probe mixture and minor modifications.32 Gene-specific left and right probes were created using AlleleID 7.70 (Premier Biosoft International, Palo Alto, CA). Probes for 1q were selected to include the minimum region of gain in 1q22-1q23 previously reported 25,30. All probe and primer target-specific sequences are provided with controls in Table 1. Universal forward and reverse sequences were attached to the left and right probes, respectively. The probe sizes were increased by using random sequences to obtain a specific amplicon size. The right probe was modified with an addition of 5′ phosphorylation. The probes were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). A probe master mix contained a final concentration of each left probe of 2.6 nM and of each right probe 2.6 nM. Approximately 250 ng of DNA in a volume of 5 μL was denatured at 98°C for 5 minutes. A mixture of 1.5 μL of MLPA buffer (1.5 M of KCl, 300 mM of Tris-HCl, pH 8.5, 1 mM of EDTA) and 1.5 μL of a probe set (6.9 pM for each hemiprobe) was added. The mixture was heated for 1 minute at 95°C followed by 16 to 18 hours at 60°C to allow the MLPA hemiprobes to hybridize. Next, 32 μL of ligase-65 mixture (dilution buffer containing 2.6 mM of MgCl2, 5 mM of Tris-HCl, pH 8.5, 0.013% nonionic detergents, 0.2 mM of nicotinamide adenine dinucleotide, and 1 U of ligase-65 enzyme) was added to each sample for ligation of hybridized hemiprobes during 15 to 20 minutes of incubation at 54°C, followed by 5 minutes of incubation at 98°C to inactivate the ligase. The amplification step was carried out in a 25 μl reaction volume using: 2.5 μl of 10xPCR Gold buffer, 4 μl of 25 mM MgCl2, 0.25 μl of AmpliTaq Gold (5U/μl) (ABI), 0.5 μl of dNTP, 1 μl of 10 μM universal forward primer and 1 μl of 10 μM universal reverse primer. The universal forward primer has a 6-carboxyfluorescein (FAM) fluorophore attached to the 5′ end which is used to detect the amplicon during peak height analysis. The qPCR cycling conditions were as follows: 37°C for 30 min; 95°C for 10 min; 60°C for 30 s; 95°C for 30 s and 67°C for 30 s (35 cycles); 72°C for 20 min. Analysis of the MLPA PCR products for each gene was carried out on an ABI 3100-Avant genetic analyzer (Applied Biosystems-ABI, Foster City, CA) in a mixture of 10 μL of deionized formamide (ABI), 1 μL of PCR product, and 0.5 μL of marker including a ROX-labeled internal size standard (ROX-500 Genescan, ABI) by using POP 4 polymer (ABI).
Table 1.
Probe and primer target-specific sequences
| Gene | Left Specific Sequence | Right Specific Sequence |
|---|---|---|
| LZIC (1p36) | TGGAGGTTTGTGCAATTTGAGACCGGTC | GGCACTGTGCAGAGATCAGAGTACTAAG |
| CAMTA1 (1p36) | CACTTGTTCATGGGCGCAGCA | AAGAAGAGGGATCCACAGAGCTGGA |
| AJAP1 (1p36) | CTTATTCCTGTGGCCTTCGTGTCTGAGA | AATGGTTTGAAATCTCCTGCTGACTGGC |
| DFFB (1p36) | TCGCGCCTTTGCTTTCCTGAG | CCTTCTGAGTAAGGTAATGTGGTGTCC |
| SETDB1 (1q21) | CGAGTTAACCGCAAGATGGGCTTTCATGTTATC | TATAAGACACCTTGTGGTCTCTGCCTTCGGACA |
| ADAM15 (1q22) | GAAAGAGGCTGGGACACCAACTCCTCCT | TGGAACTTTCACTTCCCGCTGCTGTCTT |
| SMG5 (1q22) | CCTGGCAGGCAGCAAGTACTA | TAATGTGGAAGCCATGTATTGCTACC |
| CACNA1E (1q25) | GCTGTGCGTGTCCTGCGGCCTT | TGAAGCTCGTGTCAGGGATACCTA |
| NFATC3 (16q22) | GAAGTGCAACCTAAAACTCATCATCGAG | CCCATTATGAAACTGAAGGTAGCCGAGG |
| KARS-2 (16q23) | GCATTGATCGAGTCGCCATGTTTCTCAC | GGACTCCAACAACATCAAGGTACGTAGC |
| CDH15 (16q24) | GACGCCTACGACATCAGCCAGCTG | CGTCACCCGACAGCGCTGAGCCT |
| TUBB3 (16q24) | GACCGGACGGTGAGTCAGCCTTAAG | CCCGGCACCAGACCCCTCTGAGGAT |
| FABP1 (2p11)* | CAGTGGACAGTCTGGTCGGCA | GAGCCGCAGGTCAGTCGTGAAGAGG |
| MITF (3p14)* | GTGCGGAAAATTCCATTTGGTGTTCGCC | GGCTGATGTGCAAGTAAAAGCAGGGAAT |
| TSC1 (9q34)* | GCAAGTGCAAAGGCCTTGAGCAAGAAAGAACCA | GTATTCCTGTGTTTGGGAAGACTGGGACTAGAGC |
| RET (10q11)* | GGACAGGCTAGCTAGCTGTGTTAGAAGTAGCAA | TGACAATGACCAAGGACTGCTACACCTCTGATT |
| DIABLO1 (12q34)* | CAGAGCAGACAGAACCGCGGA | GCTTCAGGGTGGAAGATTCGTGGAA |
| FMR1 (X)* | CATTACAGAATACCTCCAGTGAAGGTAGTCGGCT | GCGCACGGGTAAAGATCGTAACCAGAAGAAAGA |
Analysis
After separation by capillary electrophoresis, peaks corresponding to each probe were identified by GeneMapper analysis (ABI). Samples in which the smallest peak was <100 relative fluorescent intensity units were not analyzed. The raw peak area for each probe in each sample was divided by the average raw peak area for all probes in that sample. This normalized peak area was then divided by the normalized peak area of the reference samples. Those control probes with a coefficient of variation (CV) >20% were removed from the analysis. Only those samples with at least three control probes remaining were scored. Test probes >1.25 were considered gained and those <0.75 were considered to be lost. These levels were chosen empirically using the distribution of copy levels of control probes. Gain or loss for a chromosomal region was scored if at least two markers were gained or lost, respectively. Scoring was performed without knowledge of outcome, and without knowledge of 1p and 16q LOH status.
Statistical Analysis
The two end points were eight-year event-free survival (EFS) and overall survival (OS). EFS and OS curves were estimated using the method of Kaplan and Meier33 and compared using the log-rank test.34 Relative risks were calculated using the Cox proportional hazards model.35 Tests of correlation of 1q gain status and patient or disease characteristics were performed using the standard chi-square test for contingency tables.
Results
212 patients met the patient and sample criteria for this study; included were Stage I (82 patients, 39%), Stage II (54 patients, 25%), Stage III (46 patients, 22%), and Stage IV (30 patients, 14%). This distribution is comparable to that seen overall in the NWTSG studies.9 The eight-year EFS estimate was 87% (95% CI 82%, 91%) for the entire cohort of 212 patients. Tumors of 58/212 patients (27%) displayed evidence of gain of 1q using the methods and criteria described. The eight-year EFS was 76% (95% CI 63%, 85%) for those with 1q gain and 93% (95% CI 87%, 96%) for those who lacked 1q gain (p=0.0024, Figure 1). The OS was 89% (95% CI 78%, 95%) for those with 1q gain, and 98% (95% CI 94%, 99%) for those who lacked 1q gain (p=0.0075, Figure 2). There were too few EFS events (27 total) to analyze the effect of 1q gain within stage subsets. However, there was no indication that 1q gain correlated with disease stage, as gain of 1q was identified in 20%, 31%, 37%, and 27% of patients with overall Stages I, II, III, and IV, respectively (p=0.16). Estimating the effect of 1q gain on EFS after stratification for stage, 1q gain was associated with a significant increase in the risk of recurrence (risk ratio estimate: 2.72, p=0.0089). For OS, tumors with 1q gain trended strongly toward an increased risk of death (risk ratio estimate: 3.08). However, this did not reach statistical significance (p=0.067), possibly due to the small number of deaths overall (9 deaths total in cohort).
Figure 1.

Event-free survival stratified for 1q gain
Figure 2.
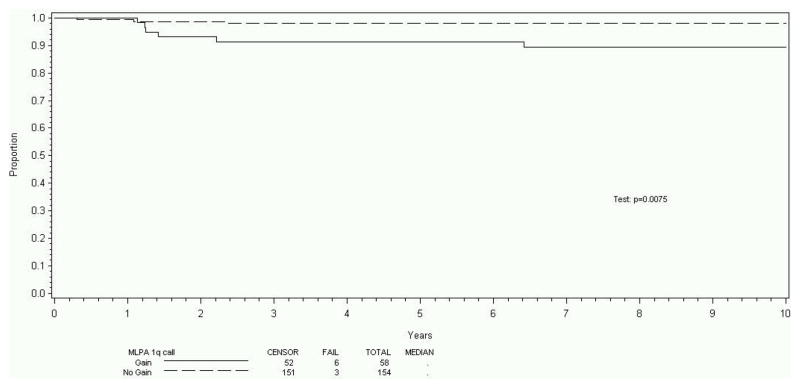
Overall survival stratified for 1q gain
Similarly, the association with outcome was determined for 1p and 16q copy number loss. Retention of both 1p and 16q (no loss at either allele) was identified in 142 patients (estimated EFS 89%, 95% CI: 83%, 93%), isolated 16q loss was identified in 25 patients (estimated EFS 84%, 95% CI: 63%, 94%), isolated 1p loss was identified in 14 patients (estimated EFS 79%, 95% CI: 47%, 93%), and 7 patients had combined 1p and 16q copy number loss (estimated EFS 71%, p=0.29, 95% CI: 26%, 92%). There were too few cases with combined 1p and 16q loss to reliably assess whether this subset was associated with poorer EFS. It is notable that in this study tumors with 1q gain were more likely to have 16q loss (42.6%) or 1p loss (32.5%) than tumors without 1q gain (7.9% and 5.4% respectively, p<0.0001 for both comparisons).
Copy number loss is not always equivalent to LOH, since LOH may not result in a net loss of genetic material. LOH resulting from somatic recombination is copy number neutral, as is LOH resulting from chromosome loss and reduplication of the remaining homologous chromosome. LOH data for 1p and/or 16q (performed using microsatellite analysis and reported previously) were available for many of the tumors analyzed in this study. LOH data were available for 27% and 98% of the samples tested in this study for 1p and 16q copy number, respectively, and the concordance was 95% and 97%, respectively. The discordance was largely due to the identification of copy neutral LOH in 2/7 cases with 1p LOH and 5/35 tumors with 16q LOH. Approximately two-thirds of tumors with 1q gain contained translocations between chromosomes 1 and another partner chromosome (Figure 3A). While a number of different chromosomal partners were seen in the cohort, the most common translocation partner was chromosome 16. The majority of the remaining patients with 1q gain contained the isochromosome 1q, i(1q), which results in gain of 1q and loss of 1p copy number (Figure 3B). Similarly, approximately 40% of patients with 16q loss demonstrate t(1q;16q), all of which retained der(16) and lost der(1) (Figure 3B). Many also contained duplication of the remaining intact chromosome 1, resulting in monosomy 16q, trisomy 1q, and two identical copies of 1p. This results in LOH for 1p (copy neutral) and 16q. Of note, in one-third of tumors, 16q loss was a result of unbalanced translocations with other partner chromosomes. Such translocations involved a mixture of duplications and losses similar to that seen in t(1;16). Twenty-one per cent demonstrated loss of the entire chromosome 16.
Figure 3.

Mechanisms responsible for 1q gain
A. Translocations involving chromosome 1 (the most common also involves chromosome 16). All tumors with t(1;16) examined thus far show loss of the der(1) and retention of the der(16). This results in loss of copy number and LOH for 1p and 16q. Tumors also variably show duplication of the normal chromosomes 1 and/or 16. While this increases the copy number for these chromosomal arms, because there are two exact copies, LOH is seen.
B. Isochromosome 1q results from the development of a derivative chromosome 1 that contains two mirror-image copies of 1q. The normal chromosome 1 remains, resulting in 1q gain and 1p LOH. Again, the normal chromosome 1 is often duplicated, resulting in copy neutral LOH for 1p.
Discussion
The central goal of the Children’s Oncology Group Renal Tumor Committee is to increase survival of patients with FHWT and minimize toxicities associated with therapy. Although the clinical features of patient age and tumor stage are valuable predictors of relapse risk in patients with FHWT, they have limited sensitivity and specificity for recurrence. Novel biomarkers are needed to enhance the current risk stratification schema, thereby allowing augmentation of therapy for patients with high risk of recurrence and reduction of therapy for patients with low risk of recurrence. This study sought to evaluate 1q gain as a possible biomarker for unfavorable prognosis in patients with FHWT.
The association of chromosomal changes with relapse in patients with FHWT has been suggested by a number of publications, all of which show similar findings. In the largest study, 127/195 tumors that subsequently relapsed had abnormal karyotypes.20 The most common changes overall were gain of chromosomes 1q (28%), 8 (24%), and 12 (38%), and loss of 1p (13%), 11q (9%), 16q (19%), and 22 (10%). A stepwise Cox proportional hazards regression demonstrated the significant independent predictors of risk to be 1q gain (relative risk (RR) 3.4, p=0.005), stage IV disease (RR 5.0, p<0.001) and monosomy 22 (RR 5.9, p<0.001). Copy number changes of 1p, 1q, and 16q were often inter-related due to recurrent unbalanced chromosomal abnormalities. This group reported similar rates of unbalanced translocation with chromosome 16 and i1q formation as was seen in the current study.
The NWTSG focused on LOH as a marker for relapse. Analysis of 232 children registered on NWTS-3 and NWTS-4 whose tumors were FHWT or WT with anaplasia demonstrated LOH for markers on 1p and 16q in 12% and 17% of patients, respectively, and each was associated with a poorer RFS and OS when adjusted for stage.12 This association was prospectively confirmed in 1727 informative FHWT registered on NWTS-5.10 A relative risk of relapse of 1.56 and 1.49 was associated with 1p and 16q LOH, respectively, stratified by stage, with similar RR for OS seen. When LOH for 1p and 16q were combined, among the 970 patients with low-stage disease the RR for death was 4.25; and among the 686 patients with advanced disease (stage III, IV) the RR for death was 2.66. On the basis of these studies, children registered on the current COG protocols are stratified according to their combined 1p and 16q LOH status. FHWT patients with combined LOH of 1p and 16q are treated on the current COG renal tumor protocols with intensified therapy with an aim to improve EFS and OS for these patients; those containing both 1p and 16q LOH are treated more aggressively for each stage. However, even if intensified therapy is shown to be beneficial for this group, this will not impact the majority of patients who will relapse, as 1p and 16q LOH is present in only 4.6% of FHWT, and is predictive of only 9.4% of all relapses. The relative risk of relapse associated with LOH of 1p alone or 16q alone was not strong enough to warrant the risks of intensifying therapy in the current COG renal protocols.
In more recent years, classic comparative genomic hybridization (CGH) confirmed the most frequent alterations in FHWT to be gain of 1q, 8, and 12, and loss of 1p, 11p, 16q, and 22.25 CGH analysis found only gain of 1q to be significantly associated with adverse outcome. Gain of 1q was observed in 27/46 relapsed FHWT (59%) compared with 5/21 (24%) 1q gain in non-relapsed FHWT (p =0.019, RR 2.5). While most tumors with 1q gain showed gain of the entire long arm, six tumors demonstrated gain of smaller regions, with the smallest region of common gain spanning 1q21-25. In eight cases (25%), 1p loss coexisted with 1q gain; and, of 27 relapse cases with 1q gain, corresponding loss of either 1p or 16q was identified in 26% and 37% of cases, respectively. These observations support a strong association between 1q gain and poor outcome, and suggest that the unbalanced chromosomal abnormalities mentioned above result in 1q gain. These observations were confirmed by the analysis of 76 FHWT using BAC array CGH, which further narrowed the recurrent region of gain to 1q22-1q23.25
A major limitation of the above studies was that none was performed in a prospectively identified patient cohort consistently treated on a cooperative group protocol. In the current study, the cohort was derived from a prospective clinical and biologic trial, and analyzed 1q gain using MLPA, which is capable of measuring both gain and loss of potential biomarkers.32 MLPA is robust, flexible, can be reliably multiplexed, and is inexpensive. It does not require a source for comparison germline DNA and can be performed on archival tissue. MLPA has become rapidly accepted in the research community, and has entered the clinical realm. This study included probes within the minimal region of gain 1q22-23 (Table 1), and was also able to include probes to 1p and 16q in order to correlate 1q gain with 1p and 16 loss.
This study reports that gain of 1q was associated with a 17% absolute reduction (p=0.0024) in eight-year RFS and a 9% absolute reduction (p=0.0075) in eight-year OS. Further, 1q gain was distributed relatively evenly among tumors of disease stages I-IV. While true within-stage analysis was not possible due to the small sample size for each disease stage, the prognostic significance of 1q gain remained (relapse RR 2.72, p=0.0089) when controlling for disease stage. The RR of death was slightly below usual significance levels (RR 3.07, p=0.067) but analysis was limited by the small number of deaths in the overall cohort (9 deaths of 212 subjects). Further, this study confirms a strong association between 1q gain and 1p and 16q loss. The important gene or genes on 1q that contribute to the reduction in survival remains an enigma.
In conclusion, these findings suggest that 1q gain may indeed be useful to stratify therapy in future therapeutic trials for FHWT. This will require validation in an independent set of tumors. This will be accomplished utilizing >1400 patients registered on NWTS-5. This larger group of patients will enable the evaluation of smaller subsets based not only within stage, but also on 1p and 16q copy number and LOH status. While the overall prognosis for FHWT is good compared to many other malignant diseases, there is significant variation among patients at higher stages of disease.36 Roughly 15% of stage III and 25% of stage IV FHWT patients will experience relapse with current standard therapy.10 Given the significant heterogeneity of patients especially within stage III and IV FHWT, the addition of 1q gain to the existing prognostic framework of clinical, pathologic, and biologic features has the potential to substantially improve the accuracy of risk stratification for appropriate therapy selection across all stages of FHWT. As a result, it may be possible to not only intensify therapy early for those patients at higher risk of relapse or death but also to decrease therapy to those patients with excellent RFS and OS who lack markers of high risk disease. Given this significantly higher percentage of patients with 1q gain compared with LOH 1p and/or 16q, 1q gain has the potential to lead to significant improvement in patient outcomes if similar results are seen in validation studies.
Acknowledgments
Funding:
Research was supported by grants from the National Institutes of Health to EP (CA155556A), to the National Wilms Tumor Study Group (CA-42326), the National Wilms Tumor Study Group Late Effects Study (CA-54498), and the Children’s Oncology Group (CA-98543 and CA-98413).
Footnotes
Disclosures:
No author has disclosures to report related to this project.
References
- 1.Bernstein L, Linet M, Smith MA, Olshan AF. Renal Tumors. In: Ries LAG, Smith MA, Gurney JG, et al., editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995, National Cancer Institute, SEER Program. NIH Pub. No. 99-4649. Bethesda, MD: 1999. pp. 79–90. [Google Scholar]
- 2.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumor. Cancer. 1978;41(5):1937–1948. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 3.Faria P, Beckwith JB, Mishra K, et al. Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol. 1996;20:909–920. doi: 10.1097/00000478-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Gratias EJ, Dome JS. Current and Emerging Chemotherapy Treatment Strategies for Wilms Tumor in North America. Pediatr Drugs. 2008;10(2):115–124. doi: 10.2165/00148581-200810020-00006. [DOI] [PubMed] [Google Scholar]
- 5.D’Angio GJ, Evans AE, Breslow NE, et al. The Treatment of Wilms’ Tumor, Results of the National Wilms Tumor Study. Cancer. 1976;38(2):633–646. doi: 10.1002/1097-0142(197608)38:2<633::aid-cncr2820380203>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 6.D’Angio GJ, Evans A, Breslow N, et al. The Treatment of Wilms’ Tumor: Results of the Second National Wilms’ Tumor Study. Cancer. 1981;47(9):2302–2311. doi: 10.1002/1097-0142(19810501)47:9<2302::aid-cncr2820470933>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 7.D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ Tumor, Results of the Third National Wilms’ Tumor Study. Cancer. 1989;64(2):349–360. doi: 10.1002/1097-0142(19890715)64:2<349::aid-cncr2820640202>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 8.Green DM, Breslow NE, Beckwith JB, et al. Comparison Between Single-Dose and Divided-Dose Administration of Dactinomycin and Doxorubicin for Patients With Wilms’ Tumor: A Report From the National Wilms’ Tumor Study Group. J Clin Oncol. 1998;16(1):237–245. doi: 10.1200/JCO.1998.16.1.237. [DOI] [PubMed] [Google Scholar]
- 9.Green DM, Breslow NE, Beckwith JB, et al. Effect of Duration of Treatment on Treatment Outcome and Cost of Treatment for Wilms’ Tumor: A Report From the National Wilms’ Tumor Study Group. J Clin Oncol. 1998;16(12):3744–3751. doi: 10.1200/JCO.1998.16.12.3744. [DOI] [PubMed] [Google Scholar]
- 10.Grundy PE, Breslow NE, Li S, et al. Loss of Heterozygosity for Chromosomes 1p and 16q Is an Adverse Prognostic Factor in Favorable Histology Wilms Tumor: A Report From the National Wilms Tumor Study Group. J Clin Oncol. 2005;23(29):7312–7321. doi: 10.1200/JCO.2005.01.2799. [DOI] [PubMed] [Google Scholar]
- 11.Breslow NE, Palmer NF, Hill LR, Buring J, D’Angio GJ. Wilms’ tumor: Prognostic factors for patients without metastases at diagnosis, Results of the National Wilms’ Tumor Study. Cancer. 1978;41(4):1577–1589. doi: 10.1002/1097-0142(197804)41:4<1577::aid-cncr2820410448>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Grundy PE, Telzerow PE, Breslow N, Moksness J, Huff V, Paterson MC. Loss of Heterozygosity for Chromosomes 16q and 1p in Wilms’ Tumors Predicts an Adverse Outcome. Cancer Res. 1994;54:2331–2333. [PubMed] [Google Scholar]
- 13.Green DM, Beckwith JB, Weeks DA, Moksness J, Breslow NE, D’Angio GJ. The Relationship between Microsubstaging Variables, Age at Diagnosis, and Tumor Weight of Children with Stage I/Favorable Histology Wilms’ Tumor. Cancer. 1994;74(6):1817–1820. doi: 10.1002/1097-0142(19940915)74:6<1817::aid-cncr2820740626>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 14.Breslow N, Sharples K, Beckwith JB, et al. Prognostic Factors in Nonmetastatic, Favorable Histology Wilms’ Tumor, Results of the Third National Wilms’ Tumor Study. Cancer. 1991;68(11):2345–2353. doi: 10.1002/1097-0142(19911201)68:11<2345::aid-cncr2820681103>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 15.Garcia M, Douglass C, Schlosser JV. Classification and prognosis in Wilms’ Tumor. Radiology. 1963;80:574–580. doi: 10.1148/80.4.574. [DOI] [PubMed] [Google Scholar]
- 16.Green DM, Grigoriev YA, Nan B, et al. Congestive Heart Failure After Treatment for Wilms’ Tumor: A Report From the National Wilms Tumor Study Group. J Clin Oncol. 2001;19(7):1926–1934. doi: 10.1200/JCO.2001.19.7.1926. [DOI] [PubMed] [Google Scholar]
- 17.Warwick AB, Kalapurakal JA, Ou SS, et al. Portal Hypertension in Children with Wilms’ Tumor: A Report From the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys. 2010;77(1):210–216. doi: 10.1016/j.ijrobp.2009.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breslow NE, Lange JM, Friedman DL, et al. Secondary malignant neoplasms after Wilms tumor: an international collaborative study. Int J Cancer. 2010;127:657–666. doi: 10.1002/ijc.25067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green DM, Lange JM, Peabody EM, et al. Pregnancy Outcome After Treatment for Wilms Tumor: A Report From the National Wilms Tumor Long-Term Follow-Up Study. J Clin Oncol. 2010;28(17):2824–2830. doi: 10.1200/JCO.2009.27.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bown N, Cotterill SJ, Roberts P, et al. Cytogenetic Abnormalities and Clinical Outcomes in Wilms Tumor: A Study by the U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. Med Pediatr Oncol. 2002;38:11–21. doi: 10.1002/mpo.1258. [DOI] [PubMed] [Google Scholar]
- 21.Williams RD, Al-Saadi R, Natrajan R, et al. Molecular Profiling Reveals Frequent Gain of MYCN and Anaplasia-Specific Loss of 4q and 14q in Wilms Tumor. Genes Chromosomes Cancer. 2011;50:982–995. doi: 10.1002/gcc.20907. [DOI] [PubMed] [Google Scholar]
- 22.MdZin R, Murch A, Charles A. Cytogenetic findings in Wilms’ tumour: a single institute study. Pathology. 2010;42(7):643–649. doi: 10.3109/00313025.2010.522171. [DOI] [PubMed] [Google Scholar]
- 23.Kullendorff, Soller M, Wiebe T, Mertens F. Cytogenetic findings and clinical course in a consecutive series of Wilms tumors. Cancer Genetics Cytogenetics. 2003;140:82–87. doi: 10.1016/s0165-4608(02)00635-0. [DOI] [PubMed] [Google Scholar]
- 24.Natrajan R, Little SE, Sodha N, et al. Analysis by array CGH of genomic changes associated with the progression or relapse of Wilms’ tumour. J Pathol. 2007;211:52–59. doi: 10.1002/path.2087. [DOI] [PubMed] [Google Scholar]
- 25.Natrajan R, Williams RD, Hing SN, et al. Array CGH profiling of favourable histology Wilms tumours reveals novel gains and losses associated with relapse. J Pathol. 2006;210:48–58. doi: 10.1002/path.2021. [DOI] [PubMed] [Google Scholar]
- 26.Huang C-C, Gadd S, Breslow N, et al. Predicting Relapse in Favorable Histology Wilms Tumor Using Gene Expression Analysis: A Report from the Renal Tumor Committee of the Children’s Oncology Group. Clin Cancer Res. 2009;15(5):1770–1778. doi: 10.1158/1078-0432.CCR-08-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams RD, Hing SN, Greer BT, et al. Prognostic Classification of Relapsing Favorable Histology Wilms Tumor Using cDNA Microarray Expression Profiling and Support Vector Machines. Genes Chromosomes Cancer. 2004;41:65–79. doi: 10.1002/gcc.20060. [DOI] [PubMed] [Google Scholar]
- 28.Rassekh SR, Chan S, Harvard C, Dix D, Qiao Y, Racjan-Separovic E. Screening for submicroscopic chromosomal rearrangements in Wilms tumor using whole-genome microarrays. Cancer Genetics Cytogenetics. 2008;182:84–94. doi: 10.1016/j.cancergencyto.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Steenman M, Redeker B, de Meulemeester M, et al. Comparative genomic hybridization analysis of Wilms tumors. Cytogenet Cell Genet. 1997;77:296–303. doi: 10.1159/000134602. [DOI] [PubMed] [Google Scholar]
- 30.Hing S, Lu Y-J, Summersgill B, et al. Gain of 1q Is Associated with adverse Outcome in Favorable Histology Wilms’ Tumors. Am J Pathol. 2001;158(2):393–398. doi: 10.1016/S0002-9440(10)63982-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Natrajan R, Little SE, Reis-Filho JS, et al. Amplification and Overexpression of CACNA1E Correlates with Relapse in Favorable Histology Wilms’ Tumors. Clin Cancer Res. 2006;12(24):7284–7293. doi: 10.1158/1078-0432.CCR-06-1567. [DOI] [PubMed] [Google Scholar]
- 32.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 34.Peto R, Peto J. Asymptotically Efficient Rank Invariant Test Procedures. J R Stat Soc Ser A. 1972;135(2):185–207. [Google Scholar]
- 35.Cox DR. Regression Models and Life-Tables. J R Stat Soc Series B Stat Methodol. 1972;34(2):187–220. [Google Scholar]
- 36.Spreafico F, Gandola L, D’Angelo P, et al. Heterogeneity of disease classified as stage III in Wilms Tumor: A Report from the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) Int J Radiat Oncol Biol Phys. 2012;82(1):348–354. doi: 10.1016/j.ijrobp.2010.09.022. [DOI] [PubMed] [Google Scholar]