

Abstract
In schizophrenia, cognitive overload is thought to reflect an inability to suppress non-salient information, a process which is studied using prepulse inhibition of the startle response. Prepulse inhibition is reduced in schizophrenia and routinely tested in animal models and pre-clinical trials of antipsychotic drugs. However the underlying neuronal circuitry is not well understood. We used a novel genetic screen in larval zebrafish to reveal the molecular identity of neurons that are required for prepulse inhibition in fish and mice. Ablation or optogenetic silencing of neurons with developmental expression of the transcription factor Gsx1 produced profound defects in prepulse inhibition in zebrafish, and prepulse inhibition was similarly impaired in Gsx1 knockout mice. Gsx1 expressing neurons reside in the dorsal brainstem and form synapses closely apposed to neurons which initiate the startle response. Surprisingly brainstem Gsx1 neurons are primarily glutamatergic despite their role in a functionally inhibitory pathway. As Gsx1 plays an important role in regulating interneuron development in the forebrain, these findings reveal a molecular link between control of interneuron specification and circuits which gate sensory information across brain regions.
Keywords: prepulse inhibition, startle, Gsx1, interneuron, glutamatic acid, zebrafish
Introduction
Schizophrenia is a severe neurological disorder whose etiology includes an important but poorly understood neurodevelopmental component 1. A direct consequence of abnormal neural development may be cognitive impairments, which are observed in young children well before the onset of psychosis 2–3. Cognitive deficits result in part from non-salient thoughts and sensory information flooding the mind with irrelevant percepts due to impaired central filtering mechanisms 4–6. Neuronal filtering mechanisms are studied using prepulse inhibition of the startle reflex (PPI), a paradigm in which a weak ‘prepulse’ occurring 10–500 ms before a sudden intense stimulus suppresses the startle response 7–8. Because disrupted PPI is observed in individuals at high risk for developing psychosis, abnormalities may reflect disturbances in neural development that predispose these individuals to disease 9.
Genetic analyses of large patient cohorts are now yielding many candidate genes which contribute to schizophrenia risk making new tools for investigating the causal role of these genes essential 10. Animal models with a well defined neuronal substrate for disease-relevant behaviors are an invaluable asset for investigating how genetic or environmental disturbances lead to neurological impairment. Supporting the use of PPI as an endophenotype which can be investigated using animal models, PPI defects in schizophrenia are present in prodromal phases of the disease and in unaffected relatives 11–12. Measurements of PPI in animal models are a standard method for analyzing the effects of genetic and pharmacological manipulations. However neuronal circuitry which mediates PPI is not fully understood, limiting the insights that these models can offer. Defining the precise identity and pattern of synaptic connectivity of neurons required for PPI would thus illuminate a substrate for investigating the effects of gene mutations which increase risk for schizophrenia and promote rational design of therapeutic interventions 13.
Systems neuroscience approaches have shown that core circuitry for PPI resides in the brainstem and that partially overlapping mechanisms mediate PPI at different interstimulus intervals (ISIs) between the prepulse and the startling stimulus 14–16. However detailed analysis of neuronal mechanisms for PPI in mammals remains a formidable challenge 17. In contrast startle circuitry in fish is relatively simple and is therefore an attractive system in which to dissect neuronal connectivity. Acoustic/vibrational stimuli elicit rapid escape responses triggered by the Mauthner cells, a single bilateral pair of giant reticulospinal neurons which are similar in position and function to the central brainstem neurons for the mammalian acoustic startle response (ASR) 18–19. Because Mauthner-driven startle responses are all-or-nothing events, PPI is manifest as a reduction in the probability of eliciting a startle response, rather than a reduction in the magnitude of the response as in mammals 20, 21. PPI is robustly induced in larval zebrafish and reproduces salient features of the response in mammals, including the effective ISI and susceptibility to drugs that alter dopaminergic and glutamatergic neurotransmission 20, 22. However the identity of neurons which inhibit the startle response during PPI remains unknown. Here we exploited the amenability to genetic screens and less complex brain structure of larval zebrafish to reveal the molecular identity of neurons required for PPI in this species. We then extended our work to behavioral analysis of a mouse model to confirm that our findings are pertinent to mammals.
Methods
Zebrafish husbandry
Construction of the transgene used to recover enhancer trap line Et(REx2-SCP1:Gal4ff)y252 has been reported 23 but briefly, this transgene contains the reduced toxicity Gal4 variant Gal4ff 24 driven by a minimal promoter consisting of tandem NRSE elements (REx2) and the supercore promoter 1 (SCP1) 25. The UAS:Kaede transgenic line is Tg(UAS:Kaede)s1999t 26. J1229 is Gt(T2KSAG)j1229a 27. 4xUAS:GFP is Tg(4xUASnr:GFP)c369 28. vGlut:DsRed is TgBAC(vglut2.1:DsRed) and glyt2:GFP is TgBAC(glyt2:GFP) 29. Tg(UAS:nfsB-mCherry) fish were generated as previously reported 26. The UAS:Arch3 transgenic line was generated from a 14xUAS-E1b:Arch3-TS-CFP-ER transgene which was constructed as follows. Zebrafish codon optimized cerulean (Genscript) was amplified by PCR, adding the Kir2.1 membrane trafficking signal (TS) at the N-terminal and an endoplasmic reticulum (ER) export signal at the C-terminal as described for eNpHR3.0 30, then cloned into pT1UMP 31. SS-PRL-Archaerhodopsin-3 32 was synthesized, codon optimized for zebrafish (Genscript) then amplified by PCR and cloned in frame with the TS-CFP-ER cassette. The plasmid was injected into one cell stage embryos with Tol1 transposase 33 and founders screened to isolate transgenic line Tg(UAS-E1b:Arch3-TS-CFP-ER)y259. The UAS:lynTagRFPT line is Tg(UAS-E1b:BGi-lynTagRFPT)y260 which was made using Tol1 transgenesis using a previously described plasmid 31. For the UAS:Synaptophysin-RFP line overlapping extension PCR was used to fuse synaptophysin, amplified from UAS:Synaptophysin-EGFP 34 and zebrafish optimized TagRFPT. The product was cloned into pT1UMP and injected as above to isolate line Tg(UAS-E1b:Synaptophysin-TagRFPT)y261. For the UAS:nls-GFP line zebrafish codon optimized emerald GFP was synthesized (Genscript), PCR amplified adding the SV40 large T antigen nuclear localization sequence (MAPKKKWKV) to the N-terminal, cloned into pT1UMP and injected to isolate line Tg(UAS-E1b:BGi-NLS-emGFP)y262. All lines were maintained on a Tubingen long fin background and embryos raised in E3 medium supplemented with 1.5 mM HEPES pH 7.3 (E3/h) at 28°C on a 14:10 light:dark cycle. Additional details of plasmid construction are available on request. All zebrafish protocols were approved by the NICHD animal care and use committee.
Mice
Gsx1 knockout mouse heterozygotes 35(kind gift of K. Campbell) were maintained on a CD1 background as in previous generations. Heterozygous mice were incrossed to non-littermates to generate litters of Gsx1+/+, Gsx1+/−, Gsx1−/− siblings for behavioral testing. Using the AIMS neonate ID system, sibling mice were genotyped at postnatal day (P)7–10 by PCR analysis of tail DNA extracted using the DirectPCR Tail buffer system and the manufacturer’s protocol (Viagen). Genotyping primers were: CGGGTGAAGCACAAGAAAGAAG; CCAATGGTCCTCTAAAAGGCG; GGTTCATCATCACTAATCACGACG; CGCTGTTCTCCCTCTTCCTCATCTC resulting in a 220 bp band for the wild type allele and a 150 bp band for the knockout allele. Litters were maintained at no more than 8 pups per dam to promote mutant survival and to match group size as an additional control. All experimental protocols were approved by the NICHD animal care and use committee.
Transgene mapping
Linker mediated PCR 36 was used to map the insertion site in y252. The site was confirmed by PCR on single embryos using chromosome 5 specific primer 5-TGCTTGTTGCTTGTTTTTGC with primer 5-TTGAGTAGCGTGTACTGGCATT, specific to one of the Tol2 arms yielding a 347 bp band for the transgene locus. A single band was present in all Kaede positive embryos from a y252; UAS:Kaede outcross to TL (N = 31). For subsequent genotyping of y252, a multiplex PCR strategy was used, with primers 5-AGCAGAAATGTGCATCAAC and 5-TTGCTTGTTTTTGCAGTTGG which produce a 328 bp band for the wildtype genomic locus and a third primer 5-CAAGAATCTCTAGTTTTCTTTCTTGCT, yielding a 221 bp band for the transgene locus.
Zebrafish behavior
For screening Gal4 lines, each line was crossed to a UAS:nfsB-mCherry transgenic line allowing cell specific and temporally controlled ablation using metronidazole treatment 37–38. Sibling larvae were sorted into strongly-expressing mCherry and non-expressing groups and both groups were treated from 3–5 dpf with 10 mM metronidazole in E3/h medium for ablation of nitroreductase-mCherry expressing neurons. Ablation was confirmed visually by checking for loss of patterned mCherry expression. Behavioral testing was performed after 24 h recovery from the treatment, at 6 dpf. Zebrafish larvae show two forms of startle response distinguished by latency and kinematics, Mauthner cell mediated short-latency c-start (SLC) and non-Mauthner long-latency c-start (LLC) responses 20, 39. SLC responses are susceptible to PPI 20 and were therefore the focus of this study. In the initial PPI screen, a 500 ms ISI was used. For Arch3 experiments larvae were embedded in 2 % low melt agarose in a glass bottom Petri dish before the area caudal to the fins was removed so that the tail was free. The dish was mounted in a custom stage printed in ABS plastic using a uPrint rapid prototyping system (Stratasys, Eden Prairie, MN) attached to a compound microscope (Axioimager.A1, Zeiss). Tail movements were imaged with a camera (μEye IDS-1545LE-M, 1stVision, Andover, MA) and objective (infiniGage CW with 0.50x mag precision spacer) mounted in place of the condenser. To determine the latency threshold for distinguishing short-latency startle responses in head fixed embryos, larvae were tested 20 times with a vibrational stimulus and responses recorded at 200 Hz. Images were scored manually to determine the earliest time of tail movement. A 10X objective was used to focus light pulses (520 nm) delivered by a high power LED (UHP-Mic-LED-520, Prizmatix, Israel) onto the head of the larva. Infra-red illumination was used to visualize the tail, with light used for the Arch3 stimulus excluded with a long pass filter in the camera objective (NT54-662, Edmunds). Stimulus timing and image acquisition was coordinated using a digital-analog card (PCI-6221, National Instruments, Austin, TX) controlled by DAQtimer event control software 20. 520 nm illumination was started 500 ms prior to the stimulus to inhibit y252 neurons. To exclude trials in which larvae were engaged in spontaneous locomotor activity during presentation of the startle stimulus, images were collected 32 and 16 ms before the startle stimulus to check for tail movements. An image was taken 20 ms after the startle stimulus to identify short latency startle responses and another image 80 ms after the pulse to identify long latency startle responses. For PPI trials, the pulse was delivered 500 ms after the prepulse. LED illumination was present over the course of the entire trial when used (642 ms for startle, 1110 ms for PPI). Inter-trial intervals were 20 s long, and conditions were presented in a pseudorandom order across the experiment. Control trials included LED alone trials and no stimulus trials. Startle response, PPI experiments, baseline spontaneous movement, dark flash responses, kinematic analysis of swimming and the visual motor response were measured as previously described 20, 40, 41. For the MK801 experiment, 6 dpf larvae were treated in groups of 20 per 6 cm dish with 30 μM MK801 (M107, Sigma) in 1% DMSO for 20 minutes before behavioral testing. Controls received 1% DMSO only.
Mouse behavior
Acoustic startle and PPI were measured using the SR-Lab Startle Response System (San Diego Instruments, San Diego, CA). P20–21 pups were transported in a standard plastic cage with the dam and littermates to a lit holding room just outside of the testing room where they adapted for 30 minutes prior to testing. Two mice were removed at a time from their home cage for analysis, one per available testing chamber. Mice were enclosed so that they could just turn around inside Plexiglas tubes within the lit and ventilated testing chambers and allowed to acclimate for 3 minutes prior to the session. Background noise level was maintained at 65 dB for the duration of the test including during acclimation. For acoustic startle testing, mice were presented with 60 trials using a variable inter trial interval of 15–25 s. Trial types included no stimulus trials and trials of 40 ms bursts of 5 to 55 db above background noise pulses spaced at 5 db increments. Trials were presented across 5 blocks of 12 trials each so that no one trial was repeated in any single block. The trial types were presented in a pseudorandom order over the entire experiment. For PPI testing mice were presented with 5 trial types over 12 blocks with a variable inter trial interval of 15–25 s. Trial types included a 115 db 40 ms pulse alone trial to measure startle amplitude, and PPI trials of 20 ms bursts of a 70 or 90 db prepulse followed by the 40 ms 115 db startling pulse. For testing long ISI PPI we used prepulses presented 100 or 500 ms prior to the pulse. The trial types were presented in pseudorandom order across the experiment except for in the first and last block which contained only 6 pulse alone trials to assess initial baseline startle reactivity and habituation. Pulse alone trials from the first and middle blocks were used to calculate average startle magnitude used in PPI calculations. PPI was calculated as: [(average startle magnitude – average PPI trial startle magnitude)/(average startle magnitude)]*100. For both acoustic startle and PPI testing, startle amplitude was measured every 1 millisecond over a 100 ms period beginning at the onset of the startle stimulus. The maximum startle amplitude over this period was taken as the definitive startle magnitude if having occurred 40 ms or less following the stimulus. In mice, startle responses are measured by whole body movement and the apparent magnitude of the startle response is thus influenced by body weight (Supplementary Fig. 6A). As mutant pups were significantly smaller (Supplementary Fig. 6B), startle magnitude was normalized to body weights (g) recorded on a scale prior to testing. Locomotor activity was assessed by placing mouse pups into the corner of a novel open field chamber measuring 35×35 cm under low room lighting of ~30 lux and monitoring activity for 5 minutes using the ANY-Maze software (SDI, San Diego, CA, U.S.A.). Mice were tested during the second half of their light period. A black cloth was placed underneath four clear chambers arranged on the floor of the testing room to generate contrast, and solid white barriers were placed in between the chambers so that 4 mice could be tested at a time without seeing each other. Each chamber was divided into 9 equal square sections within the video analysis software. The inner most section was demarcated as the ‘center’ zone while the surrounding squares were marked as ‘periphery’. Distance traveled, mobility time, and speed were analyzed across the entire chamber and in each zone. The minimum time immobile used was 5 seconds. Chambers were cleaned with 70% ethanol and dried between mice.
Histochemical techniques
The Gsx1 probe for in situ hybridization in zebrafish was as previously described 42. Antibodies used were against GFP (1:200; A-11122, Invitrogen), Kaede (1:200; PM012, MBL International), TagRFP (1:200; NC9044899, Fisher), Maguk (1:100; 73-029, NeuroMab). Image analysis was performed using ImageJ and Fluorender 43. For in situ hybridization analysis in mouse, E10.5 embryos and E13.5 embryonic brains were prepared by fixation in 4% PFA in PBS followed by cryopreservation in a sucrose gradient and embedding in OCT medium as previously described 44. Frozen sections were taken sagittally through E10.5 embryos at a thickness of 12 μm, and E13.5 brains were sectioned coronally through the brainstem at a thickness of 14 μm. Plasmids to generate in situ probes by in vitro transcription were prepared by amplification of the 3′ end (partial last exon and 3′UTR) of Atoh1 and Gsx1 from mouse genomic DNA and the product subsequently cloned into the pGEMT-easy vector (Promega). Primers are published in the Allen Brain Atlas (http://www.brain-map.org/). The in situ hybridization procedure was essentially as previously described 45. Tissue was post fixed in 4% PFA to stop the NBT/BCIP colorimetric reaction with alkaline phosphatase (AP) and counterstained using 0.005% Nuc Fast Red (Polyscientific) then dehydrated through an EtOH gradient (50, 70, 95, 100%) and Xylene washes. Coverslips were mounted on the slides using Permount (Fisher). Photographs were taken at 5X magnification for E13.5 brains and 10X magnification for E10.5 embryos on a Zeiss Axioplan2 microscope equipped with a Leica DFC490 camera.
Statistical analysis
was performed using SPSS 17.0 (IBM, Armonk, NY) and Gnumeric 1.10.16 (http://projects.gnome.org/gnumeric/). Graphs show mean and standard error. Post-hoc tests were Bonferroni corrected for multiple comparisons as appropriate and homogenous subsets calculated using the Student-Newman-Keuls test.
Results
Transgenic line y252 labels neurons which are required for PPI
In fish, PPI is manifest as a reduction in startle responsiveness when the prepulse precedes the intense stimulus at an ISI of 30–1000 ms, and as in mammals, suppression of the startle response is dependent on the ISI used and the prepulse intensity (Supplementary Fig. 1a)20–21. We used high speed video recording and computational image analysis to assess the responsiveness of zebrafish larvae to brief vibrational stimuli during startle alone and PPI trials 20. To identify neurons required for PPI, we screened a library of transgenic zebrafish enhancer trap lines which express the Gal4 transcription factor in distinct populations of neurons (Fig. 1a). These lines were generated using a technique that restricts Gal4 expression to the nervous system, allowing functional analysis of these genetically targeted neurons in physiology and behavior 23. Neurons labeled in each line were selectively ablated by using Gal4 to activate expression of a cytotoxin from a UAS promoter. After visually confirming ablation efficacy (Fig. 1b), larvae were tested for changes in PPI. After ablation, PPI was reduced by 45% at an ISI of 500 ms in transgenic line Et(REx2-SCP1:Gal4ff)y252 (y252) (Fig. 1c, Supplementary Fig. 1b). Unexpectedly, at ISIs of 30 and 120 ms, PPI was significantly increased (Fig. 1d) indicating that the change in PPI depended on the ISI. y252 labels a prominent bilateral stripe of dorso-medially positioned neurons extending through the entire hindbrain continuous with the spinal cord. Transgene expression was also observed in the optic tectum, ventral forebrain and hypothalamus (Fig. 4b). After neuronal ablation in y252, movement kinematics during the startle response were normal indicating that changes in startle were not due to motor system defects (Supplementary Fig. 2a–d). It is unlikely that larvae failed to detect the prepulse because larvae showed an increase in startle responsiveness (Fig. 1e), and a shorter response latency (Supplementary Fig. 2e). Moreover, because groups of ablated and control larvae matched for startle responsiveness retained significant differences in PPI, these differences could not be attributed to increased startle sensitivity (Fig. 1f). The differential effect of y252 neuron ablation on PPI at long and short ISI implies that different mechanisms mediate PPI during distinct temporal windows, consistent with findings from pharmacological and lesion studies in mammals 16. Ablation caused additional behavioral phenotypes including a loss of responsiveness to a light flash stimulus and reduced baseline swimming activity (Fig. 1g, Supplementary Fig. 2). Despite the low baseline activity, after ablation, larvae showed a significant increase in swim distance after a change in illumination, indicative of hyperactivity (Fig. 1h). Neurons expressing Gal4 in the y252 enhancer trap line are thus required for multiple behaviors including modulation of startle sensitivity and PPI.
Figure 1. Neurons that modulate startle sensitivity and prepulse inhibition are identified by enhancer trap line y252.

(a) Schematic of the screen used to identify neurons required for PPI. Gal4 enhancer trap lines were crossed to a UAS:nfsB-mCherry transgenic line and sorted into mCherry expressing and non-expressing groups. Both groups were treated from 3–5 dpf with metronidazole for ablation of nitroreductase-mCherry expressing neurons. Behavioral testing was performed after 24 h recovery from the treatment. For screening, a 500 ms ISI was used.
(b) y252; UAS:nfsB-mCherry larvae maintained in embryo medium (ctrl) or in medium supplemented by 10 mM metronidazole for 48 h (ablated).
(c) Dorsal confocal projection of y252; UAS:nls-GFP larva showing the distribution of neurons expressing the Gal4 transgene (green), counterstained with DAPI (blue). Inset shows the mean and standard error of %PPI values for y252 control (N=47) and ablated (N=48) larvae. * P < 0.001. Scale bar 50 μm.
(d) Changes in PPI at different ISIs after neuronal ablation in y252; UAS:nfsB-mCherry larvae (open circles) compared to sibling controls (filled). N= 15–32 larvae per group. ANOVA significant interaction between ablation status and interstimulus interval F5,257 = 10.23, p < 0.001. # p < 0.05. * p < 0.01.
(e) Startle responsiveness (percentage of fish initiating a short latency c-start response, %SLC) in y252 ablated larvae (open circles, N = 41) compared to sibling controls (filled, N = 27). Main effect of ablation F1,66 = 26.58, p < 0.001. Comparisons between ablated and controls at all stimulus intensities are significant, p < 0.001.
(f) PPI at 500 ms ISI (top) and 30 ms ISI (bottom) in subsets of y252 ablated larvae (open circles) and sibling controls (filled) with similar startle responsiveness. Larvae were binned according to startle responsiveness (%SLC) in three groups: 30–50, 51–70 and 71–90. N=16–47 larvae per group. t-test * p < 0.01.
(g) Reduced locomotor activity after y252 ablation. Larvae initiate two types of swimming movements, routine turns (R-turns) and slow swims (scoots) both of which are significantly reduced. N = 5 groups controls, 3 groups ablated larvae. * p < 0.001.
(h) Distance travelled (pixels) per swim bout in y252 ablated larvae (Abl, grey) and controls (Ctrl, black) during baseline activity (yellow background) and movement in response to sustained darkness (grey background). N=36 larvae each group. * p < 0.001.
Figure 4. y252 is an enhancer trap for Gsx1.
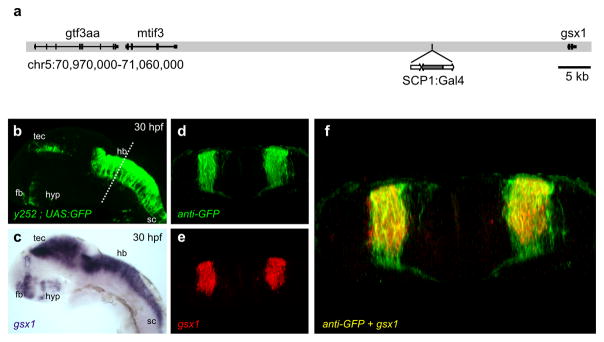
(a) Schematic of the y252 transgene integration site on chromosome 5. Genome edition Zv9.
(b) Lateral epifluorescent image of y252; UAS:GFP embryo at 30 hpf. Forebrain (fb), optic tectum (tec), hypothalamus (hyp), hindbrain (hb), spinal cord (sc).
(c) Lateral image of in situ hybridization for Gsx1 at 30 hpf.
(d–f) Coronal section through the hindbrain in y252; UAS:GFP embryos showing GFP
(d), fluorescent in situ hybridization signal for Gsx1 (e) and single confocal slice merge (f).
Acute silencing of y252 neurons produces startle and PPI defects
To test whether neurons in y252 are directly required for PPI, neuronal excitability was acutely suppressed using the green light-activated proton pump Arch3 32. For these experiments, we established a semi-restrained preparation allowing illumination of the brain through a microscope objective, while using a stage-mounted speaker to provide an acoustic/vibrational stimulus for eliciting startle responses (Fig. 2a, b; Supplementary Fig. 3). Tail movements were monitored using a second objective, allowing us to measure startle responsiveness and PPI. We used y252 to activate expression of a UAS:Arch3-CFP transgene and selected fish with strong CFP expression, and non-fluorescent siblings that lacked either the Gal4 (y252) transgene or the UAS reporter. Because acoustic startle responses are initiated with a latency of less than 20 ms after the stimulus, they were easily distinguished from locomotor responses to the sudden increments in light intensity which have broadly distributed response latencies of 100–500 ms 40. Thus in these experiments, any tail movement observed within 20 ms of the stimulus was classified as a startle response, and illumination alone did not elicit tail movement responses during this window (paired t-test t33 = 0.93, p = 0.36; Fig. 2c). When illuminated y252; UAS:Arch3 larvae showed an increase in startle responsiveness while sibling control larvae were not effected (Fig. 2b, d). We next tested PPI, using a 500 ms ISI as in our initial screen and found that illumination reduced PPI by 38% in y252; UAS:Arch3 larvae but had no effect on siblings (Fig. 2e). Acute and reversible silencing of y252 neurons thus phenocopied the effects of ablating these neurons confirming that transgenic line y252 expresses Gal4 in neurons required for PPI at long ISI and regulation of the startle response threshold.
Figure 2. Acute suppression of activity in y252 neurons reduces PPI.

(a) Apparatus for Arch3 experiments using semi-restrained larvae tested on a microscope stage fitted with a speaker.
(b) Representative example of tail movement responses for an Arch3 expressing larva under baseline conditions (top) and during LED illumination (bottom), during no stimulus trials, prepulse only trials and pulse only trials.
(c) Frequency of tail movement initiation within a 15 ms window during baseline conditions without a stimulus (top) and during LED illumination (bottom). No significant differences were found between Arch3 expressing larvae and controls (N=18, 17 respectively).
(d) Startle responsiveness in Arch3 expressing y252 larvae (N=18, Arch3+) and non-expressing siblings (N=17, Arch3−) when tested while illuminated with intense green light (green) or when not illuminated (black). Repeated measures ANOVA, interaction of light and Arch3 expression, F1,33 = 12.06, p = 0.001. paired t-test * p < 0.001.
(e) PPI during green light illumination (on) or in the dark (off) in Arch3 expressing y252 larvae (Arch3+, N=13, paired t-test t12 = 2.66, * p=0.021) and siblings not expressing the Arch3 transgene (Arch3− N=14, t13 = 1.24, p=0.24).
y252 neurons are primarily glutamatergic and form synapses adjacent to central neurons which initiate the startle response
To analyze how y252 neurons are involved in PPI, we first examined their neurotransmitter profile. Prominent y252 transgene expression was observed in a bilateral longitudinal column of dorso-medially located neurons extending along the rostro-caudal axis of the hindbrain (Fig. 3a). In the hindbrain, neurons with a defined neurotransmitter type are also arranged in distinct longitudinal columns 29, 46–47. In transverse and dorsal sections, columns of glutamatergic, GABAergic and glycinergic neurons are seen to be intercalated with columns occupying different mediolateral positions (for example Fig. 3b) specified by a code of regionally expressed transcription factors. We characterized y252 neurons using transgenic lines which express fluorescent proteins in glutamatergic and glycinergic columns 29, 47. y252 neurons were situated in the second most lateral of the four columns of glutamatergic neurons in the caudal hindbrain (Fig. b–d). Moreover, in triple transgenic y252; UAS:nls-GFP; vGlut:DsRed larvae, GFP and DsRed were strongly co-expressed throughout the rostro-caudal extent of the hindbrain (Fig. 3e–g), and single confocal slices confirmed that y252 neurons were predominantly glutamatergic (Fig. 3j–l). However, scattered y252 neurons showed co-expression with the glycinergic neuron marker, mostly in the caudal hindbrain (Fig. 3h–i and m–o).
Figure 3. Hindbrain neurons in y252 form glutamatergic synapses adjacent to the Mauthner cell.

(a) Projection of a dorsal confocal stack in y252; UAS:nlsGFP; vGlut:DsRed larva showing y252 expression (green) and glutamatergic neurons (red). Scale bar 100 μm.
(b–d) Coronal view of the caudal hindbrain, representing a 4 μm section derived from rotation of a dorsal confocal stack at the rostro-caudal level indicated in (a) showing that y252 GFP+ nuclei (c, green) are located in the second-most lateral of four mediolateral stripes of glutamatergic neurons (b, red, arrowheads). Scale bar 50 μm.
(e–i) Projection of dorsal confocal sections in the region indicated in (a) showing colocalization (g,i white) between y252 neurons (e, green) and glutamatergic neurons (f, red; vGlut:DsRed) or glycinergic neurons (h, red; glyt2:GFP, note that GFP is pseudocolored red). For (i) we used y252; UAS:nfsB-mCherry and pseudocolored the y252 pattern in green after imaging for comparison with (e–g). Scale bar 20 μm.
(j–o) Single confocal sections of the regions indicated in (f) and (h) showing colocalization between y252; UAS:nlsGFP (j, green) and vGlut:dsRed (k, red), and y252; UAS:nfsB-mCherry (m, pseudocolored green) and glyt2:GFP (n, pseudocolored red). Scale bar 10 μm.
(p) Dorsal confocal projection in y252; UAS:lynTagRFPT; J1229 larva showing the Mauthner cell (green, arrowheads) and midline projections of y252 axons (red). J1229 is an enhancer trap line that expresses GFP in the Mauthner cell and other reticulospinal neurons 27. Scale bar 50 μm.
(q) Coronal projection at the region indicated in (p) showing ventral projections from dorsally positioned y252 neurons (red, asterisk) passing beside the lateral dendrite of the Mauthner cell (arrowheads). Inset schematic shows plane of section.
(r–w) y252; UAS:Synaptophysin-RFP; J1229 larva stained with anti-Maguk revealing presynaptic puncta formed by y252 neurons (red), the Mauthner cell (blue) and glutamatergic postsynaptic densities (green).
(t) Closer view of the lateral dendrite outlined in (s). Arrows and dotted circle mark two distinct regions where synapses from y252 neurons are closely opposed to glutamatergic postsynaptic densities. Scale bar 10 μm.
(u–w) Split channels from (t). Colocalization between y252 presynaptic puncta and the Mauthner cell (u, magenta), MAGUK and the Mauthner cell (v, teal) and presynaptic y252 puncta and postsynaptic MAGUK proteins (w, yellow).
(x) PPI in larvae treated with MK-801 (open circles) or vehicle (closed circles). N=15 groups each (20 larvae per group). Main effect of treatment F1,28 = 15.8, p < 0.001; Interaction of treatment and ISI F2,56 = 40.4, p < 0.001. # p = 0.021, * p< 0.001.
(y) Startle responsiveness in larvae treated with MK-801 (open circles) or vehicle (closed circles). N=6 groups of 25 fish each. Main effect of treatment F1,10 = 180.35, p < 0.001. * p < 0.01.
(z) Reaction time (startle latency). N=334 and 343 responses for MK801 and vehicle treated larvae. * p < 0.001.
Next we examined connections between y252 neurons and the Mauthner cell which is localized in the brainstem and is required for short latency startle responses that are inhibited during PPI. Using y252 to drive a membrane-tagged RFP reporter line, we found that axons from y252 hindbrain neurons projected ventrally, running in close apposition to the lateral dendrite of the Mauthner cell and extending beneath the Mauthner cell soma and initial axon segment (Fig. 3p, q). To determine whether synaptic contacts may exist between y252 neurons and the Mauthner cells, presynaptic terminals of y252 neurons were marked using a synaptophysin-RFP reporter. The lateral dendrite of the Mauthner cell strongly colocalized with RFP puncta and additional sparse puncta were also observed on the soma (Fig. 3r). Immunostaining against post-synaptic density proteins confirmed that these puncta represent synapses (Fig. 3s). The post-synaptic side of glutamatergic synapses is demarcated by Maguk family proteins (PSD95/93, SAP-97) 48. Maguk immunofluorescence opposed RFP puncta in two areas on the lateral dendrite of the Mauthner cell (Fig. 3t–w) including a region at the distal tip of the lateral dendrite, which receives acoustic input from neurons of the statoacoustic ganglion 49. These data thus implicate glutamatergic neurons as an intrinsic part of the neuronal mechanism for PPI.
Genetic and pharmacological evidence have implicated dysregulation of glutamate signaling in schizophrenia 50–51 and glutamate receptor antagonists produce schizophrenia-like symptoms including reduced PPI 52. After bath exposure to the NMDA receptor antagonist MK801, larvae showed increased PPI at short ISIs, reduced PPI at long ISIs and increased startle responsiveness (Fig. 3x–z) similar to the phenotype of y252 ablated larvae and consistent with previous findings 20, 53. These results show that y252 neurons form synapses closely adjacent to startle initiating neurons and suggest that mechanisms involving glutamatergic neurotransmission are involved in regulating the startle response during PPI at distinct temporal windows.
y252 delineates neurons which express Gsx1 during embryonic development
Transgenic reporters in enhancer trap lines frequently recapitulate the expression pattern of adjacent genes 54. We used linker-mediated PCR 36 to map the integration site of the Gal4 transgene in y252 to a locus 21 kb upstream of genomic screen homeobox 1 (Gsx1) on chromosome 5 (Fig. 4a). Gsx1 is a homeodomain containing transcription factor with a highly dynamic pattern of expression during neural development. Expression is first detected in the hindbrain and subsequently observed in several regions of the nervous system including the spinal cord, optic tectum, hypothalamus and ventral forebrain in both fish and mice (Supplementary figure 4a–e)42, 55. Throughout development, Gsx1 showed a very similar neuroanatomical pattern of expression to the Gal4 reporter in y252 (Fig. 4b, c). In the spinal cord, Gsx1 specified neurons are reported to comprise a population of dorsally located glutamatergic interneurons as well as a late-born population of GABAergic neurons 56–57, similar to the identity of neurons in the spinal cord in y252 (Supplementary figure 4f–h). Finally, confocal analysis of hindbrain sections of y252 embryos stained for gsx1 and anti-GFP demonstrated that Gsx1 and Gal4 were co-expressed (Fig. 4d–f). Taken together, these data strongly indicate that y252 is an enhancer trap for Gsx1.
Gsx1 knockout mice show impairments in PPI
Gsx1 promotes the maturation of interneurons in the mammalian subpallium, 58 and is downregulated to allow migration of interneurons into the cortical plate 59. Cortical interneuron development is of intense interest for understanding the etiology of schizophrenia 60 and several genome-wide studies for neuropsychiatric disorders including schizophrenia have suggested the presence of vulnerability loci which map near Gsx1 (Supplementary Table 1). To determine whether our finding that neurons with developmental expression of Gsx1 are required for PPI in fish is relevant to neuronal mechanisms of PPI in mammals, we analyzed the expression of Gsx1 in mouse brain regions previously reported to be linked to PPI. We first confirmed the finding that Gsx1 is expressed in longitudinal stripes extending through the developing mouse hindbrain 55. Using in situ hybridization on coronal sections through E13.5 mouse brainstem we observed robust Gsx1 expression in discrete bilaterally symmetric domains at the ventricular zone of the neuroepithelium in the medulla (Fig. 5a–b, d). Next, we examined expression in proliferative zones which give rise to brain regions linked to PPI. Two primarily cholinergic nuclei of the mesopontine tegmentum have been implicated in PPI: the laterodorsal tegmental nucleus and the pedunculopontine tegmental nucleus (PPTg) 61–62. Progenitors for these regions include Atoh1 expressing cells which migrate from the rhombic lip at E10.5 63. In situ hybridization showed that Gsx1 is expressed in a region adjacent to the Atoh1 domain but that these transcription factors are not co-expressed (Supplementary figure 5). Two other areas that may contribute to PPI in mice are the inferior and superior colliculi 64–65. Gsx1 expression was observed at the ventricular zone of the neuroepithelium of both of these regions (Fig. 5c, e). We then analyzed PPI in Gsx1 knockout mice. Because Gsx1 homozygous knockout mice have growth retardation and a high mortality rate after post-natal day 14, it was not possible to study adult homozygous mutants 35. Instead, we analyzed responses in mice at P20–21 which is several days after PPI is first detected 66. When tested with a range of stimulus intensities, homozygotes showed similar weight-normalized startle responsiveness and latency to siblings (Fig. 5f, Supplementary fig. 6a–c), indicating that knockout mice are not hearing impaired. Mutant pups showed a significant overall reduction in PPI compared to littermates (Fig. 5g, Supplementary Figure 6d). PPI in heterozygotes was not significantly different from wildtypes at this stage, nor in 8 week old juvenile mice (Supplementary fig. 6e). In posthoc tests, at a 100 ms ISI the stronger prepulse induced significantly less PPI in homozygous knockout pups than in siblings; this reduction was also significant when mutants and siblings with similar startle reactivity were compared (Fig. 5h). Thus Gsx1 is required for normal PPI in mice suggesting that it has a conserved functional role in the development of startle modulating neurons across vertebrates. Gsx1 likely has additional conserved functions in neuronal circuit formation because in a novel open field test, mutant mice showed reduced mobility (Fig. 5i) but during movement events, moved 45% more quickly than siblings (Fig. 5j) similar to the motility phenotype of fish after ablation of Gsx1 neurons.
Figure 5. Gsx1 is required for PPI in mammals.

(a) Schematic of E13.5 mouse embryonic brain showing coronal planes of section through the brainstem for RNA in situ hybridization with Gsx1 probe in (b–e).
(b) Dorso-lateral expression domains of Gsx1 at the ventricular zone of the neuroepithelium in caudal hindbrain (arrowheads).
(c) Coronal section at the level of the aqueduct (Aq) showing expression of Gsx1 in ventricular zone of the superior colliculus (scn) and inferior colliculus (icn).
(d) Same plane of section as in (c) through the 4th ventricle (4V) showing Gsx1 expression at the ventricular zone of the pons (arrowheads).
(e) Rostral brainstem section showing Gsx1 expression in the ventricular neuroepithelium of the superior colliculus (scn). Scale bars (b–e) 100 μm.
(f) Startle magnitude in Gsx1 knockout mice and littermates, normalized by body weight (Vmax/g). Knockouts (open circles, N=10) show similar startle sensitivity to wildtype (black, N=15) and heterozygotes (grey, N=14).
(g) PPI in Gsx1 knockout mice (N=13) compared to wildtype (N=17) and heterozygous siblings (N=33) at a 100 ms ISI at the indicated prepulse intensities. Repeated measures ANOVA F1,61 = 4.99, p = 0.029. Genotypes are indicated. * p < 0.01.
(h) PPI in subgroups of mice from (g) with similar startle magnitudes (not adjusted for body weight), using mice with a mean response magnitude of between 100 and 200 (Vmax). ANOVA for genotype F1,28 = 6.49, p = 0.017. Posthoc homogeneous subsets are indicated.
(i–j) Locomotor activity in novel open field. Fraction of time (5 minute trials) spent mobile (i) and average speed during mobile periods (j) for wildtype (N=23), heterozygous (N=31) and homozygous Gsx1 knockout mice (N=16). # p < 0.05. * p < 0.01.
Discussion
Our findings demonstrate that neurons required for PPI show developmental expression of the homeodomain transcription factor Gsx1. Ablation of Gsx1 expressing neurons produced an ISI-dependent deficit in PPI in zebrafish, and Gsx1 knockout mice exhibited a robust reduction in PPI. Genetic manipulations offer improved cell type specificity over methods based on lesions or pharmacology for suppressing cell function. However deficits produced by ablation or gene knockout might perturb neuronal development or activate compensatory mechanisms. In our experiments this is unlikely, because optogenetic silencing of Gsx1 expressing neurons produced a reduction in PPI indicating that these neurons are directly required for normal PPI. Gsx1 has a dynamic pattern of expression during neuronal development, with a prominent early stripe of expression observed in the dorsal hindbrain. We found that Gsx1 hindbrain neurons are primarily glutamatergic and form synapses in close apposition to neurons which initiate startle responses, the Mauthner cells. Together, the acute requirement for Gsx1 neurons during PPI and the fact that they likely directly connect to central startle initiating neurons strongly argues that these neurons are an intrinsic part of the neuronal mechanism for inhibition of the startle response during PPI.
Screening zebrafish to identify components of functional neuronal circuits
To identify neurons required for PPI, we established a novel genetic screening method based on ablation of neurons in randomly generated enhancer trap lines which frequently report the expression of nearby genes. These lines can be rapidly generated, are easily mapped and provide a tool for visualizing and manipulating the targeted population of neurons 67. Mutagenesis screens in zebrafish have been previously used to isolate gene mutations which perturb behavior 68–69. However, it is challenging to subsequently map the underlying genetic mutation and analyze how it disrupts neural function. In invertebrates, recent studies have instead screened libraries of transgenic animals with specific patterns of reporter gene expression. The reporter is used to inactivate neurons allowing their contribution to behavior to be assessed. Like mutagenesis screens, such screens are ‘unbiased’, and therefore able to identify components of a neural pathway without a priori knowledge. This approach also recognizes that neurons rather than proteins are the fundamental unit of circuits 70.
Because vertebrates share a fundamental brain architecture, insights from studies in fish may help to elucidate circuits operating in the mammalian brain. PPI in fish and mammals is behaviorally similar and susceptible to disruption through similar pharmacological agents; however we recognize that differences in the underlying neuronal circuitry will likely exist. Neurons in the hindbrain are patterned according to a dorso-ventral transcriptional code similar to that which acts in the spinal cord 29, 71. Spatially, in fish these transcription factors are expressed in distinct mediolateral domains, while in mice, progenitor domains are organized in dorsoventral stripes. The dorso-medial expression of Gsx1 in zebrafish caudal hindbrain is therefore similar in position to expression of Gsx1 at the dorsal ventricular zone of the medulla. Thus after finding that neurons with developmental expression of Gsx1 are required for PPI in zebrafish, and noting the similar expression pattern of Gsx1 during neural development in fish and mouse, we tested whether Gsx1 is also required for PPI in mice. The PPI phenotype of Gsx1 neuron ablated zebrafish was recapitulated in Gsx1 knockout mice, however mice, unlike fish, did not show elevated startle sensitivity. This may reflect differences in circuits for startle control between species, but also could be due to the distinction between the effect of removing neurons from a circuit through ablation or silencing, and perturbing neuronal development through gene disruption. However, our finding that in both cases, PPI was disrupted supports the idea that Gsx1 plays a conserved role in specifying neurons that are a key part of the neuronal circuitry for sensorimotor gating.
Circuit mechanisms for PPI
In mammals, startle responses are triggered by giant reticulospinal neurons of the pontine nucleus caudalis (PNc) which receive short latency acoustic input and make monosynaptic inputs to motor neurons in the spinal cord 19, 72. PNc neurons show reduced electrical activity during PPI trials but the precise identity of neurons which regulate PNc neuron activity during PPI remains unclear 73–74. Pharmacological and lesion experiments suggest that multiple neuronal mechanisms mediate PPI at different ISIs 16, 75. PPI is intact after complete transection of structures anterior to the midbrain indicating that core circuitry for PPI resides in the brainstem 76. Lesions in several brainstem regions disrupt PPI, including in the inferior colliculus, the superior colliculus, the substantia nigra pars reticulata, the laterodorsal tegmental nucleus (LDTg) and the pedunculopontine tegmental nucleus (PPTg) 61–62, 64–65, 77. Intriguingly, we observed Gsx1 expression at the ventricular zones underlying the inferior and superior colliculi, and in a region adjacent to a progenitor domain for LDTg and PPTg neurons. Tracing experiments show that PPTg neurons project to the PNc 78. PPTg lesions reduce PPI at an ISI of between 100 and 500 ms and may also increase baseline startle amplitude 62, 75. In recordings from rat brain slices, stimulation of the PPTg inhibits the response of giant reticulospinal neurons in the PnC to auditory input 16. This effect can be suppressed by the application of cholinergic antagonists in the PNc, consistent with the large population of cholinergic neurons in the PPTg. Whether the PPTg directly modulates PNc reticulospinal neurons (or their presynaptic input), or acts via local interneurons in the PNc is unclear. Moreover PPTg stimulation does not inhibit PNc responses to auditory stimulation for all ISIs, and cholinergic antagonists only suppress this effect for long ISIs, indicating that the PPTg pathway and/or cholinergic signaling represents only part of the mechanism for PPI. Although most work has focused on the function of cholinergic PPTg neurons, the PPTg contains large intermingled groups of glutamatergic and GABAergic neurons which could also regulate the PNc 79–80.
Gsx1 neurons in the zebrafish hindbrain are primarily glutamatergic. There are several mechanisms by which glutamatergic neurons could regulate Mauthner cell responsiveness. We observed strong apposition of presynaptic elements from Gsx1 neurons with Maguk immunofluorescence on the distal part of the Mauthner cell lateral dendrite. This region receives input from the acoustic nerve leading to the possibility that Gsx1 neurons presynaptically regulate acoustic input to the Mauthner cell 81. Our pharmacological data suggests an important role for NMDA receptors in regulating PPI. As presynaptic NMDA receptors negatively regulate transmitter release from primary sensory neurons in rat, one possibility is that Gsx1 neurons regulate acoustic input to the Mauthner cell through presynaptic NMDA receptors 82. Alternatively Gsx1 neurons may directly inhibit the Mauthner cell through metabotropic glutamate receptors. Many Gsx1 presynaptic puncta on the Mauthner cell did not colocalize with immunostaining for Maguk, which is generally used to delineate ionotropic glutamatergic synapses 83. These synapses may therefore represent metabotropic glutamate receptors which trigger postsynaptic inhibition of the Mauthner cell through secondary messenger cascades. In addition, it should be noted that while most hindbrain y252 neurons were glutamatergic, a small population co-localized with a glycinergic marker. During neural development in fish, many glycinergic neurons co-express GABA 46 and Gsx1 has an established role in specifying both glutamatergic and GABAergic neurons in the spinal cord 56–57. Our data thus does not exclude the possibility that y252 neurons mediate PPI through direct inhibitory signaling to the Mauthner cell.
Finally, Gsx1 neurons may act indirectly by regulating activity in interneurons which regulate Mauthner excitability. One class of candidate neurons are the Passive Hyperpolarizing Potential neurons which regulate the Mauthner cell firing threshold 84. Indeed electrophysiological recordings have demonstrated that PPI in fish is in part mediated by decreased excitability of the Mauthner cell. Following the prepulse, the Mauthner cell shows shunting inhibition and loss of a non-linear current response that normally drives the membrane toward its firing threshold 21–22. However these mechanisms act on a shorter timescale than behavioral PPI indicating that additional mechanisms must be present. Because ablation produced increased startle responsiveness, increased short ISI PPI and decreased long ISI PPI, Gsx1 neurons may regulate startle responsiveness using some or all of these mechanisms for different behavioral functions.
Neurodevelopmental defects in psychiatric disorders
Many psychiatric disorders including schizophrenia, attention deficit hyperactivity disorder, and autism spectrum disorders are believed to be caused in part by abnormalities of brain development. There is converging evidence that non-specific disruptions in developmental pathways underlie shared aspects of these diseases 1. Several lines of evidence support the idea that schizophrenia has a strong neurodevelopmental origin, including early childhood cognitive and motor impairments and increased risk associated with pre and peri-natal events 2, 85–86. However specific neurodevelopmental consequences remain poorly understand in part because neuroanatomical findings in schizophrenia have been inconsistent and because progressive changes during the course of the disease make it difficult to identify developmental abnormalities 87. Nevertheless deficits have been found repeatedly in cortical interneurons which regulate the balance of excitation-inhibition suggesting that perturbations in interneuron generation during fetal development predispose the brain to psychosis 60.
Mouse studies have revealed roles for Gsx1 and its homolog Gsx2 in regulating proliferation and differentiation of neuronal progenitors in ventral telencephalic regions that generate forebrain interneurons 88, 89. Loss of Gsx1 exacerbates defects in neurogenesis of striatal and olfactory bulb neurons seen in Gsx2 mutants 89–90. Gross changes in cortical interneuron populations have not been described in double Gsx1/Gsx2 knockout mice 90, however increased Gsx1 expression profoundly disrupts progenitor domains from which these neurons are derived and suppresses interneuron migration into the cortical plate 58–59. In the spinal cord early expression of Gsx1 is involved in the specification of excitatory neurons, while late expression regulates the differentiation of both excitatory and inhibitory neuronal populations 56. Thus, perturbations in the timing of expression of Gsx1 during neural development may lead to subtle changes in the number or molecular identity of forebrain interneurons. Genome-wide association and linkage studies for several psychiatric disorders including schizophrenia 91–92, have suggested the presence of vulnerability loci near Gsx1 but no Gsx1 or Gsx2 mutations have been reported in psychiatric disorders. As knockout mice for these genes have severe postnatal defects ultimately leading to death, involvement of Gsx1 in neurological dysfunction would most likely be due to changes in the regulation of its expression 35, 88.
PPI defects and neuronal gating dysfunction in psychiatric disorders
Gating defects are posited to represent a fundamental abnormality in schizophrenia leading to cognitive processes being overwhelmed by sensory impressions and their associations 4. PPI is a robust measure of sensorimotor gating of the startle response and PPI deficits in schizophrenic patients correlate with the degree of thought disorder 93. However, the neurophysiologic basis for reduced PPI in schizophrenic patients remains unclear. It has been proposed that reduced PPI in schizophrenia is due to altered activity in a modulatory circuit comprising forebrain nuclei, acting on core PPI circuits which reside in brainstem regions 94. However PPI is impaired in unaffected relatives of schizophrenic patients, implying that PPI defects may not be secondary to psychosis but represent an independent phenotype 12, 95. It has been suggested that PPI defects in schizophrenia reflect impaired attention to the prepulse, however some (although not all) studies have found reduced PPI in schizophrenic patients at ISIs as short at 30 ms, too brief to allow attentional modulation 96–99. PPI defects may therefore not be a consequence of disrupted forebrain function, but rather represent an independent abnormality in brainstem circuitry for startle modulation.
An intriguing possibility is that a class of interneurons with common neurodevelopmental origins in brainstem and forebrain has a basic role gating information flow in multiple brain regions. Perturbations in these neurons would therefore produce independent disruptions in PPI and cortical information processing. Previous work has implicated Gsx1 as a key player in neuronal development in the telencephalon. Our study shows that neurons with developmental expression of Gsx1 have an essential role in neuronal circuitry that is involved in PPI, thus providing a molecular link between the specification of forebrain neurons and brainstem circuits which regulate the transmission of sensory information.
Supplementary Material
(a) Prepulse inhibition in zebrafish larvae. Red marker indicates the probability of short latency C-bend responses to the acoustic stimulus alone (no prepulse condition). Curves show response probability to the startle stimulus when preceded at the indicated interval by a prepulse of different magnitudes.
(b) Histogram showing the mean change in prepulse inhibition in ablated larvae compared to non-transgenic siblings for each of 46 transgenic Gal4 lines tested. ΔPPI = (%PPI in ablated larvae) - (%PPI in sibling controls), where %PPI is the percent reduction in startle responsiveness during PPI trials. For each line, the sibling controls were non-fluorescent fish that were treated with metronidazole and tested in parallel. Metronidazole treatment of Gal4; UAS:nfsB-mCherry expressing fish did not alter mean PPI (change in PPI from non-expressing controls −0.7% ± 12.3%, mean/std, p=0.70).
(a) Head orientation (relative to starting orientation, inset) during the larval startle response with annotation of kinematic measures used to compare response performance in control and ablated larvae in (b–e). C1 max AV refers to the maximal angular velocity attained during the initial C-bend.
(b–d) Movement kinematics during acoustic startle responses in control and y252 ablated larvae (N=109, 119 responses). No significant differences were observed.
(e) Reaction time (latency from stimulus to initiation of a Mauthner mediated startle response) is significantly reduced in y252 ablated larvae. * p < 0.001.
(f) Visual responsiveness to a 1 s long reduction in light intensity. Controls perform a stereotyped O-bend response within 800 ms of the light decrement. N = 5 groups controls, 3 groups ablated larvae. * p < 0.001.
(g) Baseline activity (0–20 min) and motor response to sustained darkness (20–40 min) in control (black) and ablated larvae (blue). Traces show mean distance moved (pixels) per 2.5 s bin. Illumination condition indicated on x-axis. N = 36 larvae each group.
(h) Quantification of the response in (g). Both groups show a large increased in activity at the peak of the response to darkness (grey, 23–25 min) compared to baseline activity (yellow, 0–20 min). * p < 0.001 paired t-test.
(a) Dorsal epifluorescent image of 5 dpf y252; UAS:Arch3 larva. Outline of larvae indicated. Dark patches are pigment obscuring view of fluorescence in brain.
(b) Semi-restrained preparation showing larvae head-embedded in agarose with the tail free to move.
(c) Latency histogram for responses in embedded larvae. Graph shows mean number of responses made at a given latency for individual larvae (grey, N=10) and mean of all larvae (black).
(d) Computational identification of tail movements. In each trial, three images of the tail were taken: 32 and 16 ms before and 16 ms after the stimulus. Images were thresholded, analyzed and classified as no response (left), swimming activity before the stimulus (middle) or a short-latency response (right).
(e) Confusion matrix showing accuracy of response classification by comparison of manual observation (‘Manual’) to computational analysis (‘Code’) for 256 trials.
(f) Response probability to a vibrational stimulus of increasing intensity. Both short latency (filled circles) and long latency (open circles) responses increase demonstrating that the system produces graded responses as for free swimming fish. N = 10 larvae. Main effect of intensity for SLC responses F4,36 = 16.97, p < 0.001; for LLC responses F4,36 = 14.67, p < 0.001.
(g) PPI in head-embedded larvae. Graph shows response probability to the prepulse alone, the pulse alone, or during prepulse-pulse (PPI) trials. N=6 larvae. * p = 0.011.
(a–e) RNA in situ hybridization for Gsx1 during neuronal development in zebrafish.
(a) 12 hpf. Hindbrain (bracket) and midbrain (asterisk).
(b) 20 hpf. Ventral forebrain (fb), midbrain (mb), and throughout the dorsal hindbrain (hb).
(c) 30 hpf. Dorsal spinal cord (sc), developing hypothalamus (hyp), tectum (tec).
(d) 48 hpf. Note loss of expression in spinal cord.
(e) 72 hpf. Expression in forebrain is weak. Cerebellum (cer). Eyes removed to visualize brain structures.
(f–h) Lateral confocal section of spinal cord in y252; UAS:Kaede; vGlut:DsRed. y252 neurons (green) are dorsally positioned and show partial overlap with the glutamatergic neuron marker (red, arrows) but many y252 neurons are not glutamatergic (arrowheads). Inset shows coronal projection.
(a), (c), (e) Expression of Atoh1 in the rhombic lip (arrows) in sagittal sections of an E10.5 stage embryo. Each panel is approximately 50 μm apart.
(b), (d), (f) Expression of Gsx1 in consecutive sagittal sections to (a, c, e) respectively of a stage E10.5 mouse embryo. Gsx1 expression does not extend into the Atoh1 expressing rhombic lip region (arrows). Scale bars a–f = 100 μm.
(g) Schematic of a dorsal view of the head of a E10.5 mouse embryo. Purple box shows area shown in a–f, dashed lines represent lateral to medial sections. Dark gray area is the rhombic lip (rl).
(a) Startle magnitude in wildtype P21 pups correlates with weight. Mean Vmax of responses to startle stimuli (110–120 dB) versus weight (N = 21). Measures show a moderate correlation which is significant (Pearson r = 0.426, p = 0.021).
(b) Gsx1 knockouts (P21) are significantly smaller than littermates (ANOVA main effect of genotype F2,33 = 76.06, p < 0.001; no effect of sex F1,33 = 0.196, p = 0.661). Post-hoc homogenous subsets are indicated. Numbers indicate N for each group.
(c) Latency to peak response (Tmax) to a 120 dB acoustic startle stimulus is not different in Gsx1 knockout mice (F2,36 = 0.046, p = 0.96). N for each group is shown.
(d) PPI in Gsx1 knockout mice (N=13), wildtype (N=17) and heterozygous siblings (N=33) at a 500 ms ISI. No significant differences between groups.
(e) PPI in 8 week old Gsx1 heterozygous mice (N=6) and wildtype siblings (N=5) at a 100 ms ISI. Repeated measures ANOVA F1,9 = 0.51, p = 0.49.
Acknowledgments
We thank Jennifer Strykowski for zebrafish support and Victoria Carter and Daniel Abebe for mouse support. We also thank Andres Buonanno for assistance and useful discussions. This work was supported by the Intramural Research Program of the NICHD.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry. 2012;17(12):1228–1238. doi: 10.1038/mp.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones P, Murray R, Rodgers B, Marmot M. Child developmental risk factors for adult schizophrenia in the British 1946 birth cohort. The Lancet. 1994;344(8934):1398–1402. doi: 10.1016/s0140-6736(94)90569-x. [DOI] [PubMed] [Google Scholar]
- 3.Seidman LJ, Giuliano AJ, Meyer EC, et al. Neuropsychology of the prodrome to psychosis in the napls consortium: Relationship to family history and conversion to psychosis. Archives of General Psychiatry. 2010;67(6):578–588. doi: 10.1001/archgenpsychiatry.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 5.Perry W, Braff DL. Information-Processing Deficits and Thought Disorder. Am J Psychiatry. 1994;15(1):363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- 6.Perry W, Braff DL. Information-processing deficits and thought disorder in schizophrenia. Am J Psychiatry. 1994;151(3):363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- 7.Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15 (4):339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman HS, Ison JR. Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychological Review. 1980;87(2):175–189. [PubMed] [Google Scholar]
- 9.Ziermans TB, Schothorst PF, Sprong M, Magnee MJ, van Engeland H, Kemner C. Reduced prepulse inhibition as an early vulnerability marker of the psychosis prodrome in adolescence. Schizophr Res. 2012;134(1):10–15. doi: 10.1016/j.schres.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Mowry BJ, Gratten J. The emerging spectrum of allelic variation in schizophrenia: current evidence and strategies for the identification and functional characterization of common and rare variants. Mol Psychiatry. 2013;18(1):38–52. doi: 10.1038/mp.2012.34. [DOI] [PubMed] [Google Scholar]
- 11.Quednow BB, Frommann I, Berning J, Kuhn KU, Maier W, Wagner M. Impaired sensorimotor gating of the acoustic startle response in the prodrome of schizophrenia. Biol Psychiatry. 2008;64(9):766–773. doi: 10.1016/j.biopsych.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 12.Cadenhead KS, Swerdlow NR, Shafer KM, Diaz M, Braff DL. Modulation of the startle response and startle laterality in relatives of schizophrenic patients and in subjects with schizotypal personality disorder: evidence of inhibitory deficits. Am J Psychiatry. 2000;157(10):1660–1668. doi: 10.1176/appi.ajp.157.10.1660. [DOI] [PubMed] [Google Scholar]
- 13.Swerdlow N, Weber M, Qu Y, Light G, Braff D. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199(3):331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fendt M, Li L, Yeomans JS. Brain stem circuits mediating prepulse inhibition of the startle reflex. Psychopharmacology (Berl) 2001;156(2–3):216–224. doi: 10.1007/s002130100794. [DOI] [PubMed] [Google Scholar]
- 15.Diederich K, Koch M. Role of the pedunculopontine tegmental nucleus in sensorimotor gating and reward-related behavior in rats. Psychopharmacology. 2005;179(2):402–408. doi: 10.1007/s00213-004-2052-y. [DOI] [PubMed] [Google Scholar]
- 16.Bosch D, Schmid S. Cholinergic mechanism underlying prepulse inhibition of the startle response in rats. Neuroscience. 2008;155(1):326–335. doi: 10.1016/j.neuroscience.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Nusbaum MP, Contreras D. Sensorimotor gating: startle submits to presynaptic inhibition. Curr Biol. 2004;14(6):R247–249. doi: 10.1016/j.cub.2004.02.059. [DOI] [PubMed] [Google Scholar]
- 18.Zottoli SJ, Newman BC, Rieff HI, Winters DC. Decrease in occurrence of fast startle responses after selective Mauthner cell ablation in goldfish (Carassius auratus) J Comp Physiol [A] 1999;184(2):207–218. doi: 10.1007/s003590050319. [DOI] [PubMed] [Google Scholar]
- 19.Koch M, Lingenhöhl K, Pilz PKD. Loss of the acoustic startle response following neurotoxic lesions of the caudal pontine reticular formation: Possible role of giant neurons. Neuroscience. 1992;49(3):617–625. doi: 10.1016/0306-4522(92)90231-p. [DOI] [PubMed] [Google Scholar]
- 20.Burgess HA, Granato M. Sensorimotor gating in larval zebrafish. J Neurosci. 2007;27(18):4984–4994. doi: 10.1523/JNEUROSCI.0615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neumeister H, Szabo TM, Preuss T. Behavioral and physiological characterization of sensorimotor gating in the goldfish startle response. J Neurophysiol. 2008;99(3):1493–1502. doi: 10.1152/jn.00959.2007. [DOI] [PubMed] [Google Scholar]
- 22.Medan V, Preuss T. Dopaminergic-induced changes in Mauthner cell excitability disrupt prepulse inhibition in the startle circuit of goldfish. J Neurophysiol. 2011;106(6):3195–3204. doi: 10.1152/jn.00644.2011. [DOI] [PubMed] [Google Scholar]
- 23.Bergeron SA, Hannan MC, Codore H, Fero K, Li G, Moak ZB, et al. Brain selective transgene expression in zebrafish using an NRSE derived motif. Frontiers in Neural Circuits. 2012;6:110. doi: 10.3389/fncir.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, Urasaki A, et al. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci USA. 2008;105(4):1255–1260. doi: 10.1073/pnas.0704963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods. 2006;3(11):917–922. doi: 10.1038/nmeth937. [DOI] [PubMed] [Google Scholar]
- 26.Davison JM, Akitake CM, Goll MG, Rhee JM, Gosse N, Baier H, et al. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev Biol. 2007;304(2):811–824. doi: 10.1016/j.ydbio.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess HA, Johnson SL, Granato M. Unidirectional startle responses and disrupted left-right co-ordination of motor behaviors in robo3 mutant zebrafish. Genes Brain Behav. 2009;8(5):500–511. doi: 10.1111/j.1601-183X.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akitake CM, Macurak M, Halpern ME, Goll MG. Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol. 2011;352(2):191–201. doi: 10.1016/j.ydbio.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinkhabwala A, Riley M, Koyama M, Monen J, Satou C, Kimura Y, et al. A structural and functional ground plan for neurons in the hindbrain of zebrafish. Proc Natl Acad Sci U S A. 2011;108(3):1164–1169. doi: 10.1073/pnas.1012185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, et al. Molecular and cellular approaches for diversifying and extending optogenetics. Cell. 2010;141(1):154–165. doi: 10.1016/j.cell.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokogawa T, Hannan MC, Burgess HA. The dorsal raphe modulates sensory responsiveness during arousal in zebrafish. The Journal of Neuroscience. 2012;32:15205–15215. doi: 10.1523/JNEUROSCI.1019-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463(7277):98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koga A, Cheah FS, Hamaguchi S, Yeo GH, Chong SS. Germline transgenesis of zebrafish using the medaka Tol1 transposon system. Dev Dyn. 2008;237(9):2466–2474. doi: 10.1002/dvdy.21688. [DOI] [PubMed] [Google Scholar]
- 34.Appelbaum L, Wang G, Yokogawa T, Skariah GM, Smith SJ, Mourrain P, et al. Circadian and Homeostatic Regulation of Structural Synaptic Plasticity in Hypocretin Neurons. Neuron. 2010;68(1):87–98. doi: 10.1016/j.neuron.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Zeitler PS, Valerius MT, Small K, Potter SS. Gsh-1, an orphan Hox gene, is required for normal pituitary development. EMBO J. 1996;15(4):714–724. [PMC free article] [PubMed] [Google Scholar]
- 36.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436(7048):221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 37.Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, Stainier DY. Conditional targeted cell ablation in zebrafish: a new tool for regeneration studies. Dev Dyn. 2007;236(4):1025–1035. doi: 10.1002/dvdy.21100. [DOI] [PubMed] [Google Scholar]
- 38.Pisharath H, Rhee JM, Swanson MA, Leach SD, Parsons MJ. Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mechanisms of Development. 2007;124(3):218–229. doi: 10.1016/j.mod.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Issa FA, O’Brien G, Kettunen P, Sagasti A, Glanzman DL, Papazian DM. Neural circuit activity in freely behaving zebrafish (Danio rerio) J Exp Biol. 2011;214(Pt 6):1028–1038. doi: 10.1242/jeb.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess HA, Granato M. Modulation of locomotor activity in larval zebrafish during light adaptation. J Exp Biol. 2007;210(14):2526–2539. doi: 10.1242/jeb.003939. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes AM, Fero K, Arrenberg AB, Bergeron SA, Driever W, Burgess HA. Deep Brain Photoreceptors Control Light-Seeking Behavior in Zebrafish Larvae. Curr Biol. 2012 doi: 10.1016/j.cub.2012.1008.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheesman SE, Eisen JS. gsh1 demarcates hypothalamus and intermediate spinal cord in zebrafish. Gene Expression Patterns. 2004;5(1):107–112. doi: 10.1016/j.modgep.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 43.FluoRender: An application of 2D image space methods for 3D and 4D confocal microscopy data visualization in neurobiology research. Proceedings of the Pacific Visualization Symposium (PacificVis), 2012; Feb. 28 2012–March 2 2012; IEEE; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes L, Zhang Z, Albert P, Zervas M, Ahn S. Timing of Sonic hedgehog and Gli1 expression segregates midbrain dopamine neurons. The Journal of Comparative Neurology. 2011;519(15):3001–3018. doi: 10.1002/cne.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayes L, Ralls S, Wang H, Ahn S. Duration of Shh signaling contributes to mDA neuron diversity. Dev Biol. 2013;374(1):115–126. doi: 10.1016/j.ydbio.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higashijima S, Mandel G, Fetcho JR. Distribution of prospective glutamatergic, glycinergic, and GABAergic neurons in embryonic and larval zebrafish. J Comp Neurol. 2004;480(1):1–18. doi: 10.1002/cne.20278. [DOI] [PubMed] [Google Scholar]
- 47.Koyama M, Kinkhabwala A, Satou C, Higashijima S, Fetcho J. Mapping a sensory-motor network onto a structural and functional ground plan in the hindbrain. Proc Natl Acad Sci U S A. 2011;108(3):1170–1175. doi: 10.1073/pnas.1012189108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer MP, Trimmer JS, Gilthorpe JD, Smith SJ. Characterization of zebrafish PSD-95 gene family members. J Neurobiol. 2005;63(2):91–105. doi: 10.1002/neu.20118. [DOI] [PubMed] [Google Scholar]
- 49.Kimmel CB, Sessions SK, Kimmel RJ. Morphogenesis and synaptogenesis of the zebrafish Mauthner neuron. J Comp Neurol. 1981;198(1):101–120. doi: 10.1002/cne.901980110. [DOI] [PubMed] [Google Scholar]
- 50.Moghaddam B, Javitt D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and its Implication for Treatment. Neuropsychopharmacology. 2012;37(1):4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geyer MA, Ellenbroek B. Animal behavior models of the mechanisms underlying antipsychotic atypicality. Prog Neuro-Psychopharmacol Biol Psychiatry. 2003;27 (7):1071–1079. doi: 10.1016/j.pnpbp.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Wolman MA, Jain RA, Liss L, Granato M. Chemical modulation of memory formation in larval zebrafish. Proc Natl Acad Sci USA. 2011;108(37):15468–15473. doi: 10.1073/pnas.1107156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balciunas D, Davidson AE, Sivasubbu S, Hermanson SB, Welle Z, Ekker SC. Enhancer trapping in zebrafish using the Sleeping Beauty transposon. BMC Genomics. 2004;5(1):62. doi: 10.1186/1471-2164-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valerius MT, Li H, Stock JL, Weinstein M, Kaur S, Singh G, et al. Gsh-1: a novel murine homeobox gene expressed in the central nervous system. Dev Dyn. 1995;203(3):337–351. doi: 10.1002/aja.1002030306. [DOI] [PubMed] [Google Scholar]
- 56.Mizuguchi R, Kriks S, Cordes R, Gossler A, Ma Q, Goulding M. Ascl1 and Gsh1/2 control inhibitory and excitatory cell fate in spinal sensory interneurons. Nat Neurosci. 2006;9(6):770–778. doi: 10.1038/nn1706. [DOI] [PubMed] [Google Scholar]
- 57.Satou C, Kimura Y, Hirata H, Suster ML, Kawakami K, Higashijima S. Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development. 2013;140(18):3927–3931. doi: 10.1242/dev.099531. [DOI] [PubMed] [Google Scholar]
- 58.Pei Z, Wang B, Chen G, Nagao M, Nakafuku M, Campbell K. Homeobox genes Gsx1 and Gsx2 differentially regulate telencephalic progenitor maturation. Proc Natl Acad Sci U S A. 2011;108(4):1675–1680. doi: 10.1073/pnas.1008824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang B, Long JE, Flandin P, Pla R, Waclaw RR, Campbell K, et al. Loss of Gsx1 and Gsx2 function rescues distinct phenotypes in Dlx1/2 mutants. J Comp Neurol. 2013;521(7):1561–1584. doi: 10.1002/cne.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13(2):107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 61.Jones CK, Shannon HE. Lesions of the laterodorsal tegmental nucleus disrupt prepulse inhibition of the acoustic startle reflex. Pharmacol Biochem Behav. 2004;78 (2):229–237. doi: 10.1016/j.pbb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Prepulse inhibition of acoustic startle in rats after lesions of the pedunculopontine tegmental nucleus. 1993. [Accessed Date Accessed 1993]. [DOI] [PubMed] [Google Scholar]
- 63.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48(1):17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 64.Li L, Korngut LM, Frost BJ, Beninger RJ. Prepulse inhibition following lesions of the inferior colliculus: prepulse intensity functions. Physiol Behav. 1998;65(1):133–139. doi: 10.1016/s0031-9384(98)00143-7. [DOI] [PubMed] [Google Scholar]
- 65.Fendt M, Koch M, Schnitzler HU. Sensorimotor gating deficit after lesions of the superior colliculus. Neuroreport. 1994;5(14):1725–1728. doi: 10.1097/00001756-199409080-00009. [DOI] [PubMed] [Google Scholar]
- 66.Nakamura K, Koyama Y, Takahashi K, Tsurui H, Xiu Y, Ohtsuji M, et al. Requirement of tryptophan hydroxylase during development for maturation of sensorimotor gating. J Mol Biol. 2006;363(2):345–354. doi: 10.1016/j.jmb.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 67.Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, Xiao T, et al. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods. 2007;4(4):323–326. doi: 10.1038/nmeth1033. [DOI] [PubMed] [Google Scholar]
- 68.Granato M, van Eeden FJ, Schach U, Trowe T, Brand M, Furutani-Seiki M, et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development. 1996;123:399–413. doi: 10.1242/dev.123.1.399. [DOI] [PubMed] [Google Scholar]
- 69.Neuhauss S, Biehlmaier O, Seeliger M, Das T, Kohler K, Harris W, et al. Genetic Disorders of Vision Revealed by a Behavioral Screen of 400 Essential Loci in Zebrafish. J Neurosci. 1999;19(19):8603–8615. doi: 10.1523/JNEUROSCI.19-19-08603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pfeiffer BD, Jenett A, Hammonds AS, Ngo TT, Misra S, Murphy C, et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci USA. 2008;105(28):9715–9720. doi: 10.1073/pnas.0803697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gray PA. Transcription Factors Define the Neuroanatomical Organization of the Medullary Reticular Formation. Frontiers in Neuroanatomy. 2013:7. doi: 10.3389/fnana.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leitner DS, Powers AS, Hoffman HS. The neural substrate of the startle response. Physiol Behav. 1980;25(2):291–297. doi: 10.1016/0031-9384(80)90219-x. [DOI] [PubMed] [Google Scholar]
- 73.Lingenhohl K, Friauf E. Giant neurons in the rat reticular formation: a sensorimotor interface in the elementary acoustic startle circuit? J Neurosci. 1994;14 (3 Pt 1):1176–1194. doi: 10.1523/JNEUROSCI.14-03-01176.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carlson S, Willott JF. Caudal Pontine Reticular Formation of C57BL/6J Mice: Responses to Startle Stimuli, Inhibition by Tones, and Plasticity. J Neurophysiol. 1998;79(5):2603–2614. doi: 10.1152/jn.1998.79.5.2603. [DOI] [PubMed] [Google Scholar]
- 75.Diederich K, Koch M. Role of the pedunculopontine tegmental nucleus in sensorimotor gating and reward-related behavior in rats. Psychopharmacology. 2005;179(2):402–408. doi: 10.1007/s00213-004-2052-y. [DOI] [PubMed] [Google Scholar]
- 76.Davis M, Gendelman PM. Plasticity of the acoustic startle response in the acutely decerebrate rat. Journal of Comparative and Physiological Psychology. 1977;91 (3):549–563. doi: 10.1037/h0077345. [DOI] [PubMed] [Google Scholar]
- 77.Koch M, Fendt M, Kretschmer BD. Role of the substantia nigra pars reticulata in sensorimotor gating, measured by prepulse inhibition of startle in rats. Behav Brain Res. 2000;117(1–2):153–162. doi: 10.1016/s0166-4328(00)00299-0. [DOI] [PubMed] [Google Scholar]
- 78.Koch M, Kungel M, Herbert H. Cholinergic neurons in the pedunculopontine tegmental nucleus are involved in the mediation of prepulse inhibition of the acoustic startle response in the rat. Exp Brain Res. 1993;97(1):71–82. doi: 10.1007/BF00228818. [DOI] [PubMed] [Google Scholar]
- 79.Clements JR, Grant S. Glutamate-like immunoreactivity in neurons of the laterodorsal tegmental and pedunculopontine nuclei in the rat. Neurosci Lett. 1990;120(1):70–73. doi: 10.1016/0304-3940(90)90170-e. [DOI] [PubMed] [Google Scholar]
- 80.Wang H-L, Morales M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci. 2009;29(2):340–358. doi: 10.1111/j.1460-9568.2008.06576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eaton R, Farley R, Kimmel C, Schabtach E. Functional development in the Mauthner cell system of embryos and larvae of the zebra fish. J Neurobiol. 1977;8(2):151–172. doi: 10.1002/neu.480080207. [DOI] [PubMed] [Google Scholar]
- 82.Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24(11):2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gardoni F, Marcello E, Di Luca M. Postsynaptic density-membrane associated guanylate kinase proteins (PSD-MAGUKs) and their role in CNS disorders. Neuroscience. 2009;158(1):324–333. doi: 10.1016/j.neuroscience.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 84.Weiss SA, Preuss T, Faber DS. A role of electrical inhibition in sensorimotor integration. Proc Natl Acad Sci U S A. 2008;105(46):18047–18052. doi: 10.1073/pnas.0806145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Devel Neurobio. 2012;72(10):1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clarke MC, Harley M, Cannon M. The Role of Obstetric Events in Schizophrenia. Schizophrenia Bulletin. 2006;32(1):3–8. doi: 10.1093/schbul/sbj028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewis DA, Levitt P. SCHIZOPHRENIA AS A DISORDER OF NEURODEVELOPMENT. Annu Rev Neurosci. 2002;25(1):409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- 88.Szucsik JC, Witte DP, Li H, Pixley SK, Small KM, Potter SS. Altered forebrain and hindbrain development in mice mutant for the Gsh-2 homeobox gene. Dev Biol. 1997;191(2):230–242. doi: 10.1006/dbio.1997.8733. [DOI] [PubMed] [Google Scholar]
- 89.Toresson H, Campbell K. A role for Gsh1 in the developing striatum and olfactory bulb of Gsh2 mutant mice. Development. 2001;128(23):4769–4780. doi: 10.1242/dev.128.23.4769. [DOI] [PubMed] [Google Scholar]
- 90.Yun K, Garel S, Fischman S, Rubenstein JL. Patterning of the lateral ganglionic eminence by the Gsh1 and Gsh2 homeobox genes regulates striatal and olfactory bulb histogenesis and the growth of axons through the basal ganglia. J Comp Neurol. 2003;461(2):151–165. doi: 10.1002/cne.10685. [DOI] [PubMed] [Google Scholar]
- 91.Sullivan PF, Lin D, Tzeng JY, van den Oord E, Perkins D, Stroup TS, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008;13(6):570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong KS, Won H-H, Cho E-Y, Jeun HO, Cho S-S, Lee Y-S, et al. Genome-widely significant evidence of linkage of schizophrenia to chromosomes 2p24. 3 and 6q27 in an SNP-Based analysis of Korean families. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009;150B(5):647–652. doi: 10.1002/ajmg.b.30884. [DOI] [PubMed] [Google Scholar]
- 93.Perry W, Geyer MA, Braff DL. Sensorimotor gating and thought disturbance measured in close temporal proximity in schizophrenic patients. Arch Gen Psychiatry. 1999;56(3):277–281. doi: 10.1001/archpsyc.56.3.277. [DOI] [PubMed] [Google Scholar]
- 94.Kumari V, Gray JA, Geyer MA, ffytche D, Soni W, Mitterschiffthaler MT, et al. Neural correlates of tactile prepulse inhibition: a functional MRI study in normal and schizophrenic subjects. Psychiatry Research: Neuroimaging. 2003;122(2):99–113. doi: 10.1016/s0925-4927(02)00123-3. [DOI] [PubMed] [Google Scholar]
- 95.Kumari V, Das M, Zachariah E, Ettinger U, Sharma T. Reduced prepulse inhibition in unaffected siblings of schizophrenia patients. Psychophysiology. 2005;42(5):588–594. doi: 10.1111/j.1469-8986.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 96.Dawson ME, Schell AM, Hazlett EA, Nuechterlein KH, Filion DL. On the clinical and cognitive meaning of impaired sensorimotor gating in schizophrenia. Psychiatry Research. 2000;96(3):187–197. doi: 10.1016/s0165-1781(00)00208-0. [DOI] [PubMed] [Google Scholar]
- 97.Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49(3):206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- 98.Kumari V, Soni W, Sharma T. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999;156(7):1046–1051. doi: 10.1176/ajp.156.7.1046. [DOI] [PubMed] [Google Scholar]
- 99.Hong LE, Summerfelt A, Wonodi I, Adami H, Buchanan RW, Thaker GK. Independent domains of inhibitory gating in schizophrenia and the effect of stimulus interval. Am J Psychiatry. 2007;164(1):61–65. doi: 10.1176/ajp.2007.164.1.61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Prepulse inhibition in zebrafish larvae. Red marker indicates the probability of short latency C-bend responses to the acoustic stimulus alone (no prepulse condition). Curves show response probability to the startle stimulus when preceded at the indicated interval by a prepulse of different magnitudes.
(b) Histogram showing the mean change in prepulse inhibition in ablated larvae compared to non-transgenic siblings for each of 46 transgenic Gal4 lines tested. ΔPPI = (%PPI in ablated larvae) - (%PPI in sibling controls), where %PPI is the percent reduction in startle responsiveness during PPI trials. For each line, the sibling controls were non-fluorescent fish that were treated with metronidazole and tested in parallel. Metronidazole treatment of Gal4; UAS:nfsB-mCherry expressing fish did not alter mean PPI (change in PPI from non-expressing controls −0.7% ± 12.3%, mean/std, p=0.70).
(a) Head orientation (relative to starting orientation, inset) during the larval startle response with annotation of kinematic measures used to compare response performance in control and ablated larvae in (b–e). C1 max AV refers to the maximal angular velocity attained during the initial C-bend.
(b–d) Movement kinematics during acoustic startle responses in control and y252 ablated larvae (N=109, 119 responses). No significant differences were observed.
(e) Reaction time (latency from stimulus to initiation of a Mauthner mediated startle response) is significantly reduced in y252 ablated larvae. * p < 0.001.
(f) Visual responsiveness to a 1 s long reduction in light intensity. Controls perform a stereotyped O-bend response within 800 ms of the light decrement. N = 5 groups controls, 3 groups ablated larvae. * p < 0.001.
(g) Baseline activity (0–20 min) and motor response to sustained darkness (20–40 min) in control (black) and ablated larvae (blue). Traces show mean distance moved (pixels) per 2.5 s bin. Illumination condition indicated on x-axis. N = 36 larvae each group.
(h) Quantification of the response in (g). Both groups show a large increased in activity at the peak of the response to darkness (grey, 23–25 min) compared to baseline activity (yellow, 0–20 min). * p < 0.001 paired t-test.
(a) Dorsal epifluorescent image of 5 dpf y252; UAS:Arch3 larva. Outline of larvae indicated. Dark patches are pigment obscuring view of fluorescence in brain.
(b) Semi-restrained preparation showing larvae head-embedded in agarose with the tail free to move.
(c) Latency histogram for responses in embedded larvae. Graph shows mean number of responses made at a given latency for individual larvae (grey, N=10) and mean of all larvae (black).
(d) Computational identification of tail movements. In each trial, three images of the tail were taken: 32 and 16 ms before and 16 ms after the stimulus. Images were thresholded, analyzed and classified as no response (left), swimming activity before the stimulus (middle) or a short-latency response (right).
(e) Confusion matrix showing accuracy of response classification by comparison of manual observation (‘Manual’) to computational analysis (‘Code’) for 256 trials.
(f) Response probability to a vibrational stimulus of increasing intensity. Both short latency (filled circles) and long latency (open circles) responses increase demonstrating that the system produces graded responses as for free swimming fish. N = 10 larvae. Main effect of intensity for SLC responses F4,36 = 16.97, p < 0.001; for LLC responses F4,36 = 14.67, p < 0.001.
(g) PPI in head-embedded larvae. Graph shows response probability to the prepulse alone, the pulse alone, or during prepulse-pulse (PPI) trials. N=6 larvae. * p = 0.011.
(a–e) RNA in situ hybridization for Gsx1 during neuronal development in zebrafish.
(a) 12 hpf. Hindbrain (bracket) and midbrain (asterisk).
(b) 20 hpf. Ventral forebrain (fb), midbrain (mb), and throughout the dorsal hindbrain (hb).
(c) 30 hpf. Dorsal spinal cord (sc), developing hypothalamus (hyp), tectum (tec).
(d) 48 hpf. Note loss of expression in spinal cord.
(e) 72 hpf. Expression in forebrain is weak. Cerebellum (cer). Eyes removed to visualize brain structures.
(f–h) Lateral confocal section of spinal cord in y252; UAS:Kaede; vGlut:DsRed. y252 neurons (green) are dorsally positioned and show partial overlap with the glutamatergic neuron marker (red, arrows) but many y252 neurons are not glutamatergic (arrowheads). Inset shows coronal projection.
(a), (c), (e) Expression of Atoh1 in the rhombic lip (arrows) in sagittal sections of an E10.5 stage embryo. Each panel is approximately 50 μm apart.
(b), (d), (f) Expression of Gsx1 in consecutive sagittal sections to (a, c, e) respectively of a stage E10.5 mouse embryo. Gsx1 expression does not extend into the Atoh1 expressing rhombic lip region (arrows). Scale bars a–f = 100 μm.
(g) Schematic of a dorsal view of the head of a E10.5 mouse embryo. Purple box shows area shown in a–f, dashed lines represent lateral to medial sections. Dark gray area is the rhombic lip (rl).
(a) Startle magnitude in wildtype P21 pups correlates with weight. Mean Vmax of responses to startle stimuli (110–120 dB) versus weight (N = 21). Measures show a moderate correlation which is significant (Pearson r = 0.426, p = 0.021).
(b) Gsx1 knockouts (P21) are significantly smaller than littermates (ANOVA main effect of genotype F2,33 = 76.06, p < 0.001; no effect of sex F1,33 = 0.196, p = 0.661). Post-hoc homogenous subsets are indicated. Numbers indicate N for each group.
(c) Latency to peak response (Tmax) to a 120 dB acoustic startle stimulus is not different in Gsx1 knockout mice (F2,36 = 0.046, p = 0.96). N for each group is shown.
(d) PPI in Gsx1 knockout mice (N=13), wildtype (N=17) and heterozygous siblings (N=33) at a 500 ms ISI. No significant differences between groups.
(e) PPI in 8 week old Gsx1 heterozygous mice (N=6) and wildtype siblings (N=5) at a 100 ms ISI. Repeated measures ANOVA F1,9 = 0.51, p = 0.49.